

Abstract
We have developed two new continuous, coupled assays for ornithine-δ-aminotransferase (OAT) that are more sensitive than previous methods, measure activity in real time, and can be carried out in multi-well plates for convenience and high throughput. The first assay is based on the reduction of Δ1-pyrroline-5-carboxylate (P5C), generated from ornithine by OAT, using human pyrroline 5-carboxylate reductase 1 (PYCR1), which results in the concomitant oxidation of NADH to NAD+. This procedure was found to be three times more sensitive than previous methods, and is suitable for the study of small molecules as inhibitors or inactivators of OAT or as a method to determine OAT activity in unknown samples. The second method involves the detection of L-glutamate, produced during the regeneration of the cofactor PLP of OAT by an unamplified modification of the commercially available Amplex® Red L-glutamate detection kit (Life Technologies). This assay is recommended for the determination of the substrate activity of small molecules against OAT; measuring the transformation of L-ornithine at high concentrations by this assay is complicated by the fact that it also acts as a substrate for the L-glutamate oxidase (GluOx) reporter enzyme.
Keywords: continuous assay, coupled assay, ornithine-δ-aminotransferase, Δ1-pyrroline-5-carboxylate, Δ1-pyrroline-5-carboxylate reductase 1, Amplex® Red, L-glutamate oxidase
Introduction
Ornithine-δ-aminotransferase (OAT) is a mitochondrial enzyme that catalyzes the pyridoxal 5′-phosphate (PLP)-dependent transformation of L-ornithine to L-glutamate 5-semialdehyde, which spontaneously cyclizes to form Δ1-pyrroline-5-carboxylate (P5C, Scheme 1) [1].
Scheme 1.
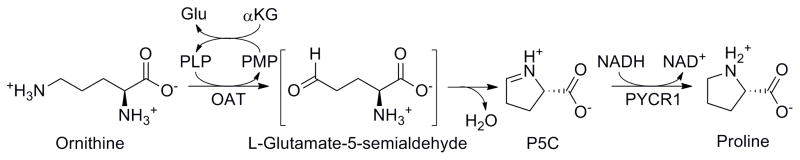
Transformation of L-ornithine into Δ1-pyrroline-5-carboxylate catalyzed by ornithine-δ-aminotransferase, followed by reduction to L-proline by Δ1-pyrroline-5-carboxylate reductase
OAT is involved in the synthesis of L-proline from L-ornithine, as well as regulating the levels of L-ornithine in the cell. Deficiency of this enzyme is known to cause gyrate atrophy of the choroid and retina [1], which leads to total blindness, and its inhibition is proposed to mediate the visual field defects (VFDs) caused by the antiepileptic vigabatrin (Sabril®) [2]. At the other end of the spectrum, in certain tissues, high levels of OAT deplete intracellular levels of L-ornithine, impairing the detoxification of ammonia by ornithine carbamoyltransferase, through the urea cycle [1].
Aside from radioisotopic methods [3], the activity of OAT is measured through the reaction of P5C with different reagents, mainly o-aminobenzaldehyde [4] or ninhydrin [5]. Both of these methods have been useful, but have serious drawbacks. o-Aminobenzaldehyde is expensive, decomposes slowly at room temperature, and displays poor solubility in aqueous solutions. The ninhydrin-based assay is more sensitive, but requires heating, centrifugation, decantation, and redissolution of the precipitate in an organic solvent [5]. Most importantly, though, neither of these methods can measure OAT activity continuously; the reactions must be quenched at various time points in order to make a measurement. As a corollary to this inconvenience, obtaining an activity curve for OAT requires substantial amounts of enzyme, considering that every time point requires aliquots of the biomolecule.
In this paper, we describe two new continuous, coupled assay methods for monitoring the catalytic activity of ornithine-δ-aminotransferase. First, we have developed a coupled assay with human pyrroline 5-carboxylate reductase 1, a NADH-dependent enzyme that catalyzes the last step in L-proline biosynthesis [6]. As P5C is produced, it is reduced to L-proline by PYCR1, with the concomitant oxidation of NADH to NAD+; this change can be monitored spectrophotometrically by the decrease in absorbance at 340 nm. This procedure is suitable for studying turnover of L-ornithine by OAT, as well as for inhibition and inactivation of the enzyme by small molecules. Although similar enzyme combinations have been used in the past to study L-proline production in plants, to our knowledge this is the first time it has been used to analyze the activity of OAT [7].
The second assay we have developed is based on L-glutamate detection. After catalysis, OAT must regenerate the PLP cofactor from its pyridoxamine 5′-phosphate (PMP) form; this is achieved by transamination, with α-ketoglutarate being simultaneously transformed into L-glutamate. To detect turnover by the enzyme, L-glutamate is measured in real time using a modified version of a commercially available detection kit. In essence, the L-glutamate is oxidized by L-glutamate oxidase (from Streptomyces sp.), producing α-ketoglutarate, ammonia, and hydrogen peroxide. The latter is then used with horseradish peroxidase to oxidize Amplex® Red to resorufin, which can be detected by fluorescence (Scheme 2). This assay is suited for measuring the turnover of varied substrates by the enzyme. This includes the natural substrate, L-ornithine, although the response becomes more error-prone at higher concentrations of this particular compound; this is because it also acts as a substrate for the reporter enzyme L-glutamate oxidase.
Scheme 2.
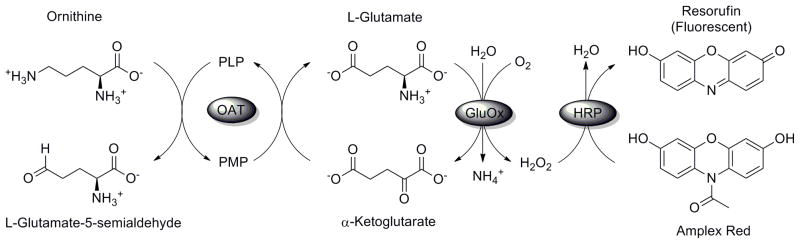
Detection of L-glutamate produced by turnover of OAT, mediated by L-glutamate oxidase and horseradish peroxidase. During this process, Amplex® Red is oxidized to the fluorescent resorufin.
Materials and Methods
Materials
L-Glutamate oxidase, L-ornithine, GABA, perchloric acid, NADH, and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Human recombinant ornithine-δ-aminotransferase and human recombinant pyrroline 5-carboxylate reductase were purchased from US Biologicals (Salem, MA, USA). Amplex® Red and horseradish peroxidase (HRP) were purchased from Life Technologies (Grand Island, NY, USA).
UV-Vis/Fluorescence spectroscopy
UV absorption and fluorescence were measured using a Synergy H1 hybrid multi-mode microplate reader (Biotek, USA) with transparent 96-well plates (Greiner bio-one, USA) and black 96-well plates (Brand, Germany), respectively.
Assay of Ornithine-δ-Aminotransferase with Pyrroline 5-carboxylate Reductase 1 (PYCR1)
A microplate well was loaded with 100 μL of an assay mixture containing 100 mM potassium pyrophosphate (pH 8.0), 10 mM α-ketoglutarate, 0.4 mM NADH, 0.025 mM PLP, and 20 mM L-ornithine. After pre-heating the mixture to 37 °C for 10 min, 1 μL of PYCR1 (0.5 mg/mL) and 1 μL of OAT (1 mg/mL) were added. The plate was shaken at 37 °C for 1 min, and the absorbance was measured at 340 nm every 5 sec for 30 min. All assays were performed in triplicate.
Determination of Km and Vmax for L-ornithine
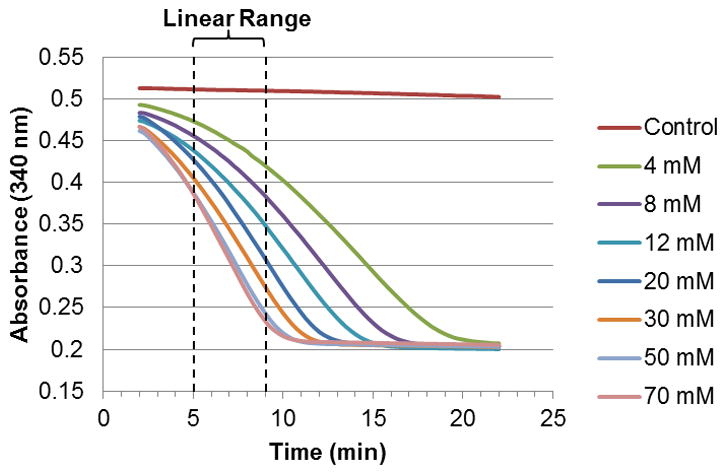
The Km for L-ornithine with OAT was determined using varying concentrations of the substrate. Assays were performed in triplicate as mentioned above, with the different concentrations of L-ornithine (0, 1, 4, 8, 12, 20, 30, 50, and 70 mM) being achieved by adding an appropriate amount of a 200 mM stock solution of L-ornithine (in buffer) to the corresponding well. The reaction rates were calculated from the linear portion of the time-dependent absorbance curve (5–9 min) using Microsoft Excel, and the values were plotted against the corresponding substrate concentrations. GraphPad Prism V 5.0 was used to determine the Km, using standard Michaelis-Menten non-linear fitting. To determine the Vmax value, the L-ornithine concentration of 1 mM was used, but adopting a different time frame so that the linear portion of the absorbance curve was long enough to be deemed constant (i.e., the enzyme had reached its maximum activity at said concentration). This determination was made by analyzing the rate (estimated by the difference between measured absorbance values at two consecutive time points) vs. time; a flat region of this curve was found between 15 and 24 min. Vmax was calculated from the Michaelis-Menten equation, and was converted to concentration using the extinction coefficient for NADH (6220 M−1 cm−1) through the Beer-Lambert equation. The cell path length was obtained by determining the ratio between the 96-well plate and a fixed path length (10 mm) cuvette, of the difference in absorbance at 975 nm and 900 nm; the value obtained was 2.98 mm [8].
Assay of Ornithine-δ-Aminotransferase with Ninhydrin
The activity of OAT was determined by the ninhydrin method as described by Kim [5], with some modifications. Briefly, the assay mixture contained 100 mM potassium pyrophosphate (pH 8.0), 0.025 mM PLP, 10 mM α-ketoglutarate, 4 mM L-ornithine, and 1 μL of OAT (1 mg/mL) in a total volume of 100 μL, and was incubated at 37 °C for the appropriate time. The reaction was quenched by adding 30 μL of perchloric acid (3N) and 20 μL of 2% aqueous ninhydrin. The resulting mixture was heated in a boiling water bath for 5 min, and centrifugation was performed for 5 min at 3,000 rpm. The supernatant was discarded, and the precipitate was redissolved in 150 μL of 95% ethanol, with further centrifugation for 5 min. The supernatant (140 μL) was transferred to a clear 96-well plate and measured for absorbance at 510 nm.
Assay of OAT by Detection of L-Glutamate
The assay mixture contained 40 mM potassium pyrophosphate (pH 8.0), 3 mM L-ornithine, 10 mM α-ketoglutarate, 0.025 mM PLP, 0.05 mM Amplex® Red, HRP (12.5 mU), L-glutamate oxidase (4 mU), and 0.1 μL of OAT (1 mg/mL) in a total volume of 100 μL. The solution was incubated at 37 °C for 30 min with gentle shaking. Fluorescence was then measured with excitation at 530 nm and emission at 590 nm. Controls lacking OAT were measured simultaneously, to determine the amount of fluorescence due to the direct reaction of L-ornithine with L-glutamate oxidase. A standard curve with varying concentrations of L-glutamate (0, 0.4, 0.8, 1.2, 1.6, 2.0, 2.4, and 2.8 μM) was obtained at the same time. All assays were performed in triplicate.
Results and Discussion
Coupled assay of OAT with PYCR1
As a prerequisite for this assay, we needed to make sure that PYCR1 was in excess relative to OAT. For this purpose, we decided to test various concentrations of OAT with a fixed amount of PYCR1 in the reaction mixture, following the conditions discussed above. As can be seen in Figure 1, the increase in reaction rate was linearly correlated to the amount of OAT added up to 1.5 μL. On the basis of this observation, we were satisfied that PYCR1 was not the limiting enzyme in this mixture up to this value, and decided to use 1 μL of OAT per well in future experiments.
Figure 1.
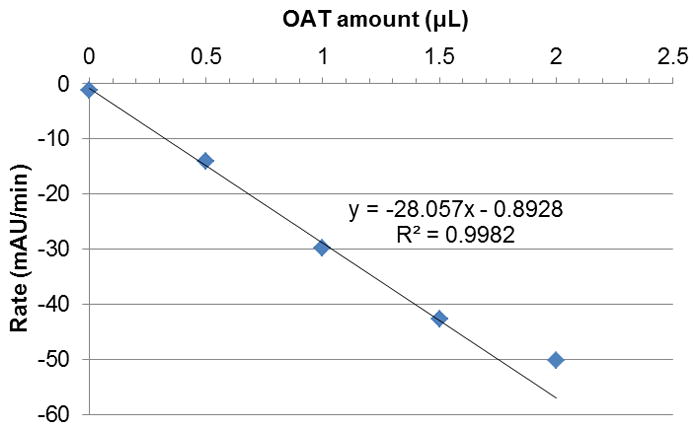
Changes in reaction rate with increasing volume of OAT added, at a fixed concentration (4.9 μg/mL) of PYCR1 (5–9 min). OAT is the rate-limiting enzyme up to 1.5 μL.
Following the conditions discussed above, we were able to obtain curves representing the turnover of L-ornithine by OAT over time. The rate of catalysis is correlated to the decrease in absorbance at 340 nm, corresponding to the oxidation of NADH to NAD+ by PYCR1. Baseline values are in the range of 0.5 AU, whereas the activity of PYCR1 seems to stop at around 0.2 AU, indicating depletion of NADH (as evidenced by controls). In order to increase the duration of the experiment, the initial concentration of NADH can be increased without detrimental effects. Of note, we observed that NADH slowly decomposed over time; this phenomenon is known to occur in phosphate buffers and by UV irradiation [9, 10]. However, the rate of decrease was constant, and was easily subtracted from the observed catalytic rates (Figure 2).
Figure 2.
Decrease in absorbance at 340 nm corresponding to turnover of ornithine by OAT in the presence of PYCR1. The 5–9 minute range was used to estimate the rates by linear regression.
As a practical application of this assay, we decided to determine the Km of L-ornithine against OAT. For this purpose, we performed the assay at various concentrations of the substrate. After analyzing the curves, we found that the observed catalytic rates for all concentrations were generally constant in the range of 5–9 min. The Km obtained from the curve (Figure 3), 10.50 ± 0.84 mM, was within the expected range [11].
Figure 3.
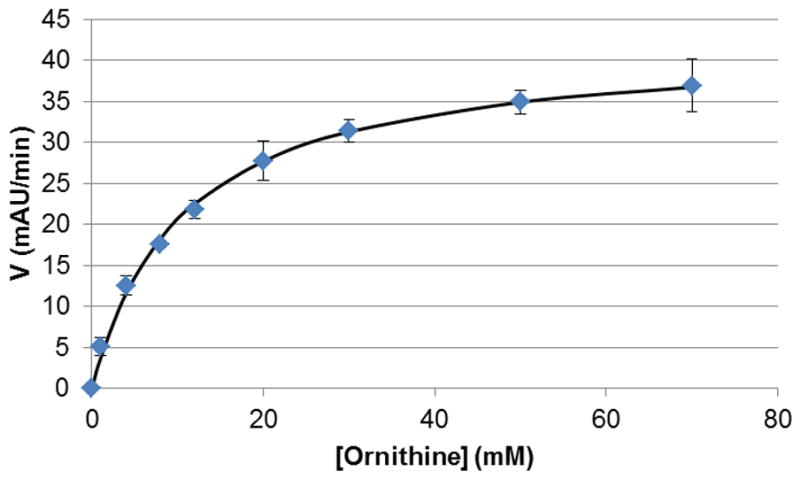
Km curve for L-ornithine by OAT (5–9 min). After fitting to the Michaelis-Menten equation, a Km value of 10.50 mAU min−1 was obtained.
The Vmax value was calculated from a different time frame, because the enzyme did not show maximal activity at all the concentrations in the 5–9 minute range. To ensure that we would observe optimal enzyme activity, a lower concentration of substrate (1 mM) was used. By plotting the rate of absorbance decrease vs. time (Figure 4), we were able to see a flat region of the curve, between 15 and 24 min, which corresponded to the maximum rate at said concentration. Through the use of the Michaelis-Menten equation, the Vmax value was found to be 94.9 ± 1.0 mAU min−1, or 5.22 ± 0.06 nmol min−1. The specific activity of 5.22 ± 0.06 units mg−1 was similar to the value reported previously [11].
Figure 4.
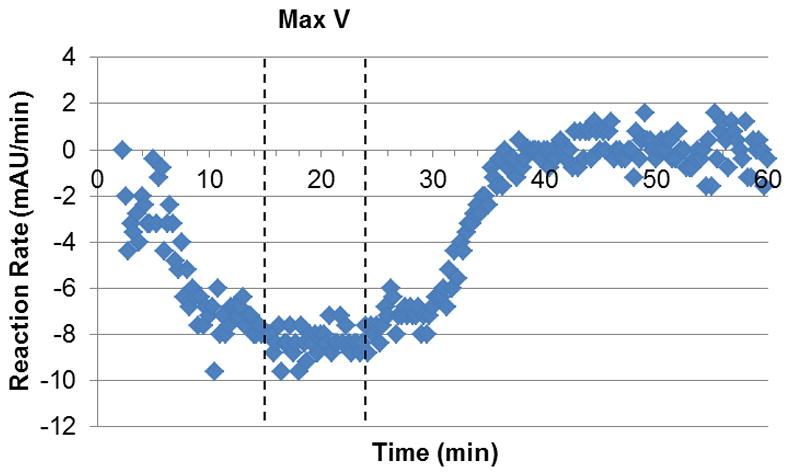
Reaction rate vs. time (1 mM L-ornithine). The curve plateaus at a maximum reaction rate of 8.32 mAU min−1 between 15 and 24 min.
A comparison of this newly-developed assay was made with the previously discussed ninhydrin method, which is the most sensitive of the currently used spectrophotometric methods for assaying OAT [5]. As can be seen in Figure 5, after the initial ramp-up, the PYCR1 assay is about three times more sensitive than the known method, i. e., the change in absorbance by varying the concentration of L-ornithine using our method is roughly triple that observed for the ninhydrin derivative.
Figure 5.
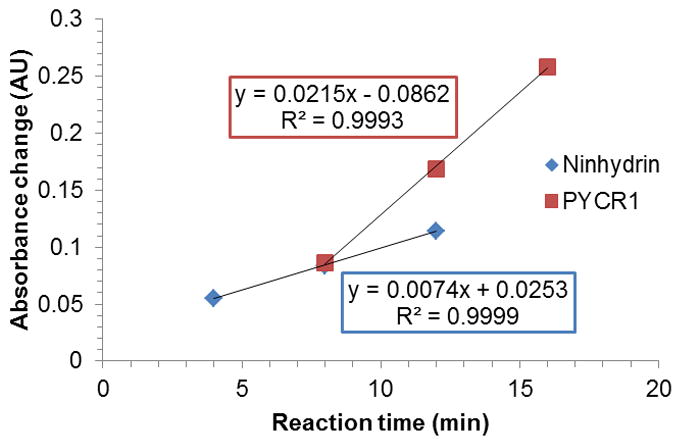
Sensitivity of the PYCR1-based assay versus the ninhydrin method (1 mM L-ornithine). A change in the concentration of L-ornithine results in a three-fold higher absorbance change, when comparing the newly developed method to the previously used one.
Possible applications of this assay are: testing for inhibition and inactivation assays for synthetic molecules (with or without preincubation), characterization of different OATs and/or their mutants, and determination of OAT activity in crude samples. To control for endogenous Δ1-pyrroline-5-carboxylate, the assays should also be carried out in the absence of OAT. For applicability to plant samples, the presence of ornithine-α-aminotransferase must be accounted for; however, we do not expect the assay to be affected by this enzyme, as long as a suitable excess of PYCR1 exists in the mixture to offset inhibition by its product, Δ1-pyrroline-2-carboxylate. This caveat also applies to crude mixtures that might include other PYCR1 inhibitors.
Assay of OAT by Detection of L-Glutamate
To test for substrate activity of small molecules against OAT, we took a similar approach to the one we have previously developed for GABA-AT [12]. In essence, we wanted to detect the L-glutamate produced by OAT from α-ketoglutarate per turnover event (Scheme 2).
As an application of our assay, we again attempted to obtain a Km for L-ornithine with OAT using the developed protocol. However, after many attempts, we found that we could not saturate the system with increasing amounts of L-ornithine, as high as 80 mM (Figure 6). We found that the divergence from our previous results stemmed from L-ornithine being a substrate for L-glutamate oxidase, part of the L-glutamate reporter system. This fact was confirmed by a standalone assay; a mixture of L-ornithine, HRP, and Amplex Red displayed fluorescence only in the presence of GluOx (data not shown). This is not the case when performing substrate assays for GABA-AT.
Figure 6.
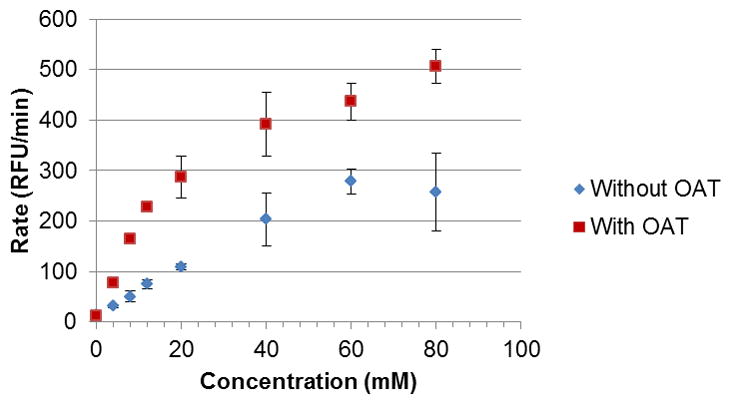
Changes in fluorescence at increasing concentrations of L-ornithine, with and without addition of OAT. The blue points (lower curve) represent the baseline fluorescence, caused by the direct reaction of L-ornithine with the reporter enzyme L-glutamate oxidase. The fluorescence caused by OAT activity is the difference between the higher values (reaction with OAT) and the lower values (reaction without OAT).
By controlling for the contributions to the fluorescence by nonspecific reactions (i.e., subtracting the rates from the control wells, which did not contain OAT), we found that a Km value can, in fact, be obtained by this method. However, because of the increasing magnitude of the non-specific fluorescence response at higher L-ornithine concentrations, the errors increase concurrently (Figure 7). The Km and Vmax values obtained by this method are 8.97 ± 3.85 mM and 4.01 ± 0.48 nmol min−1 (238.8 ± 28.3 RFU min−1), respectively. The specific activity was determined to be 4.01 ± 0.48 units mg−1; all of these values are in agreement with our previous results. The Km and Vmax values for the oxidation of L-ornithine by L-glutamate oxidase were found to be 94.6 ± 4.3 mM and 153 ± 4 nmol min−1, using the previously discussed control curve.
Figure 7.
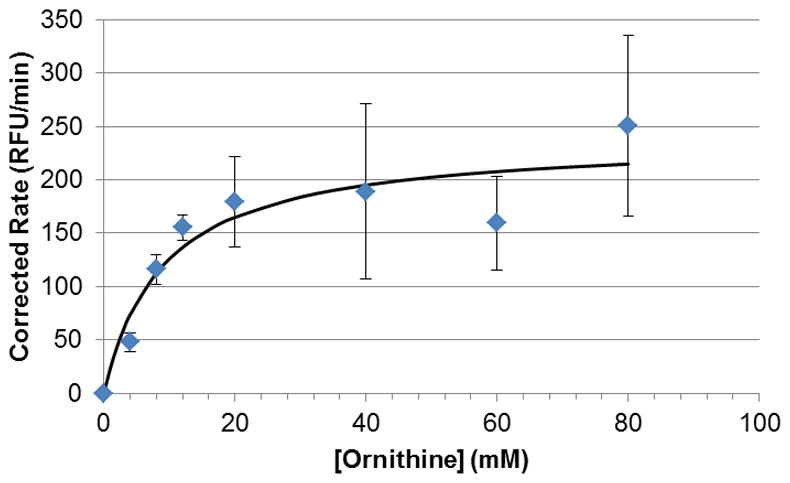
Michaelis-Menten curve for L-ornithine with OAT, on the basis of L-glutamate detection. After fitting to the Michaelis-Menten equation, the Km and Vmax values obtained were 8.97 ± 3.85 mM and 4.01 ± 0.48 nmol min−1 (238.8 ± 28.3 RFU min−1), respectively.
As with the other assay, we made sure that OAT was the rate-limiting enzyme in the system. In this case, we varied the amount of GluOx until it was in excess. We chose a value of 12.5 mU GluOx (0.8 μL of a 5 U mL−1 stock solution), which was more than twice the amount at which OAT became rate-limiting (around 0.25 μL, Figure 8).
Figure 8.
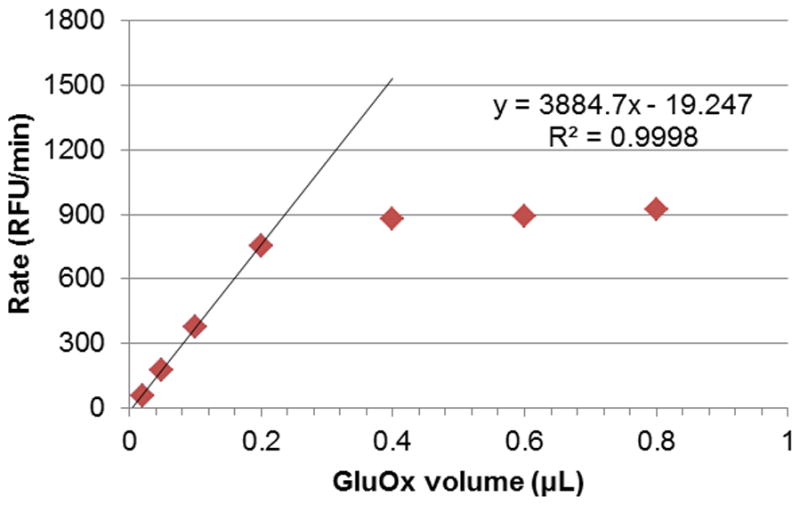
Changes in reaction rate with increasing volume of GluOx added, at a fixed concentration of OAT (0.8 μL of 1 mg mL−1 stock). GluOx is the rate-limiting enzyme up to approximately 0.25 μL of stock solution (5 U mL−1); at 0.4 μL and higher, GluOx is no longer rate-limiting, and is therefore in excess.
As mentioned earlier, this assay is directed at studying the substrate activity of small molecules against OAT, including the natural substrate (L-ornithine), because the detection method does not depend on the structure of the direct enzymatic product, only on the production of glutamate. In the specific case of L-ornithine, the assay is applicable as long as the concentrations used are lower than ~20 mM, roughly twice the Km value; at higher values, the results may be confounded by the magnitude of the errors. Because L-ornithine is also a substrate for L-glutamate oxidase, the baseline fluorescence response increases with the concentration, and the significance of the (test)-(baseline) difference will diminish concomitantly. It is not likely that this will be a problem in other cases, however, because GluOx has been found to be a fairly selective enzyme [13].
Conclusions
We have developed and optimized two coupled assays for measuring the catalytic activity of OAT continuously. Our first assay was based on monitoring the consumption of OAT’s product by PYCR1 through changes in the level of NADH; this procedure was found to be three times more sensitive than the best spectrophotometric method in use, based on ninhydrin. This assay was designed to study processes through which L-ornithine is transaminated by the enzyme, such as determining enzyme properties, and inhibition or inactivation of OAT by small molecules. Our second assay was focused on measuring the L-glutamate produced by the regeneration of OAT’s cofactor after turnover, using a modification of a commercially available detection kit. This method was proposed mainly to study substrate activity of small molecules at OAT; because L-ornithine itself reacts with the L-glutamate reporter system, assays with this particular substrate should remain under a concentration of ~20 mM. In addition to the increased sensitivity, these assays are an improvement on current methods by eliminating the need to quench the reaction in order to perform measurements; this fact makes the entire procedure much faster and more convenient, while at the same time significantly reducing the amount of enzyme required for each determination.
List of Abbreviations
- AU
Absorbance units
- GABA-AT
γ-Aminobutyric acid aminotransferase
- GluOx
Glutamate oxidase from Streptomyces sp
- Km
Concentration of substrate that leads to half maximum reaction rate
- NAD+
Nicotinamide adenine dinucleotide (oxidized form)
- NADH
Nicotinamide adenine dinucleotide (reduced form)
- OAT
Ornithine-δ-aminotransferase
- P5C
Δ1-Pyrroline-5-carboxylate
- PLP
Pyridoxal 5′-phosphate
- PMP
Pyridoxamine 5′-phosphate
- PYCR1
Human Δ1-pyrroline-5-carboxylate reductase 1
- RFU
Reference fluorescence units
- VFD
Visual field defect
- Vmax
Maximum reaction rate achieved by the system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Seiler N. Ornithine aminotransferase, a potential target for the treatment of hyperammonemias. Current drug targets. 2000;1(2):119–53. doi: 10.2174/1389450003349254. [DOI] [PubMed] [Google Scholar]
- 2.Sorri I, Brigell MG, Malyusz M, Mahlamaki E, de Meynard C, Kalviainen R. Is reduced ornithine-δ-aminotransferase activity the cause of vigabatrin-associated visual field defects? Epilepsy Res. 2010;92(1):48–53. doi: 10.1016/j.eplepsyres.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 3.Ohura T, Kominami E, Katunuma N. A New Sensitive and Convenient Assay of Ornithine Aminotransferase. J Nutr Sci Vitaminol. 1983;29(2):123–128. doi: 10.3177/jnsv.29.123. [DOI] [PubMed] [Google Scholar]
- 4.Strecker HJ. Purification and Properties of Rat Liver Ornithine δ-Transaminase. J Biol Chem. 1965;240(3):1225–1230. [PubMed] [Google Scholar]
- 5.Kim HR, Rho HW, Park JW, Park BH, Kim JS, Lee MW. Assay of Ornithine Aminotransferase with Ninhydrin. Anal Biochem. 1994;223(2):205–207. doi: 10.1006/abio.1994.1574. [DOI] [PubMed] [Google Scholar]
- 6.Meng Z, Lou Z, Liu Z, Li M, Zhao X, Bartlam M, Rao Z. Crystal structure of human pyrroline-5-carboxylate reductase. Journal of molecular biology. 2006;359(5):1364–77. doi: 10.1016/j.jmb.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 7.da Rocha IMA, Vitorello VA, Silva JS, Ferreira-Silva SL, Viegas RA, Silva EN, Silveira JAG. Exogenous ornithine is an effective precursor and the delta-ornithine amino transferase pathway contributes to proline accumulation under high N recycling in salt-stressed cashew leaves. J Plant Physiol. 2012;169(1):41–49. doi: 10.1016/j.jplph.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Lampinen JRM, Perala A, Oranen H, Harinen R-R. Microplate based pathlength correction method for photometric DNA quantification assay. [accessed 03/05/13];Application Notes [Online] 2012 http://www.thermoscientific.de/eThermo/CMA/PDFs/Various/File_60220.pdf.
- 9.Rover L, Jr, Fernandes JCB, de Oliveira Neto G, Kubota LT, Katekawa E, Serrano SHP. Study of NADH stability using ultraviolet-visible spectrophotometric analysis and factorial design. Analytical biochemistry. 1998;260(1):50–55. doi: 10.1006/abio.1998.2656. [DOI] [PubMed] [Google Scholar]
- 10.Kiianitsa K, Solinger JA, Heyer W-D. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Analytical biochemistry. 2003;321(2):266–271. doi: 10.1016/s0003-2697(03)00461-5. [DOI] [PubMed] [Google Scholar]
- 11.Ohura T, Kominami E, Tada K, Katunuma N. Crystallization and Properties of Human-Liver Ornithine Aminotransferase. J Biochem-Tokyo. 1982;92(6):1785–1792. doi: 10.1093/oxfordjournals.jbchem.a134108. [DOI] [PubMed] [Google Scholar]
- 12.Juncosa JI, Jr, Groves AP, Xia G, Silverman RB. Probing the steric requirements of the γ-aminobutyric acid aminotransferase active site with fluorinated analogues of vigabatrin. Bioorganic & medicinal chemistry. 2013;21(4):903–11. doi: 10.1016/j.bmc.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arima J, Ashiuchi M, Inagaki K, Kusakabe H, Yagi T. U.S. Patent 7,109,008. L-Glutamate Oxidase. 2008 filed April 19, 2001; and issued September 19, 2006.