

Abstract
Ethidium bromide (EB) has been extensively used in the rat as a model of spinal cord demyelination. However, this lesion has not been addressed in the adult mouse, a model with unlimited genetic potential. Here we characterize behavioral function, inflammation, myelin status and axonal viability following bilateral injection of 0.20 mg/mL ethidium bromide or saline into the ventral white matter (VWM) of female C57Bl/6 mice. EB-induced VWM demyelination significantly reduced spared VWM and Basso Mouse Scale (BMS) scores persisting out to 2 months. Chronic hindlimb dysfunction was accompanied by a persistent inflammatory response (demonstrated by CD45+ immunofluorescence) and axonal loss (demonstrated by NF-M immunofluorescence and electron microscopy; EM). These cellular responses differ from the rat where inflammation resolves by 3–4 weeks and axon loss is minimal following EB demyelination. As these data suggest that EB-injection in the mouse spinal cord is a non-remyelinating lesion, we sought to ask whether wheel running could promote recovery by enhancing plasticity of local lumbar circuitry independent of remyelination. This did not occur as BMS and Treadscan® assessment revealed no significant effect of wheel running on recovery. However, this study defines the importance of descending ventral motor pathways to locomotor function in the mouse as VWM loss results in a chronic hindlimb deficit.
Keywords: Ethidium bromide, Demyelination, Spinal cord injury, Locomotion, Spared white matter, Inflammation
INTRODUCTION
EB1 has been used extensively in the rat as a demyelinating model to assess endogenous remyelination (Blakemore & Franklin, 2008), remyelination by engrafted cells (Tetzlaff et al., 2011; Enzmann et al., 2006) and to identify white matter tracts responsible for locomotion (Loy et al., 2002a,b). Both oligodendrocyte and astrocyte loss are hallmarks within the epicenter of an EB lesion while axons remain unaffected (Blakemore, 2005). The mechanism of selective glial death has been suggested to occur through EB’s action as a minor-groove DNA intercalator (Blakemore, 1982; 1984). However, other evidence suggests that while EB does intercalate both chromosomal and mtDNA2, it only affects transcription of mtDNA (Hayakawa et al., 1979; Desjardins et al., 1985; Hayashi et al., 1990). Therefore, it is likely that EB injection into white matter compromises mtDNA transcription in all cells local to the lesion with neurons and endothelial cells being preferentially less sensitive than glia in rat models.
Past literature has reported the acute use of EB into the dorsal columns of the adult mouse, but the EB concentrations were low and no chronic time-points were used to assess remyelination status (Fushimi & Shirabe, 2002; 2004). Considering the potential of genetic mouse models in assessing mechanism(s) of remyelination, we set out to characterize the cellular and behavioral responses to EB injection into the adult mouse spinal cord. To understand how ventral motor pathways modulate/control hindlimb locomotion in the mouse, demyelination was targeted to the VLF3 and VC4, together referred to as VWM5. In the rat, multiple ventral and dorsal pathways contain descending information involved in locomotion and functional deficits are only observed when more than one pathway is lost (Jordan, 1986; Jordan, 1991; Loy et al., 2002a,b; Schucht et al., 2002; Noga et al., 1991; Steeves and Jordan, 1980). Here, we additionally asked whether the organization of ventral motor pathways in the mouse is similar to the rat.
MATERIALS AND METHODS
Animals
All animal procedures were performed in strict accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, 1996), and with the approval of the University of Louisville Institutional Animal Care and Use Committee. A total of 116 female C57Bl/6 mice 6–8 weeks of age were used for all experiments (Charles River; Wilmington, MA). For the initial injection parameter pilot experiment, each group contained 4 mice (0.15 mg/mL EB, 0.20 mg/mL EB, 0.25 mg/mL EB and saline). To characterize the effect of 0.20 mg/mL EB or saline, a total of 48 mice were used and allowed to recover for 2 weeks (n=11 EB, n=9 saline) or 8 weeks post-injection (n=16 EB, n=12 saline). A total of 10 mice were used for EM imaging (n=2/group; saline or 1, 2, 3, 4 or 6 weeks post 0.20 mg/mL EB).
Surgery
Mice were anaesthetized with 2.0% avertin (2,2,2-tribromoethenol in 1.25% 2-methyl-2-butanol in saline), treated with 1 cc sterile saline (s.c.) and lacrilube for the eyes. All surgery and post-op procedures were performed on a 37°C heating pad. Injections were performed using a 45° beveled glass micropipette (internal diameter ~25 μm) driven by a Parker II picospritzer (Pine Brook, NJ). After a laminectomy and durectomy at the T10 vertebral level, mice were placed in a stereotaxic device and two 0.25 μl boluses of either EB (at concentrations of 0.15 mg/mL, 0.20 mg/mL, 0.25 mg/mL EB in saline) or saline spaced 1 minute apart were injected into 2 locations separated by 1.5 mm rostro-caudally within the laminectomy. Injections were bilateral, approached at an 18° angle to a depth of 1.1 mm and centered 0.45 mm lateral from midline Following injections, the needle was left in place for 3 minutes to allow the EB or saline to diffuse. Following post-op suturing and care, mice were treated prophylactically with gentamycin and a second 1 cc bolus of sterile saline (s.c.) and placed in heated recovery cages with alpha-dry bedding. Buprenorphine (0.05 mg/kg) was administered twice daily for 48 hours and bladders were expressed as needed (up to 2–3 times/day). All mice experienced identical housing and recovery conditions prior to and 1 week following EB.
Characterization of behavioral and cellular responses to EB
Previous studies showed the medial VLF carries signals important for locomotion in the rat spinal cord (Loy et al., 2002a,b; Cao et al., 2009). Pilot studies revealed a steep does-response curve for axon sensitivity to EB in mouse VWM as 0.25 mg/mL EB completely destroy axons 2 weeks post-injection whereas spinal cords injected with 0.15 mg/mL display little to no demyelination (data not shown). 0.20 mg/mL EB consistently destroyed glia within the lesion epicenter while leaving axons apparently intact at 2 weeks (Fig. 1C). To confirm accuracy of the injections, serial cross sections throughout the lesion were stained with EC6 to visualize myelinated and demyelinated areas (Fig. 1B).
Figure 1.

(A–C) Longitudinal sections stained with Hoechst and GFAP to identify cell nuclei (A) and astrocytes (B), respectively, display GFAP immunoreactivity restricted to the perimeter of the lesion at 2 weeks post-0.20 mg/mL EB into the mouse VWM (C). Hoechst staining reveals a large number of nuclei within the lesion epicenter (A), which were later confirmed to be infiltrating inflammatory cells (Fig. 3). (D–G) EC stained cross sections at the level of injection: Saline (D) and 0.20 mg/mL EB −1 mm (E), at the epicenter (F) and +1 mm (G). (H–J) EM images of (H) naïve axons which display thick compact myelin sheaths and 0.20 mg/mL EB-injected VWM which reveal spared, demyelinated axons at 1 (I) and 2 (J) weeks post-injection. Scale bars in C and J = 1000 μm and 10 μm, respectively.
BMS7
The BMS (Basso et al., 2006; Engesser-Cesar et al., 2005) was used to assess hindlimb function at baseline, and weekly for 5 weeks following EB injection. Mice were scored by two raters (trained by Dr. Basso and colleagues at the Ohio State University) blinded to the experimental condition.
Treadscan®
We utilized the Treadscan® system (Clever Sys Inc., Reston, VA) to assess gait, coordination and paw rotation during forced locomotion (Beare et al., 2009; Saraswat Ohri et al., 2011). Treadscan® related measures analyzed were: run speed, hindlimb rotation, RI8, total coupling and body rotation. Run speed was the maximum speed mice were able to maintain optimally coordinated locomotion. Hindlimb rotation and body rotation were calculated as absolute degrees from midline. Total coupling was calculated as follows: the percent deviation (absolute value) from naïve mice from homolateral, homologous and diagonal coupling measures summed together. Hindlimb rotation, body rotation and RI values were obtained manually. Treadscan® analysis was performed terminally, immediately prior to tissue processing.
Tissue Processing
Mice were sacrificed with an overdose of avertin (2.0%) and transcardially perfused first with cold PBS9 until the liver was clear and subsequently with cold 4% PFA10 in PBS. Spinal cords were dissected and allowed to post-fix for 24 hours in 4% PFA in PBS, then cryoprotected in 30% sucrose in PBS for 72 hours, both at 4°C. Spinal cords were blocked in tissue freezing medium (Triangle Biomedical Sciences, Durham, NC) and frozen at −20°C prior to sectioning with a Leica cryostat (Buffalo Grove, IL) at 20 μm. Sections were transferred to coated slides and stored at −20°C.
EC Staining
Tissue sections were stained with EC to distinguish myelinated from unmyelinated spinal cord axons. The percentage of SWM11 surface area from an entire cross-section at the level of injection from EB injected mice relative to saline injected control mice was calculated as described previously (Magnuson et al., 2005; Cao et al., 2010; Saraswat Ohri et al., 2011). Brightfield images were captured with a Nikon Eclipse TE 300 inverted microscope and a spot CCD camera. White matter was traced with Nikon Elements software and epicenters were defined as the segment(s) with the least SWM. Percent SWM was calculated by comparing these numbers to identically processed control spinal cord at the identical spinal level. For ventral SWM analysis, ROIs12 were defined as the ventral half of the cord with the equatorial line centered on the central canal (Fig. 2B).
Figure 2.

Functional and histological consequences of EB lesions. (A) BMS scores reveal a significant hindlimb dysfunction which persists chronically following injection of 0.20 mg/mL EB into the mouse VWM (saline n=9; 0.20 mg/mL EB n=11). (B,C) A positive correlation (C;R2=0.2591; p<0.05; n=19) exists when terminal BMS scores were plotted against ventral spared VWM (B) highlighting the importance of ventral white matter to hindlimb function following injection of 0.20 mg/mL EB (C). Data in A are means ± SD; *p < 0.05, **p < 0.01.
IHC13
To characterize the cellular effect of EB or saline injections following 2 or 8 weeks of recovery, animals were sacrificed, perfused, fixed and spinal cord tissue processed as described above. To determine the extent of inflammation within the lesion epicenter, IHC against the common leukocyte antigen CD45 was used at 2 weeks (n=11 EB, n=9 saline) and 8 weeks (n=9 EB, n=8 saline). Hoechst™ was used to identify nuclei. To determine the epicenter ROI on IHC sections, an adjacent EC stained section (<100 μM away) was superimposed over the IHC section in Photoshop™ and made partially transparent so the EC epicenter ROI could be traced over the appropriate area. To determine the effect of 0.20 mg/mL EB on axonal patency, sections were stained with primary antibodies against NF-M to assess axon number and diameter within the epicenter. Staining protocols were as follows: tissue sections were blocked with 5% BSA14, 10% NDS15, and 0.1% Triton-X-100 in TBS16 for 1 h at room temperature. Primary polyclonal antibodies NF-M17 (rabbit, 1:250, Millipore; Billerica, MA), GFAP18 (rabbit, 1:500, Dako; Carpinteria, CA), CD45 (rat, 1:500, Millipore) and MBP19 (rat, 1:250, Millipore) were incubated for 24 hrs at 4°C with 5% BSA, 5% NDS, and 0.1% Triton-X-100 in TBS. Sections were then washed at room temperature (x 3) with TBS and incubated with fluorescein (FITC; 1:200) and rhodamine (TRITC; 1:200)-conjugated F(ab′)2 secondary antibodies (donkey, Jackson ImmunoResearch; West Grove, PA) for 1 h at room temperature. Species-specific IgG isotype controls were used to account for any non-specific binding or other cellular protein interactions. IHC imaging was done on a Nikon Eclipse TE 300 inverted microscope with a spot CCD camera. Exposure times remained identical across all images. To define the EB lesion epicenter ROI on IHC sections, an adjacent EC stained section (<100 μM away) was superimposed over the IHC section in PhotoshopTM and made partially transparent so that both the EC and IHC images could be visualized simultaneously to accurately line up the sections. Using the EC section as a guide, epicenter ROIs were traced and ROIs saved. Finally, the traced ROI file was opened along with its respective IHC image to allow for area and intensity analysis. A subset of mice at the 8 week time-point were sacrificed and total RNA from the epicenter collected (n=7 EB, n=4 saline).
EM20
Tissue was fixed in buffered 2% glutaraldehyde followed by 1% buffered osmium oxide, dehydrated in ethanol and embedded in LX 112 (Ladd Research Industries, Williston, VT). Appropriate areas were selected for examination and thin sectioned for EM. Thin sections were stained with uranyl acetate and lead citrate for viewing under a Philips CM-12 transmission electron.
qRT-PCR21
Total RNA was extracted from whole spinal cord epicenters of EB mice (n=7) and saline mice (n=4) at 8 weeks post injection with the RNeasy Mini-Kit (Qiagen; Valencia, CA) according to manufacturer’s protocol. Extracted RNA was quantified with a Thermo Scientific NanoDrop 2000 (Cole-Parmer, Vernon Hills, IL) and cDNA was synthesized with 1 μg of total RNA (RT2 First-Strand Kit, SA Biosciences; Valencia, CA) and used as a template on an ABI 7900HT real-time PCR instrument (Applied Biosystems; Foster City, CA) using a RT2Sybr® Green/Rox qPCR Mastermix (SA Biosciences) in technical replicates of 4. Target and reference gene amplification was performed separately using RT2 qPCR Primer Assays (Qiagen) as follows: Fn122 (Mm.193099) and Gapdh23 (Mm.343110). The RNA levels were quantified using the ΔΔCT method. Expression values obtained Fn1 primer assays were normalized to Gapdh expression values. Transcript levels are expressed as fold change respective to saline injected controls.
RW24
Spontaneous wheel running has been demonstrated to improve outcome following contusive thoracic SCI25 (Engesser-Cesar et al., 2005; 2007). To assess potential segmental lumbar plasticity, EB-injected mice were allowed free access to a running wheel for 5 weeks post-EB. Mice were initially exposed to the wheel for 3 days and returned to non-RW cages for 1 week prior to baseline testing to allow any pre-lesion treatment effects from the RW to dissipate. All EB-injected mice received identical post-operative care and housing conditions for 1 week of recovery at which point they were randomly assigned to one of 3 groups: EB with a RW provided ad libitum (EB+RW; n=12), EB no RW single housed (EB single; n=9) and EB no RW multiple housed (EB multiple; n=10). The EB single group was included to control for potential effects of housing condition on recovery as EB+RW mice were individually housed. Saline treated mice were multiple housed throughout the experiment (n=7). Wheel turns were magnetically monitored and a digital counter recorded data using a VitalView acquisition system and ER4000 Transponders (MiniMitter; Bend, OR) and data exported to a spreadsheet where total distance per unit time per mouse were calculated. The inside of the wheels were modified with clear vinyl (Home Depot) to provide a flat running surface and attached with two bolts. This modification is necessary to ensure that the rungs on the wheel do not exacerbate post-injury dysfunction (Engesser-Cesar et al., 2005).
Statistics
Left-right side differences were analyzed using paired t-tests where appropriate before proceeding with further statistical analyses with the right-left mean values. A repeated measures ANOVA with the group factor on averaged right-left values was employed for BMS and modified BMS subscore data across time. All significant group effects were followed by Tukey HSD post-hoc t-tests. For all other statistical analyses, a one-way ANOVA was used followed by Tukey HSD post-hoc t-tests where appropriate. Correlations were calculated with Pearson’s coefficient.
RESULTS
EB Dose Response
At 2 weeks post injection, EC staining of longitudinal sections revealed that 0.20 mg/mL EB was the optimal dose for demyelination. The 0.15 mg/mL EB dose exhibited little to no demyelination and the 0.25 mg/mL dose severely damaged axons (data not shown). GFAP immunoreactivity at 2 weeks is restricted to the perimeter of the lesion (Figs. 1A–C) while EC stained cross sections confirmed the accuracy of the injections into the VWM (Figs. 1D–G). EM images reveal demyelinated axons which appear intact 2 weeks post-injection (Figs. 1H–J).
Behavioral response to 0.20 mg/mL EB injection into the adult mouse VWM
Bilateral injections of 0.20 mg/mL EB into the mouse VWM induces a chronic hindlimb deficit observed as a significant reduction in BMS scores compared to controls (Fig. 2A). At 1 week post-EB, scores centered around 4 to 5, indicating the ability to plantar step with little to no coordination. This deficit recovered slightly until week 3 when BMS scores plateaued at approximately 6, indicating hindlimb coordination during overground locomotion. Subscores of all mice achieving a BMS score of 5 or above were also significantly lower in EB-injected mice as compared to controls (data not shown). EC staining revealed that at 2 weeks and 8 weeks post-EB, SWM at the lesion epicenter was 28.00% and 24.12% of saline injected mice, respectively. Terminal BMS scores plotted against SWM (% of saline) at the epicenter for animals receiving 0.15 (n=6), 0.20 (n=11) or 0.25 mg/mL (n=5) EB showed a positive correlation (R2=0.7674; p< 0.001, n=22). However, BMS scores and total SWM for the animals that received 0.2 mg/ml EB showed no correlation due to the narrow range of BMS scores represented. Previous work in the rat has shown that demyelination within the VWM (VLF+VC) induces a chronic hindlimb impairment despite recovery of the ability to consistently weight support whereas complete loss of ventral white matter results in flaccid paralysis of the hindlimbs (Loy et al., 2002a,b). To specifically assess the role of ventral white matter in the mouse (Fig. 2B), regression analyses of all 0.20 mg/mL EB injected mice revealed a significant positive correlation between terminal BMS scores and spared VWM (R2 = 0.2521, p < 0.05, n=19; Fig. 2C).
Inflammatory responses within the EB-lesioned VWM
At 8 weeks post-injection, there was a significant increase in epicenter inflammatory cells as compared to saline, suggesting that EB induces a chronic inflammatory response that remains unresolved for at least 2 months (Figs. 3A–C). This was accompanied by a significant 4.2-fold increase in epicenter Fn1 mRNA transcript levels at 8 weeks as compared to saline injected VWM (Fig. 3D).
Figure 3.

Chronic inflammatory response following EB lesions. (A–C) There was a significant increase in CD45+ expression within the EB-injected epicenter (A) as compared to saline (B) suggesting that EB induces a chronic inflammatory response that persists for at least 2 months post-injection (saline n=8, 0.20 mg/mL EB n=9; C). (D) This was accompanied by a significant 4.29-fold increase in epicenter Fn1 mRNA levels (normalized to Gapdh) at 8 weeks as compared to saline injected VWM (saline n=4, 0.20 mg/mL EB n=7). Adjacent EC images (upper right corner of A and B) were used to select ROIs within the VWM for CD45 IHC (A and B). Data in C and D are mean ± SD; *p < 0.05; **p < 0.01. Scale bar in B = 50 μm
Chronic axonal patency after EB lesions in the VWM
NF-M immunoreactivity of cross-sections was performed to quantify axon number and diameter at the injury epicenter. At 8 weeks there was a significant reduction in the number of axons/mm2 within the VWM to approximately 36% of saline controls (Figs. 4A–C). The mean equivalent diameters of the remaining NF-M labeled axons were significantly larger in EB-injected epicenters suggesting a preferential sparing of larger diameter axons (Fig. 4D). However, ultrastructural analysis of the lesion epicenter revealed that surviving axons were severely swollen at 6 weeks post-EB suggesting that swelling, as opposed to preferential sparing of large diameter axons, is a more likely interpretation of why the remaining axons appear larger (Figs. 4E–G). These data suggest that in the mouse, remyelination does not occur as a majority of axons are lost with remaining axons appearing swollen.
Figure 4.

Chronic axonal integrity following VWM EB lesions. (A–C) NF-M expression at 8 weeks revealed a significant reduction in axons/mm2 within the VWM to approximately 36% of saline controls (saline n=8, 0.20 mg/mL EB n=9). (D) Of the remaining NF-M labeled axons, mean equivalent diameters were significantly larger in EB-injected epicenters. (E–G) Ultrastructural analysis revealed that compared to (E) saline-injected epicenters surviving axons after EB were severely swollen and much of the remaining myelin disorganized at both (F) 3 and (G) 6 weeks post-EB, accounting for the significant increase in spared axon diameter observed in D. Data in C and D are mean ± SD; *p < 0.05. Scale bars in B and G = 50 μm and 10 μm, respectively.
Effect of spontaneous wheel running on hindlimb function post-EB
Both saline and EB-injected mice averaged approximately 2.4 miles/day on the running wheel and this was no different from the average daily distance run during the pre-injury habituation period which is similar to the 2.4–3.8 miles/day previously reported (Adlard et al., 2005). There were also no differences in weight gained/lost or SWM with running (data not shown). The BMS scores at 1 week post-injection for EB+RW mice were higher than either the EB single or multiple housed mice indicating that the EB+RW group received a less severe lesion (Fig. 5A). Therefore, we determined the change in BMS scores between week 1 and week 5 (BMS difference score), which revealed no significant difference between groups (Fig. 5B). Moreover, Treadscan® analysis revealed no significant RW-induced recovery. These data suggest that spontaneous wheel running does not improve functional outcome following chronic VWM demyelination in the mouse.
Figure 5.
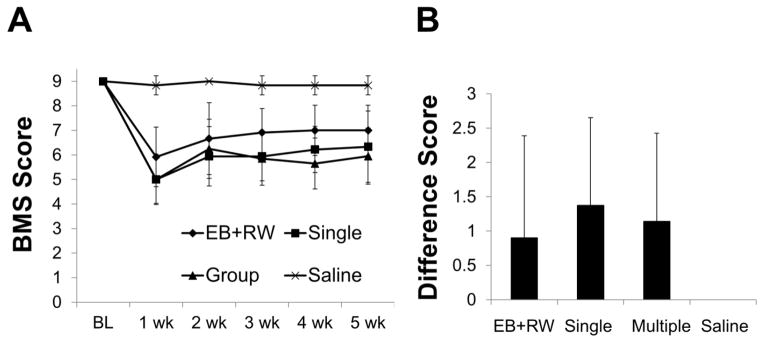
Neither spontaneous wheel running nor group housing improves hindlimb function following injections of 0.20 mg/mL EB (). (A) EB+RW mice displayed less severe deficits prior to assignment to experimental condition (i.e. EB+RW, single or multiple housed). (B) Difference scores (absolute value of week 1 BMS score – week 5 BMS score) similarly revealed no hindlimb recovery between EB-injected groups (B). Data are means ± SD, EB+RW, n=12; EB single, n=9; EB multiple, n=10; Saline, n=7.
DISCUSSION
EB-injection into the adult mouse VWM resulted in persistent inflammation and demyelination concomitant with axonal loss resulting in hindlimb dysfunction. RW-induced hindlimb recovery was not observed. Although the inflammatory response and axonal loss differ in the rat and mouse, the mouse is similar to other mammals in the functional redundancy of descending locomotor pathways.
EB lesions in the mouse exhibit a much steeper dose-response curve as there is only a 0.1 mg/mL difference in concentration between little to no demyelination (0.15 mg/mL) and complete axon loss (0.25 mg/mL), whereas the rat exhibits a much wider range of concentrations between little demyelination and axon loss (Yajima and Suzuki, 1979; Graca and Blakemore, 1986; Crang and Blakemore, 1991; Penderis et al., 2003). In the rat, EB demyelination of the VLF induced no behavioral deficits detectable using the BBB26 Scale (Loy et al., 2002a). However, when VLF demyelination was combined with either dorsal column/corticospinal tract, ventral column or dorsolateral funiculus demyelination, mean BBB scores dropped to 15.9, 12.7 and 11.1, respectively. Consistent with those data, in the mouse, a significant reduction in BMS scores was observed 1 week post VWM EB-injection (VLF+VC) and this deficit persisted out to at least 2 months (Fig. 2A). Talbott et al. (2005) found that the epicenter of a rat VLF EB-lesion exhibited a transient inflammatory response that was completely resolved by 1 month. In contrast, the current study found increased CD45 immunofluorescence (Figs. 3A–C), increased Fn1 mRNA levels (Fig. 3D) and increased CD11b immunofluorescence (data not shown) at the EB lesion epicenter in mice out to at least 2 months suggesting a persistent inflammatory response. This Is a phenotypic characteristic of fibrocytes, blood-borne immune cells which express CD45, CD11b and FN1 while lacking other lymphocyte specific markers (Sroga et al., 2003; Chesney and Bucala, 1997). Although differences in the inflammatory response between rats and mice have been previously suggested following spinal cord contusion (Sroga et al., 2003), this is the first evidence of this following EB-injection.
In the rat, there is no evidence of axon loss following dorsal column EB-injection at concentrations appropriate for demyelination, despite repeated episodes of focal demyelination (Franklin and Blakemore, 2008; Penderis et al., 2003). However, NF-M immunofluorescence, as well as ultrastructural analysis of epicenter cross sections confirms that EB induces axon loss in the mouse VWM (Fig. 3A) and without remaining axons, remyelination ultimately fails. Axon loss is due to one of two possibilities. The first is that EB directly affected axons resulting in axonal death and subsequent inflammation independent of EB-induced oligodendrocyte loss. The second possibility is that EB-induced glial loss resulted in inflammation which, in combination with loss of glial-derived axonal support, resulted in axonal loss. It is likely that both possibilities contribute to axonal loss in a reciprocal manner, but these data do not provide insight into which situation arises first. A better understanding of why axonal loss occurs would aid in explaining why this phenomenon is not seen in rats following VWM demyelination. Taken together, our findings of a chronic inflammatory response, axon loss and the lack of a SWM difference between 2 and 8 weeks post-injection suggests that EB lesions in the mouse VWM are non-remyelinating. It cannot be determined if remyelination would have still failed without axon loss. However, this scenario is unlikely using this model considering the steep dose-response curve for EB in the mouse. Henceforth, establishing a demyelinating lesion which consistently eliminates glia without damaging axons may not be possible in the mouse using EB.
Engesser-Cesar et al. (2005) demonstrated that spontaneous wheel running resulted in significant improvements in BMS scores following a dorsal thoracic spinal contusion. Given the permanent loss of descending ventral motor pathways and its concomitant behavioral deficit in the mouse, we hypothesized that spontaneous wheel running might partially restore hindlimb function of EB-injected mice by enhancing plasticity of intrinsic lumbar circuitry independent of remyelination. This was not observed in either the BMS or Treadscan® which is more sensitive to milder injuries (Beare et al., 2009) suggesting differences in recovery between contusive SCI and VWM demyelination. The lack of additional improvement in the RW group may be due to inherent differences in the injury model (dorsal contusion vs ventral demyelination) and/or there may be a ceiling effect rendering RW training unable to provide recovery additional to the recovery provided by in-cage activity alone (Fouad et al., 2000; Kuerzi et al., 2010). If the latter explanation is correct, this suggests that the greater initial (i.e. 1 week) hindlimb function in EB lesioned mice allowed for high levels of in-cage activity regardless of RW status. Taken together, these data suggest two interpretations. The RW does not facilitate recovery in mice following EB-induced VWM demyelination. Alternatively, the RW is unable to improve hindlimb dysfunction in mice beyond what in-cage training alone achieves following ventral lesions which do not limit the initial capacity for extensive in-cage activity. A similar ceiling for in-cage training in the rat has been suggested with multiple additional training regimens (Fouad et al., 2000; Magnuson et al., 2009; Alluin et al., 2011; Kuerzi et al., 2010).
These data suggest that mice are similar to the rat and cat in the anatomical distribution and functional redundancy of descending locomotor pathways. There are two main polysynaptic pathways regulating the initiation of locomotion. The primary locomotor pathway originates in the MLR27, projects to the MedRF28 and subsequently descends through the VLF where it terminates at the spinal locomotor centers (Noga et al., 1991; Noga et al., 2003; Jordan et al., 2008). The second originates in the PLR29 and is suggested to integrate sensory trigeminal information from the propriospinal system with the spinal locomotor system (Noga et al., 1991). The PLR projects to the spinal locomotor centers either by converging with the MedRF-VLF pathway or via the DLF30 (Noga et al., 1991; Noga et al., 2003). The work by Loy et al. (2002a) also suggests a role for the VC in locomotion as combined VLF-VC demyelination induced a significant behavioral deficit in rats which was greater than that seen following VLF demyelination alone, potentially by disrupting long propriospinal interneurons. Those data are consistent with the VWM lesions described here which resulted in chronic behavioral deficits suggesting that multiple locomotor pathways have been lost. Moreover, once BMS scores plateaued at approximately week 3 (Fig. 2A), locomotion was mostly coordinated (as defined by BMS and Treadscan® analysis) suggesting the presence of secondary / tertiary locomotor pathways located within the dorsal and dorsolateral region of the spinal cord.
Therefore, as in the rat and cat, the mouse has functionally redundant locomotor pathways able to compensate when other locomotor pathways are lost. However, as more descending motor pathways are lost, compensation becomes limited and locomotor impairments are observed. This is not surprising considering the conservation of these pathways across mammals and in birds (Jordan et al., 2008; Noga et al., 1991; Loy et al., 2002a; Vilensky et al., 1992; Schucht et al., 2002; Sholomenko and Steeves, 1987) and supports the importance of ventral pathways to locomotion suggesting that at least some VWM must be spared for compensation to occur. To our knowledge, this is the first description of ventral locomotor pathway function in mice. Given that infarction to the ventral portion of the cord (ACS31) is far more common than infarction to the dorsal portion of the cord (PCS32) following human SCI (Fehlings et al., 2012; Fulk et al., 2007; Schneider et al., 2010), the functional deficits observed with this VWM lesion may better-reflect what is observed clinically. Moreover, recovery from PCS is common while ACS related deficits are chronic as patients experience little recovery (Fehlings et al., 2012; Kirshblum and O’Conner, 1998; McKinley et al., 2007). Although these data support the use of vental lesions in modeling human SCI, EB-induced VWM demyelination in the mouse may not be the most optimal model to achieve this.
Highlights.
Ethidium bromide (EB)-induced demyelination leads to chronic inflammation and axon loss in mice.
Ventral white matter (VWM) demyelination induces persistent hindlimb dysfunction in mice.
Wheel running does not restore behavioral deficit following VWM demyelination in mice.
Distribution and functional redundancy of ventral mouse locomotor pathways are similar to rat and cat.
Acknowledgments
We thank Darlene Burke for assistance with statistical analyses, Kariena Andres for animal care, Christine Yarberry for surgical support, Kim Cash for post-operative animal care, Johnny Morehouse for assistance with Treadscan® analysis and William DeVries and Minh Tran for assistance with tissue processing, histology and image quantification. Supported by NS054708, RR15576/GM103507, Norton Healthcare, Friends for Michael, Kentucky Spinal Cord and Head Injury Research Trust, and the Commonwealth of Kentucky Challenge for Excellence.
Footnotes
EB: ethidium bromide,
mtDNA: mitochondrial DNA,
VLF: ventrolateral funiculus,
VC: ventral column,
VWM: ventral white matter,
EC: iron eriochrome cyanine,
BMS: Basso mouse scale,
RI: regularity index,
PBS: phosphate buffered saline,
PFA: paraformaldehyde,
SWM: spared white matter,
ROI: regions of interest,
IHC: Immunohistochemistry,
BSA: bovine serum albumin,
NDS: normal donkey serum,
TBS: tris buffered saline,
NF-M: neurofilament M,
GFAP: glial fibrilary acidic protein,
MBP: myelin basic protein,
EM: electron microscopy,
qRT-PCR: quantitative real-time polymerase chain reaction,
Fn1: fibronectin 1,
Gapdh: glyceraldehyde 3-phosphate dehydrogenase,
RW: running wheel,
SCI: spinal cord injury,
BBB: Basso, Beattie, and Bresnahan,
MLR: mesencephalic locomotor region,
MedRF: pontomedullary reticular formation,
PLR: pontomedullary locomotor region,
DLF: dorsolateral funiculus,
ACS: anterior cord syndrome,
PCS: posterior cord syndrome
Authors declare no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adlard PA, Perreau VM, Cotman CW. The exercise-induced expression of BDNF within the hippocampus varies across life-span. Neurobiol Aging. 2005;26:511–520. doi: 10.1016/j.neurobiolaging.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Alluin O, Karimi-Abdolrezaee S, Delivet-Mongrain H, Leblond H, Fehlings MG, Rossignol S. Kinematic study of locomotor recovery after spinal cord clip compression injury in rats. J Neurotrauma. 2011;28:1963–1981. doi: 10.1089/neu.2011.1840. [DOI] [PubMed] [Google Scholar]
- 3.Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- 4.Beare JE, Morehouse JR, DeVries WH, Enzmann GU, Burke DA, Magnuson DS, Whittemore SR. Gait analysis in normal and spinal contused mice using the TreadScan system. J Neurotrauma. 2009;26:2045–2056. doi: 10.1089/neu.2009.0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blakemore WF. Ethidium bromide induced demyelination in the spinal cord of the cat. Neuropathol Appl Neurobiol. 1982;8:365–375. doi: 10.1111/j.1365-2990.1982.tb00305.x. [DOI] [PubMed] [Google Scholar]
- 6.Blakemore WF. The response of oligodendrocytes to chemical injury. Acta Neurol Scand Suppl. 1984;100:33–38. [PubMed] [Google Scholar]
- 7.Blakemore WF. The case for a central nervous system (CNS) origin for the Schwann cells that remyelinate CNS axons following concurrent loss of oligodendrocytes and astrocytes. Neuropathol Appl Neurobiol. 2005;31:1–10. doi: 10.1111/j.1365-2990.2005.00637.x. [DOI] [PubMed] [Google Scholar]
- 8.Blakemore WF, Franklin RJ. Remyelination in experimental models of toxin-induced demyelination. Curr Top Microbiol Immunol. 2008;318:193–212. doi: 10.1007/978-3-540-73677-6_8. [DOI] [PubMed] [Google Scholar]
- 9.Cao Q, He Q, Wang Y, Cheng X, Howard RM, Zhang Y, DeVries WH, Shields CB, Magnuson DS, Xu XM, Kim DH, Whittemore SR. Transplantation of ciliary neurotrophic factor-expressing adult oligodendrocyte precursor cells promotes remyelination and functional recovery after spinal cord injury. J Neurosci. 2010;30:2989–3001. doi: 10.1523/JNEUROSCI.3174-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chesney J, Bucala R. Peripheral blood fibrocytes: novel fibroblast-like cells that present antigen and mediate tissue repair. Biochem Soc Trans. 1997;25:520–524. doi: 10.1042/bst0250520. [DOI] [PubMed] [Google Scholar]
- 11.Crang AJ, Blakemore WF. Remyelination of demyelinated rat axons by transplanted mouse oligodendrocytes. Glia. 1991;4:305–313. doi: 10.1002/glia.440040308. [DOI] [PubMed] [Google Scholar]
- 12.Desjardins P, Frost E, Morais R. Ethidium bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts. Mol Cell Biol. 1985;5:1163–1169. doi: 10.1128/mcb.5.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engesser-Cesar C, Anderson AJ, Basso DM, Edgerton VR, Cotman CW. Voluntary wheel running improves recovery from a moderate spinal cord injury. J Neurotrauma. 2005;22:157–171. doi: 10.1089/neu.2005.22.157. [DOI] [PubMed] [Google Scholar]
- 14.Engesser-Cesar C, Ichiyama RM, Nefas AL, Hill MA, Edgerton VR, Cotman CW, Anderson AJ. Wheel running following spinal cord injury improves locomotor recovery and stimulates serotonergic fiber growth. Eur J Neurosci. 2007;25:1931–1939. doi: 10.1111/j.1460-9568.2007.05469.x. [DOI] [PubMed] [Google Scholar]
- 15.Enzmann GU, Benton RL, Talbott JF, Cao Q, Whittemore SR. Functional considerations of stem cell transplantation therapy for spinal cord repair. J Neurotrauma. 2006;23:479–495. doi: 10.1089/neu.2006.23.479. [DOI] [PubMed] [Google Scholar]
- 16.Fehlings MG, Vaccaro AR, Boakye M, Rossignol S, Ditunno J, Burns AS. Essentials in Spinal Cord Injury: Basic Research to Clinical Practice. Thieme Medical Publishers, Inc; New York, NY: 2012. pp. 70–79. [Google Scholar]
- 17.Fouad K, Metz GA, Merkler D, Dietz V, Schwab ME. Treadmill training in incomplete spinal cord injured rats. Behav Brain Res. 2000;115:107–113. doi: 10.1016/s0166-4328(00)00244-8. [DOI] [PubMed] [Google Scholar]
- 18.Fulk G, Schmidt T, Behrman A. Traumatic Spinal Cord Injury. In: O’Sullivan S, editor. Physical Rehabilitation. F.A. Davis; Philadelphia, PA: 2007. pp. 937–996. [Google Scholar]
- 19.Fushimi S, Shirabe T. The reaction of glial progenitor cells in remyelination following ethidium bromide-induced demyelination in the mouse spinal cord. Neuropathology. 2002;22:233–242. doi: 10.1046/j.1440-1789.2002.00459.x. [DOI] [PubMed] [Google Scholar]
- 20.Fushimi S, Shirabe T. Expression of insulin-like growth factors in remyelination following ethidium bromide-induced demyelination in the mouse spinal cord. Neuropathology. 2004;24:208–218. doi: 10.1111/j.1440-1789.2004.00561.x. [DOI] [PubMed] [Google Scholar]
- 21.Graca DL, Blakemore WF. Delayed remyelination in rat spinal cord following ethidium bromide injection. Neuropathol Appl Neurobiol. 1986;12:593–605. doi: 10.1111/j.1365-2990.1986.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 22.Hayakawa T, Noda M, Yasuda K, Yorifuji H, Taniguchi S, Miwa I, Sakura H, Terauchi Y, Hayashi J, Sharp GW, Kanazawa Y, Akanuma Y, Yazaki Y, Kadowaki T. Ethidium bromide-induced inhibition of mitochondrial gene transcription suppresses glucose-stimulated insulin release in the mouse pancreatic beta-cell line betaHC9. J Biol Chem. 1998;273:20300–20307. doi: 10.1074/jbc.273.32.20300. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi J, Tanaka M, Sato W, Ozawa T, Yonekawa H, Kagawa Y, Ohta S. Effects of ethidium bromide treatment of mouse cells on expression and assembly of nuclear-coded subunits of complexes involved in the oxidative phosphorylation. Biochem Biophys Res Commun. 1990;167:216–221. doi: 10.1016/0006-291x(90)91753-f. [DOI] [PubMed] [Google Scholar]
- 24.Hill RL, Zhang YP, Burke DA, Devries WH, Zhang Y, Magnuson DS, Whittemore SR, Shields CB. Anatomical and functional outcomes following a precise, graded, dorsal laceration spinal cord injury in C57BL/6 mice. J Neurotrauma. 2009;26:1–15. doi: 10.1089/neu.2008.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan L. Initiation of Locomotion from the Mammalian Brainstem. In: Grillner S, Stein PSG, Stuart DG, Forssberg H, Herman RM, editors. Neurobiology of Vertebrate Locomotion. Macmillan Press; London: 1986. pp. 21–37. [Google Scholar]
- 26.Jordan LM. Brainstem and Spinal Cord Mechanisms for the Initiation of Locomotion. In: Shimamura M, Grillner S, Edgerton VR, editors. Neurobiological Basis of Human Locomotion. Japan Scientific Series; Tokyo: 1991. pp. 3–20. [Google Scholar]
- 27.Kirshblum SC, O’Connor KC. Predicting neurologic recovery in traumatic cervical spinal cord injury. Arch Phys Med Rehabil. 1998;79:1456–1466. doi: 10.1016/s0003-9993(98)90244-1. [DOI] [PubMed] [Google Scholar]
- 28.Kuerzi J, Brown EH, Shum-Siu A, Siu A, Burke D, Morehouse J, Smith RR, Magnuson DS. Task-specificity vs. ceiling effect: step-training in shallow water after spinal cord injury. Exp Neurol. 2010;224:178–187. doi: 10.1016/j.expneurol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linden RD, Zhang YP, Burke DA, Hunt MA, Harpring JE, Shields CB. Magnetic motor evoked potential monitoring in the rat. J Neurosurg. 1999;91:205–210. doi: 10.3171/spi.1999.91.2.0205. [DOI] [PubMed] [Google Scholar]
- 30.Loy DN, Magnuson DS, Zhang YP, Onifer SM, Mills MD, Cao QL, Darnall JB, Fajardo LC, Burke DA, Whittemore SR. Functional redundancy of ventral spinal locomotor pathways. J Neurosci. 2002;22:315–323. doi: 10.1523/JNEUROSCI.22-01-00315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loy DN, Talbott JF, Onifer SM, Mills MD, Burke DA, Dennison JB, Fajardo LC, Magnuson DS, Whittemore SR. Both dorsal and ventral spinal cord pathways contribute to overground locomotion in the adult rat. Exp Neurol. 2002;177:575–580. doi: 10.1006/exnr.2002.7959. [DOI] [PubMed] [Google Scholar]
- 32.Magnuson DS, Lovett R, Coffee C, Gray R, Han Y, Zhang YP, Burke DA. Functional consequences of lumbar spinal cord contusion injuries in the adult rat. J Neurotrauma. 2005;22:529–543. doi: 10.1089/neu.2005.22.529. [DOI] [PubMed] [Google Scholar]
- 33.Magnuson DS, Trinder TC, Zhang YP, Burke D, Morassutti DJ, Shields CB. Comparing deficits following excitotoxic and contusion injuries in the thoracic and lumbar spinal cord of the adult rat. Exp Neurol. 1999;156:191–204. doi: 10.1006/exnr.1999.7016. [DOI] [PubMed] [Google Scholar]
- 34.McKinley W, Santos K, Meade M, Brooke K. Incidence and outcomes of spinal cord injury clinical syndromes. J Spinal Cord Med. 2007;30:215–224. doi: 10.1080/10790268.2007.11753929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noga BR, Kriellaars DJ, Brownstone RM, Jordan LM. Mechanism for activation of locomotor centers in the spinal cord by stimulation of the mesencephalic locomotor region. J Neurophysiol. 2003;90:1464–1478. doi: 10.1152/jn.00034.2003. [DOI] [PubMed] [Google Scholar]
- 36.Noga BR, Kriellaars DJ, Jordan LM. The effect of selective brainstem or spinal cord lesions on treadmill locomotion evoked by stimulation of the mesencephalic or pontomedullary locomotor regions. J Neurosci. 1991;11:1691–1700. doi: 10.1523/JNEUROSCI.11-06-01691.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohri SS, Maddie MA, Zhao Y, Qiu MS, Hetman M, Whittemore SR. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia. 2011;59:1489–1502. doi: 10.1002/glia.21191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Penderis J, Shields SA, Franklin RJ. Impaired remyelination and depletion of oligodendrocyte progenitors does not occur following repeated episodes of focal demyelination in the rat central nervous system. Brain. 2003;126:1382–1391. doi: 10.1093/brain/awg126. [DOI] [PubMed] [Google Scholar]
- 39.Schneider GS. Anterior spinal cord syndrome after initiation of treatment with atenolol. J Emerg Med. 2010;38:e49–52. doi: 10.1016/j.jemermed.2007.08.061. [DOI] [PubMed] [Google Scholar]
- 40.Schucht P, Raineteau O, Schwab ME, Fouad K. Anatomical correlates of locomotor recovery following dorsal and ventral lesions of the rat spinal cord. Exp Neurol. 2002;176:143–153. doi: 10.1006/exnr.2002.7909. [DOI] [PubMed] [Google Scholar]
- 41.Sholomenko GN, Steeves JD. Effects of selective spinal cord lesions on hind limb locomotion in birds. Exp Neurol. 1987;95:403–418. doi: 10.1016/0014-4886(87)90148-8. [DOI] [PubMed] [Google Scholar]
- 42.Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol. 2003;462:223–240. doi: 10.1002/cne.10736. [DOI] [PubMed] [Google Scholar]
- 43.Steeves JD, Jordan LM. Localization of a descending pathway in the spinal cord which is necessary for controlled treadmill locomotion. Neurosci Lett. 1980;20:283–288. doi: 10.1016/0304-3940(80)90161-5. [DOI] [PubMed] [Google Scholar]
- 44.Talbott JF, Loy DN, Liu Y, Qiu MS, Bunge MB, Rao MS, Whittemore SR. Endogenous Nkx2.2+/Olig2+ oligodendrocyte precursor cells fail to remyelinate the demyelinated adult rat spinal cord in the absence of astrocytes. Exp Neurol. 2005;192:11–24. doi: 10.1016/j.expneurol.2004.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tetzlaff W, Okon EB, Karimi-Abdolrezaee S, Hill CE, Sparling JS, Plemel JR, Plunet WT, Tsai EC, Baptiste D, Smithson LJ, Kawaja MD, Fehlings MG, Kwon BK. A systematic review of cellular transplantation therapies for spinal cord injury. J Neurotrauma. 2011;28:1611–1682. doi: 10.1089/neu.2009.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vilensky JA, Moore AM, Eidelberg E, Walden JG. Recovery of Locomotion in Monkeys With Spinal Cord Lesions. J Mot Behav. 1992;24:288–296. doi: 10.1080/00222895.1992.9941624. [DOI] [PubMed] [Google Scholar]
- 47.Yajima K, Suzuki K. Demyelination and remyelination in the rat central nervous system following ethidium bromide injection. Lab Invest. 1979;41:385–392. [PubMed] [Google Scholar]