

Abstract
Proteins of the BCL2 family provide a survival mechanism in many human malignancies including chronic lymphocytic leukemia (CLL). The BCL2 inhibitor ABT-263 (navitoclax) is active in clinical trials for lymphoid malignancies, yet resistance is expected based on preclinical models. We recently demonstrated that vinblastine can dramatically sensitize several leukemia cell lines to ABT-737 (the experimental congener of ABT-263). The goal of these experiments was to determine the impact of vinblastine on ABT-737 sensitivity in CLL cells isolated from peripheral blood and to define the underlying mechanism. Freshly isolated CLL cells from 35 patients, as well as normal lymphocytes and platelets, were incubated with various microtubule disrupting agents plus ABT-737 to assess sensitivity to the single agents and the combination. ABT-737 and vinblastine displayed a range of sensitivity as single agents, and vinblastine markedly sensitized all CLL samples to ABT-737 within 6 h. Vinblastine potently induced the pro-apoptotic protein PMAIP1 (NOXA) in both a time- and dose-dependent manner and this was required for the observed apoptosis. Combretastatin A4, which dissociates microtubules by binding a different site, had the same effect confirming that interaction of these agents with microtubules is the initial target. Similarly, vincristine and vinorelbine induced NOXA and enhanced CLL sensitivity to ABT-737. Furthermore, vinblastine plus ABT-737 overcame stroma-mediated resistance to ABT-737 alone. Apoptosis was induced with clinically achievable concentrations, with no additional toxicity to normal lymphocytes or platelets. These results suggest that vinca alkaloids may improve the clinical efficacy of ABT-263 in patients with CLL.
Keywords: vinblastine, ABT-737, chronic lymphocytic leukemia, NOXA, vinca alkaloid
INTRODUCTION
Although low grade hematopoietic malignancies respond to initial chemotherapy regimens, they remain incurable, requiring additional rounds of increasingly toxic chemotherapy. Ultimately many patients die of the disease within a few to several years. The only current curative therapy for these diseases is allogenic transplantation. However, this strategy is not applicable to most patients as they tend to be elderly, have co-morbidities or lack a suitable donor.
Vinca alkaloids have been used for many years as part of treatment regimens for patients with hematopoietic malignancies including chronic lymphocytic leukemia (CLL). At first glance, it seems highly unlikely that vincristine or vinblastine should have any impact in CLL because, following dissociation of microtubules, their mechanism of action is traditionally considered to be due to mitotic arrest which progresses to apoptosis. As the proportion of cycling cells in CLL is very low, particularly in the peripheral circulation, vinca alkaloids should be inactive. However, we have recently discovered that vinblastine can induce very rapid apoptosis in primary CLL cells despite the fact they are non-proliferative (1, 2). Additionally we have been able to induce this rapid apoptosis in cell lines of other leukemia types by combining vinblastine with various approaches that target the anti-apoptotic proteins MCL1 or BCL2 (1–3). The effects of vinca alkaloids still appear to be initiated by disruption of microtubules as we show similar effects herein with combretastatin A4, which dissociates microtubules through binding to a different site on tubulin.
The intrinsic apoptotic pathway is regulated by the BCL2 family of proteins, in which the balance of pro- and anti-apoptotic proteins determines cell survival. When in excess, the BH3-only pro-apoptotic proteins [e.g. Bad, PMAIP1 (NOXA)] bind the BH3 groove of anti-apoptotic proteins, allowing for the release of other pro-apoptotic proteins [e.g. BCL2L11 (BIM)] and activation of BAX and BAK. BAX and BSK then form pores in the outer mitochondrial membrane releasing cytochrome c, leading to downstream caspase activation and apoptosis (4). Expression of the anti-apoptotic family members, BCL2, BCL2L1 (BCLX), BCL2L2 (BCLW), MCL1 and BCL2A1 (BFL1) contribute to drug resistance in many human malignancies (5, 6). The balance of these proteins is also important for normal hematopoiesis, and their dysregulation can lead to the development of cancer (e.g. BCL2 upregulation in CLL). Thus there is a need to develop targeted treatments for cancer that can overcome BCL2 protein-mediated protection.
ABT-737 is a BH3 mimetic that preferentially binds the BH3 groove of anti-apoptotic proteins BCL2, BCLXL and BCLW, inducing BAX/BAK-dependent apoptosis (7). The success of ABT-737 to induce apoptosis was first documented in CLL and small cell lung cancer (8) and has been further tested in numerous malignancies. The orally bioavailable analog, ABT-263 (navitoclax), has a similar profile of binding to BCL2 family proteins and the same effective concentrations (9). Navitoclax is currently in Phase I/II clinical trials as a single agent (NCT01557777, NCT00481091, NCT00406809) and in combination therapies (NCT00868413, NCT00788684, NCT01009073). Resistance to ABT-737 is caused by expression of MCL1 and BFL1, which can sequester BH3-only pro-apoptotic proteins (10, 11). Targeting MCL1 with siRNA or drugs has increased malignant tumor sensitivity to ABT-737 (3, 10, 12–15). Similarly, NOXA, which preferentially binds MCL1 and to a lesser degree BFL1, has been shown to aid in ABT-737-induced lethality (16–18). Thrombocytopenia is a side effect of ABT-263 exposure due to the dependence of platelets on BCLXL for survival (11, 19).
We have previously reported that vinblastine can rapidly kill CLL cells as a single agent, independent of cell cycle phase and can also acutely sensitize a panel of hematopoietic cell lines and freshly-isolated CLL cells to the cyclin-dependent kinase inhibitors flavopiridol and dinaciclib (2). Through inhibition of CDK9, these agents prevent global transcription resulting in rapid decline of proteins whose mRNA and protein both have a short half-life, in particular MCL1. Vinblastine greatly increased the levels of apoptosis in both dinaciclib-sensitive and dinaciclib-resistant CLL cells within 6 h. We hypothesized that other cells might be more dependent on BCL2 and this hypothesis was supported by the observation that NB4 leukemia cells are acutely sensitized to vinblastine by ABT-737 (3). The data presented here shows that vinblastine induces expression of NOXA, which is required to sensitize NB4 and CLL cells to ABT-737. Furthermore, vinblastine dramatically and rapidly enhances ABT-737-induced apoptosis in CLL without increasing toxicity to normal lymphocytes or platelets.
MATERIALS AND METHODS
Reagents
Vincristine and vinblastine were purchased from Millipore (Billerica, MA), vinorelbine and combretastatin A4 from Sigma (St. Louis, MO). The siRNA constructs were purchased from Ambion (Austin, TX) and the nucleofector kit from Lonza (Basel, Switzerland). Hoechst 33342 was purchased from Molecular Probes (Life Technologies; Grand Island, NY) and DNA primers from Integrated DNA Technologies (Coralville, IA). ABT-737 was provided by Abbott Laboratories (Abbott Park, IL). Unless otherwise stated, all other reagents were obtained from Sigma. Antibodies were as follows: PARP, phospho-JNK, BCLXL (Cell Signaling Technology; Beverly, MA); NOXA and actin (EMD Millipore; Billerica, MA); gelsolin and MCL1 (BD Pharmingen; San Diego, CA); BCL2 (Dako; Carpentaria, CA); BFL1 (courtesy of Jannie Borst, The Netherlands Cancer Institute); and secondary antibodies (BioRad; Hercules, CA). NB4 cells, derived from a patient with acute promyelocytic leukemia, were obtained from Ethan Dmitrovsky (Geisel School of Medicine at Dartmouth, Hanover, NH). THP-1 cells, derived from a patient with acute monocytic leukemia, were purchased from the American Type Culture Collection (ATCC; Manassas, VA). L4.5 cells expressing CD154 (CD40L) were obtained from Sonia Neron at Hema-Quebec (Quebec, Canada). No re-authentification of cell lines was performed.
Patient Samples
Blood samples were obtained from subjects at the Norris Cotton Cancer Center using a Dartmouth College IRB-approved protocol and informed consent documents. Cells from 10 mL of blood were diluted in phosphate buffered saline (PBS) and purified by centrifugation in Ficoll-Paque PLUS. Lymphocytes were collected, washed three times in PBS containing 2 mM EDTA and plated in RPMI 1640 plus 10% serum at 1 × 106 cells/mL alone or on top of 2 × 105 of L4.5 stromal cells. Cells were incubated immediately with drugs or after 24 h incubation on L4.5 stromal cells. Platelets were isolated by mixing 10 mL of blood with 1 mL sodium citrate (3% w/vol). This mixture was centrifuged for 10 min at 1,000 rpm, and the top layer containing the platelet rich plasma was collected. Purified platelets were plated in RPMI 1640 plus 10% serum and treated immediately with drugs.
Determination of apoptosis
Chromatin condensation is a hallmark of apoptosis, correlates with the cleavage of poly(ADP-ribose) polymerase (PARP), and is readily quantifiable. Both these endpoints occur much earlier than most other markers (e.g., Annexin V staining), and are therefore used here to demonstrate the rapidity with which apoptosis is induced. The apoptosis observed here was also inhibited by the pan-caspase inhibitor QVD-OPh (data not shown). To assess chromatin condensation, cells were incubated with 2 μg/mL Hoechst 33342 for 15 min at 37°C and visualized with a fluorescent microscope. At least 200 cells were scored from each sample and data were expressed as the percentage of cells with condensed chromatin.
Cell transfection and western blotting
Cells were maintained in RPMI-1640 containing 10% fetal bovine serum and incubated at 37°C in 5% CO2/95% humidified air. NB4 cells (2×106) were transfected with 2 μg NOXA (s10709) or scrambled siRNA using Nucleofector Kit V and program X-001. Forty-eight hours later, cells were treated with drugs for 6 h and then harvested for protein analysis. CLL cells (1×107) were transfected with 3 μg NOXA (s10709) or scrambled siRNA using B Cell Nucleofector Kit and program X-005. Twenty-four hours later, CLL cells were treated with drugs for 6 h and then harvested for protein analysis.
For protein analysis, cells were lysed in urea lysis buffer and boiled for 5 min (2). Proteins were separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Millipore). Membranes were blocked with 5% nonfat milk in TBS and 0.1% Tween 20, and were probed with the appropriate primary antibody overnight. Subsequently, membranes were washed in TBS and 0.1% Tween 20, and incubated with secondary antibody conjugated to horseradish peroxidase. Proteins were visualized by enhanced chemiluminescence (Amersham; GE Healthcare Bio-sciences; Piscataway, NJ). When comparing different CLL samples, lysates from a constant number of cells were loaded on each gel. Lysates from THP-1 cells were used as a positive control for protein expression (1). All platelet samples were loaded with 20 μg of protein per lane. Densitometry was performed using an HP psc1350v scanner and ImageJ software.
Quantitative Real-Time PCR
Total mRNA from 1×107 cells was isolated with TRI Reagent (Molecular Research Center; Cincinnati, OH) according to the manufacturer's instructions and reverse transcribed using iScript cDNA synthesis kit (BioRad). The cDNA was analyzed by quantitative reverse transcription PCR using the iQ SYBR Green Supermix (BioRad) according to the manufacturer's instructions. The expression ratio for NOXA relative to GAPDH was calculated according to the equation of Pfaffl (20) using untreated cells as a reference.
RESULTS
CLL cells are variably sensitive to ABT-737 and vinblastine as single agents
We have previously reported that ABT-737 sensitizes NB4 cells to vinblastine (3). This is an acute response that occurs within 6 h and at all phases of the cell cycle. To determine if this might be clinically relevant, we analyzed freshly-isolated cells from 35 CLL patients for their acute (6 h) sensitivity to ABT-737 alone or in combination with vinblastine. The concentrations of drugs used are clinically achievable as peak plasma concentrations for ABT-263 approach 4–6 μM (11, 21), while vinblastine reaches 0.1–0.6 μM (22, 23). Supplemental Table S1 displays the diagnostic and clinical features of the CLL patients included in this study. As expected, many of the CLL samples were acutely sensitive to ABT-737 alone, although a subset was relatively resistant (Fig. 1A). The median EC50 of ABT-737 was 0.01 μM, although at this concentration sensitivity still covered the entire range of 0–97% survival (boxed in Fig. 1A). CLL samples with greater than 50% survival at 0.01 μM ABT-737 were classified as resistant (n=14), while CLL samples with equal to or less than 50% survival were classified as sensitive (n=21).
Figure 1.

Vinblastine enhances sensitivity of CLL cells to ABT-737. Freshly-isolated lymphocytes from 35 CLL patients were incubated with 0–0.1 μM ABT-737 (A) or 0–1 μM vinblastine (B) for 6 h and apoptosis was assessed by chromatin condensation. Data are expressed as a percentage of cells surviving with the median sensitivity for each drug concentration shown. The samples were stratified as ABT-737 resistant or sensitive based on the sensitivity of CLL samples at 0.01 μM ABT-737 (A, boxed) or as vinblastine resistant or sensitive based on sensitivity of CLL samples at 1 μM vinblastine (B, boxed). (C) The percentage survival of CLL samples at 0.01 μM ABT-737 was compared to 1 μM vinblastine. The linear regression line is shown, Pearson correlation coefficient r(27)=0.471, p=0.0099. The shaded area emphasizes a potentially different subpopulation of samples. (D) Freshly-isolated lymphocytes from CLL patients were incubated with 0–0.1 μM ABT-737 in combination with 0–1 μM vinblastine for 6 h. Apoptosis was assessed by chromatin condensation and the percentages of cells surviving are shown for ABT-737 sensitive and resistant cells. The right panel labeled “ABT-737 very resistant” reflects the 5 most resistant samples from the resistant subset (middle panel). Standard error bars are shown. (E) Lysates from CLL cells of each ABT-737 sensitivity class were analyzed for protein expression. CLL samples 23, 19, 31 were sensitive to single agent vinblastine (<50% survival at the highest dose of 1 μM vinblastine), while all other CLL samples were resistant to single agent vinblastine (>50% survival at 1 μM vinblastine). THP-1 cells were used as a positive control for protein expression.
The same CLL samples were incubated with vinblastine alone for 6 h. These samples displayed a range of sensitivity with a median survival of 65% at the highest concentration of vinblastine (1 μM, boxed) (Fig. 1B). Samples with greater than 50% survival at 1 μM vinblastine were classified as resistant (n=19), while samples with less than 50% survival were classified as sensitive (n=10). Fig. 1C shows a significant correlation between the sensitivity to vinblastine and ABT-737 as single agents. These data suggest three subsets of CLL with different patterns of sensitivity: vinblastine resistant/ABT-737 resistant (upper right quadrant), vinblastine sensitive/ABT-737 sensitive (lower left quadrant) and vinblastine resistant/ABT-737 sensitive (upper left, shaded quadrant). Interestingly there were no vinblastine sensitive/ABT-737 resistant CLL samples as indicated by the empty lower right quadrant in Fig. 1C. These results suggest that there is a common underlying mechanism of sensitivity to ABT-737 and vinblastine for two-thirds of the samples, but there is a subset of samples that have an alternate mechanism of resistance to vinblastine (i.e. BCL2 independent). Repeat samples were obtained from several patients at different times, and the sensitivity to vinblastine or ABT-737 appeared to be a stable phenotype, although insufficient samples were available for a statistical analysis.
Vinblastine sensitizes CLL cells to ABT-737
When vinblastine and ABT-737 were combined, all of the CLL samples demonstrated increased apoptosis (Fig. 1D). To better visualize the impact of vinblastine in combination, the CLL cells were stratified as either ABT-737 sensitive or resistant as described above. In the ABT-737 resistant and sensitive cells, vinblastine reduced the EC50 of ABT-737 by 19- and 11-fold respectively. We noted that 5 samples were very resistant to ABT-737 alone (greater than 40% survival at 0.1 μM ABT-737), and therefore reanalyzed this subset. These very resistant CLL samples had an EC50 for ABT-737 of >0.1 μM in the absence of vinblastine, while the addition of 1 μM vinblastine reduced this EC50 >30-fold. These results suggest that adding vinblastine to an ABT-263 treatment regimen may significantly increase efficacy against CLL.
CLL prognostic markers and protein expression do no correlate with drug sensitivity
Due to the range of response of CLL samples to ABT-737 or vinblastine as single agents, we sought a biomarker as a predictor of response. Typically ZAP70 and CD38 status are determined at the time of patient diagnosis and can be used as prognostic factors for CLL. We therefore compared the status of these factors to the sensitivity of each CLL sample for each of our drugs. Sensitivity to ABT-737 and vinblastine was defined as discussed above and is summarized for each patient in Supplemental Table S1. There was no correlation between ZAP70/CD38 status and sensitivity to either ABT-737 or vinblastine. Additionally, patient gender and age did not significantly correlate with response to ABT-737 or vinblastine. Previous reports indicate MCL1 and BFL1 expression as mechanisms of ABT-737 resistance (10). We found no correlation of MCL1 or BFL1 expression with ABT-737 sensitivity (Fig. 1E). Lysates from THP-1 cells were used as a positive control as they express each of these BCL2 proteins (1). The BCL2 protein expression in most of the CLL samples was greater than that of THP-1 cells, with a trend of higher BCL2 expression correlating with ABT-737 sensitivity. The MCL1 level was variable in CLL and much lower than THP-1 cells. There was no detectable expression of BFL1 or NOXA (not shown) in any of the CLL samples.
Vinblastine-induced NOXA is required for apoptosis
The acute apoptosis induced in cell lines and CLL by vinblastine alone or when MCL1 is concurrently suppressed is dependent on the ability of vinblastine to potently activate c-Jun N-terminal kinase (JNK) (1, 2). However, acute apoptosis induced by vinblastine combined with ABT-737 is JNK-independent (Supplemental Fig. S1). This suggests that JNK may be required to inhibit BCL2 as it is redundant in the presence of ABT-737. Accordingly we propose that vinblastine has a second function that is required to inhibit MCL1. Vinblastine rapidly induced NOXA transcript (Fig. 2A) and protein (Fig. 2B) in CLL cells. NOXA induction was both time and concentration dependent; NOXA appeared at 1–2 h while PARP cleavage was evident at 4–6 h (Fig. 2B). No other BH3-only proteins were induced in CLL by vinblastine (data not shown) consistent with previous results in a panel of leukemia cell lines (1). These results are consistent with NOXA playing a role in vinblastine-mediated apoptosis in CLL.
Figure 2.
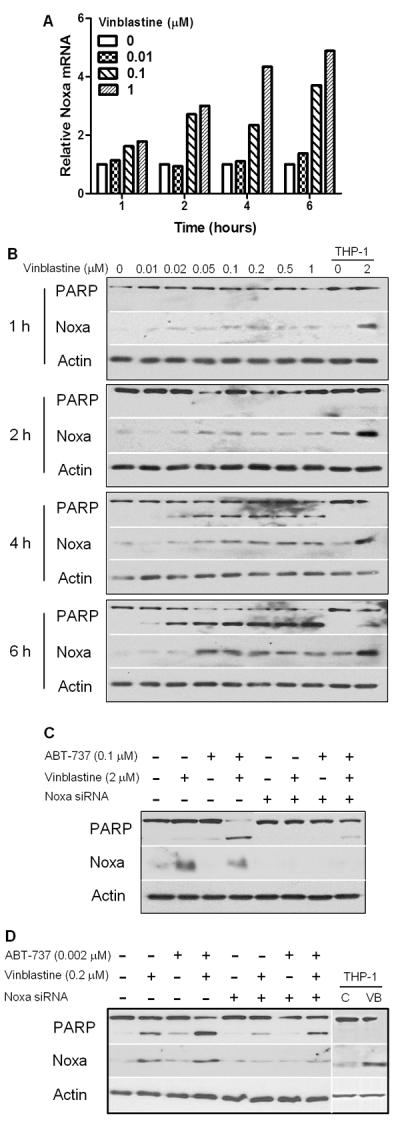
Noxa is required for apoptosis induced by the combination of vinblastine plus ABT-737. (A, B) Freshly-isolated lymphocytes from CLL patients were incubated with 0–1 μM vinblastine for various times and then harvested for total RNA or protein. (A) Quantitative RT-PCR was performed for Noxa, using GAPDH as a control. (B) Immunoblotting was used to determine protein expression; lysates from THP-1 cells were used as a control. Data shown is a representative from three patients. (C) NB4 or (D) CLL cells were transfected with scrambled or Noxa siRNA, and incubated with drugs for 6 h prior to harvest. A representative of three separate Noxa-targeting experiments for each cell type is shown. Noxa siRNA significantly reduced the percentage of cleaved PARP induced by vinblastine + ABT-737; the reduction in NB4 cells averaged 54%, range 40–68% (p<0.005); the reduction in CLL cells averaged 24%, range 17–36% (p=0.05). The THP-1 control lanes were graphically pasted from a different section of the same western blot.
To determine whether NOXA is required for vinblastine plus ABT-737-mediated apoptosis, we initially used NB4 cells because they express a similar pattern of BCL2 proteins (BCL2 high, MCL1 low, Bcl-X undetectable) as CLL, are also sensitized to ABT-737 by vinblastine and are readily transfectable with siRNA. The NOXA siRNA reduced basal NOXA levels and prevented induction of NOXA protein by vinblastine (Fig. 2C). Furthermore, NOXA siRNA significantly protected NB4 cells from apoptosis induced by ABT-737 in combination with vinblastine. We repeated this experiment in CLL cells that are more difficult to transfect (average of 40% transfection efficiency using a pmaxGFP control vector) and consequently the changes in NOXA were not as dramatic. However, the NOXA siRNA partially prevented the vinblastine-mediated induction of NOXA protein and partially protected from vinblastine and ABT-737 treatments (Fig. 2D). Together, these results demonstrate that NOXA induction is required to sensitize these hematopoietic cells to vinblastine plus ABT-737.
Other microtubule disrupting agents sensitize CLL cells to ABT-737
CLL cells were further tested for sensitivity to other vinca alkaloids commonly used in the clinic and an alternative microtubule disrupting agent, combretastatin A4, which binds to a different site on tubulin. Vincristine, vinorelbine and combretastatin A4 all activated JNK and induced NOXA in a concentration- and time- dependent manner similar to vinblastine (Fig. 3A and data not shown). However, a 10–50-fold higher concentration of vinorelbine was required to exhibit the same effects as the other two vinca alkaloids. The correlation between activation of JNK, induction of NOXA and PARP cleavage by each agent is consistent with these effects being the consequence of inhibiting a single target (i.e., microtubule disruption). However, while a correlation with JNK activation is evident, Supplemental Figure S1 demonstrates that this apoptosis is JNK-independent.
Figure 3.
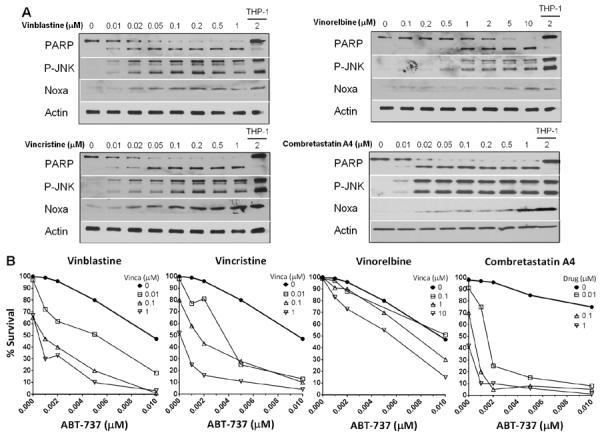
Other microtubule disrupting drugs induce Noxa and sensitize CLL cells to ABT-737. Freshly-isolated lymphocytes from CLL patients were incubated with vinblastine, vincristine, vinorelbine or combretastatin A4 alone (A) or in combination with ABT-737 (B) for 6 h. (A) Cell lysates were analyzed for protein expression, using THP-1 cells as a positive control for protein expression. Representative western blots for each drug are shown (vinca alkaloids n=13, combretastatin A4 n=6). (B) Apoptosis was assessed by chromatin condensation and the percentage of surviving cells is shown. Representative data from one of two patients is shown.
The sensitivity of CLL cells to the combination of ABT-737 and various microtubule disrupting drugs was tested in ABT-737 resistant CLL cells. Vincristine sensitized CLL cells to ABT-737 at comparable concentrations as vinblastine (Fig. 3B), while vinorelbine was less effective at lower concentrations but was still able to enhance apoptosis in the presence of ABT-737. These results are not surprising based on the ability of vincristine and vinblastine to induce NOXA at lower concentrations. Combretastatin A4 also enhanced apoptosis in combination with ABT-737. Overall, this indicates that alternative microtubule disrupting agents that induce NOXA could be used instead of vinblastine to sensitize CLL cells to ABT-737.
Vinblastine does not enhance ABT-737 toxicity in normal lymphocytes or platelets
Normal lymphocytes isolated from donors were resistant to ABT-737 and vinblastine as single agents (Fig. 4A). Incubation with 0.1 μM ABT-737 alone was significantly different from vehicle or vinblastine incubation, but only induced an average of 10% apoptosis in these lymphocyte samples. The combination of vinblastine plus ABT-737 did not increase apoptosis over single agent ABT-737 (Fig. 4A). These results suggest that this drug combination is selective for malignant lymphocytes.
Figure 4.
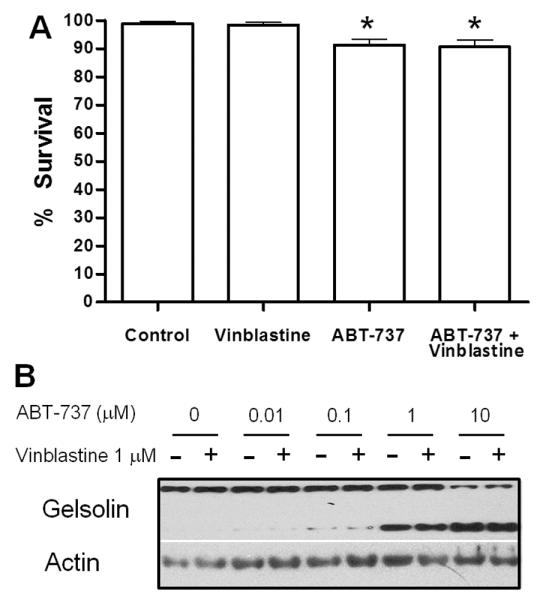
Vinblastine does not enhance ABT-737 toxicity to normal lymphocytes or platelets. (A) Freshly-isolated lymphocytes from normal donors were incubated with 0.1 μM ABT-737, 1 μM vinblastine or a combination of both drugs for 6 h. Apoptosis was scored by chromatin condensation with the percentage of cells surviving shown. Standard error bars are shown (n=10). * p value < 0.05 compared to control. (B) Freshly-isolated platelets from cancer-free patients were incubated with 0–10 μM ABT-737 with or without 1 μM vinblastine for 6 h. Cell lysates were analyzed for protein expression, and gelsolin cleavage was used as a marker for platelet apoptosis. A representative sample from 3 experiments is shown.
As thrombocytopenia is a known consequence of ABT-263 treatment, we tested the impact of our drug combination on platelet survival. Gelsolin cleavage was used as a marker of platelet apoptosis (24, 25). ABT-737 alone at concentrations ≥1 μM induced platelet apoptosis (Fig. 4B). These concentrations are much higher than those required to induce apoptosis in CLL cells. Vinblastine did not induce gelsolin cleavage as a single agent, nor did it enhance cleavage induced by ABT-737 (Fig. 4B).
Vinblastine plus ABT-737 overcomes stroma-mediated resistance
The tumor microenvironment, including T cells expressing CD154 (CD40L), is known to protect malignant cells from spontaneous and drug-induced apoptosis (26–29). Therefore we utilized L4.5 cells expressing CD154 to stimulate CLL survival (30). In the absence of stroma, low concentrations of ABT-737 (0.1 μM) induced apoptosis (Fig. 1). However, CLL cells pre-incubated with L4.5 cells for 24 h were protected from subsequent incubation with high concentrations of ABT-737 (10 μM; Fig. 5A). The L4.5 stromal cells also protected CLL cells from single agent vinblastine-mediated apoptosis. However, the combination of vinblastine plus ABT-737 overcame much of this resistance.
Figure 5.

Vinblastine plus ABT-737 overcomes stroma-mediated drug resistance. Freshly-isolated lymphocytes from CLL patients were incubated with L4.5 stromal cells for 24 h. (A) CLL cells were then incubated with 0–10 μM ABT-737 with or without 1 μM vinblastine for 6 h. Cell lysates were analyzed for protein expression, PARP cleavage was measured by densitometry, and survival is expressed as the percentage uncleaved PARP (n=4). Vinblastine significantly enhanced apoptosis compared to ABT-737 alone, *p<0.05. (B) Untreated CLL cells incubated alone or with L4.5 stromal cells were analyzed for protein expression, with THP-1 cells serving as a positive control. Sample CLL53 was analyzed on a different western blot but the level of the control bands in THP-1 cells were comparable.
CD154 exposure has previously been reported to cause induction of BFL1 and BCLXL in CLL cells (31–33). Fig. 5B shows that L4.5 cells cause a dramatic induction of BCLXL in CLL cells, while 4/5 CLL samples induced some BFL1, albeit to a low level compared to the positive control. There was a very low level of MCL1 that may have increased in 1/5 CLL samples. In contrast, BCL2 protein levels were reduced in 3/5 CLL samples in response to stroma, showing a reciprocal response that still maintains high levels of anti-apoptotic protein expression. These results are consistent with the paradigm of the tumor microenvironment causing upregulation of anti-apoptotic proteins in CLL, protecting them from ABT-737 and other anticancer drugs.
DISCUSSION
Expression of BCL2 proteins is a mechanism of drug resistance for many tumors, providing a rationale for developing targeted drug therapies that can overcome this barrier. This has led to the development of the BCL2 inhibitors ABT-737 and ABT-263. Here we show that many freshly-isolated primary CLL cells are highly sensitive to low concentrations of ABT-737 within an acute time frame of 6 h (Fig. 1). This is in agreement with previous reports showing sensitivity of CLL ex vivo to ABT-737 in both acute and chronic time frames (34, 35). Additional reports show that ABT-737 can induce apoptosis in multiple myeloma and several lymphomas at concentrations similar to those used here (34, 36). The Phase I study of ABT-263 in CLL revealed reduced lymphocytosis and 35% of patients had a partial response (11). Furthermore, ABT-737 has been shown to overcome resistance to several chemotherapy drugs such as doxorubicin, topotecan, etoposide and bortezomib in preclinical models of hematopoietic tumors and solid carcinomas (16, 36–41). Our results reveal a novel opportunity to increase the sensitivity of various leukemias by combining ABT-737 with vinca alkaloids.
As only a small fraction of CLL cells are thought to be actively cycling at any time, this disease should not be sensitive to a mitosis-specific drug such as vinblastine. However, CLL cells are variably sensitive to vinblastine as a single agent, and can undergo apoptosis within a few hours from all phases of the cell cycle. We have previously demonstrated that vinblastine strongly activates JNK and that this is required for apoptosis induced by vinblastine alone or when combined with the CDK9 inhibitor dinaciclib which prevents expression of MCL1 (1, 2). We report here that CLL cells are highly sensitive to the combination of vinblastine and ABT-737 and that this also occurs within only a few hours; yet, activation of JNK is not required for this sensitization. This observation suggests that vinblastine-induced JNK activation may directly or indirectly target BCL2, a function that is redundant in the presence of ABT-737. We previously demonstrated that incubation of NB4 cells with ABT-737 dissociates BIM from BCL2 which then bound to MCL1, preventing apoptosis (3). However, vinblastine also induces NOXA, a pro-apoptotic protein that binds to MCL1 but not BCL2 and this second mechanism explains how it sensitizes cells to ABT-737 (Fig. 2).
CLL cells incubated with CD154-expressing stromal cells demonstrated only low level induction of BFL1, and little if any MCL1, but there was a much more dramatic increase in BCLXL. These results are consistent with previous reports of strong BCLXL induction and variable levels of MCL1 and BFL1 induction in response to CD40L stimulation (42–44). It is interesting that the concentrations of ABT-737 required to kill these resistant CLL cells is similar to the concentration of ABT-737 required to kill platelets which are also dependent on BCLXL. This suggests that ABT-737 may be far less effective at inhibiting BCLXL than BCL2 in cells, and this consideration is also consistent with a recent report of the lower efficacy of ABT-737 against BCLXL (45, 46). NOXA was previously thought to inhibit only MCL1 and BFL1, yet recent reports have suggested it may also inhibit BCLXL (47–49), and our observations are consistent with this possibility. This does not contradict previous reports of sensitization to ABT-737 being mediated by an increase in the NOXA:MCL1 ratio (50), as the induced NOXA could bind to other BCL2 family members in addition to MCL1.
Although a majority of CLL samples were innately sensitive to ABT-737 alone, stratification of these samples enabled us to determine the impact of vinblastine on ABT-737-induced apoptosis in resistant CLL populations. Vinblastine, at a clinically relevant concentration of 0.1 μM (22, 23), reduced the EC50 of ABT-737 by greater than 10-fold for the very resistant CLL subset, and to a lesser extent for those more sensitive to ABT-737 alone. Importantly, vinblastine plus ABT-737 did not sensitize normal lymphocytes, nor did it increase ABT-737-mediated platelet apoptosis at the concentrations that effectively target CLL cells. This suggests that it may be possible to combine the maximally tolerated dose of both drugs for improved efficacy, particularly to target those CLL cells that are very resistant to ABT-737.
Other microtubule destabilizing agents induced NOXA and enhanced apoptosis in combination with ABT-737. The impact of combining vincristine with ABT-737 has been previously reported; synergy was shown in acute lymphocytic leukemia (ALL) cell lines and xenograft models, with ABT-737 plus vincristine delaying tumor growth and increasing overall survival time (38, 51). Additionally, ABT-737 plus vincristine was tested in glioblastoma cell lines, showing an increase in apoptosis albeit at much higher concentrations of ABT-737 (≥ 50 μM) (52). The treatment times and drug concentrations used previously far exceeded those reported here. However, combined with our data, these reports support the concept of improved efficacy by combining a BH3 mimetic with a vinca alkaloid in cancer therapy. The microtubule stabilizing agent paclitaxel, however, did not elicit the same response as vinca alkaloids as it was unable to acutely induce NOXA or sensitize CLL cells alone or in combination with ABT-737 (data not shown), suggesting that all microtubule-targeting agents do not function the same way. This suggests that microtubule destabilization is an initiating event for the induction of NOXA and apoptosis. We have previously implicated the integrated stress response through eIF2-alpha and transcription factors ATF4 and ATF3 in the induction of NOXA by several putative BH3 mimetics (3), but initial experiments suggest only ATF3 may be involved in the induction of NOXA following microtubule disruption.
We show that most CLL have very low MCL1 protein expression compared to cell lines, in agreement with most CLL being highly sensitive to ABT-737 alone (Fig. 1; ref. 8, 35). In the search for predictors of response, several groups have stratified CLL patient samples based on ABT-737 sensitivity, and no correlation between the expression of individual BCL2 proteins and sensitivity was found (14, 39). Similarly, we found no correlation between ABT-737 or vinblastine sensitivity with patient age, gender, status of ZAP70 and CD38 prognostic factors, or basal expression of MCL1 or BFL1 proteins.
Clinical trials of ABT-263 as a single agent in small cell lung cancer and lymphoid malignancies have been reported (11, 21, 53). While some tumor types or subpopulations may be inherently sensitive to ABT-737, many tumor types have variable sensitivity to the single agent and combination therapies are likely necessary. Furthermore, the interaction of tumors with stromal cells within their microenvironment can lead to resistance to single agent drug therapy. Upon oral administration of ABT-263, the time to maximum plasma concentration is about 9 h with a plasma half-life of 17 h. As a consequence, the drug is being administered on a daily basis. Shown here, cell death in response to the vinblastine and ABT-737 combination was acute (within 6 hours) and independent of the cell cycle, reflecting the ability to sensitize tumors with a low growth fraction, such as CLL. The rapid induction of apoptosis also suggests that brief exposure to drugs might be clinically effective. Additionally, the combination of vinblastine and ABT-737 overcame stroma-mediated resistance in CLL cells suggesting that this combination may be more effective than single agent therapies in the clinical setting. Considering the speed with which CLL cells undergo apoptosis when exposed to ABT-737, our data suggest that combining ABT-263 with vinblastine in an acute treatment setting might greatly enhance the therapeutic outcome in patients with hematopoietic malignancies.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by grants from the National Institute of Health (CA50224; A.Eastman), (T32 09658; D.J.P.Bates), Cancer Center Support (CA23108), and a translational research award from the Leukemia & Lymphoma Society (A. Eastman) Support to A.V. Danilov was provided by a National Cancer Institute new faculty award (3P30CA023108-31S4) to the Norris Cotton Cancer Center.
Abbreviation List
- ALL
acute lymphocytic leukemia
- CLL
chronic lymphocytic leukemia
- JNK
c-Jun N-terminal kinase
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate buffered saline
Footnotes
Conflicts of Interest: None of the authors have any conflicts to declare.
Authorship Contribution: Darcy J. P. Bates designed and executed all experiments and wrote the manuscript. Alexey V. Danilov and Christopher H. Lowrey wrote the clinical protocol and recruited CLL patients for this study. Alan Eastman designed and supervised all work and edited the manuscript.
REFERENCES
- 1.Salerni BL, Bates DJ, Albershardt TC, Lowrey CH, Eastman A. Vinblastine induces acute, cell cycle phase-independent apoptosis in some leukemias and lymphomas and can induce acute apoptosis in others when Mcl-1 is suppressed. Mol Cancer Ther. 2010;9:791–802. doi: 10.1158/1535-7163.MCT-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bates DJ, Salerni BL, Lowrey CH, Eastman A. Vinblastine sensitizes leukemia cells to cyclin-dependent kinase inhibitors, inducing acute, cell cycle phase-independent apoptosis. Cancer Biol Ther. 2011;12:314–25. doi: 10.4161/cbt.12.4.16909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albershardt TC, Salerni BL, Soderquist RS, Bates DJ, Pletnev AA, Kisselev AF, et al. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem. 2011;286:24882–95. doi: 10.1074/jbc.M111.255828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998;91:3379–89. [PubMed] [Google Scholar]
- 6.Wong WW, Puthalakath H. Bcl-2 family proteins: the sentinels of the mitochondrial apoptosis pathway. IUBMB Life. 2008;60:390–7. doi: 10.1002/iub.51. [DOI] [PubMed] [Google Scholar]
- 7.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 9.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 10.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–13. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J Clin Oncol. 2011;30:488–96. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 13.Kang MH, Wan Z, Kang YH, Sposto R, Reynolds CP. Mechanism of synergy of N-(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst. 2008;100:580–95. doi: 10.1093/jnci/djn076. [DOI] [PubMed] [Google Scholar]
- 14.Al-Harbi S, Hill BT, Mazumder S, Singh K, Devecchio J, Choudhary G, et al. An anti-apoptotic Bcl-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood. 2011;118:3579–90. doi: 10.1182/blood-2011-03-340364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hikita H, Takehara T, Shimizu S, Kodama T, Shigekawa M, Iwase K, et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology. 2010;52:1310–21. doi: 10.1002/hep.23836. [DOI] [PubMed] [Google Scholar]
- 16.Zall H, Weber A, Besch R, Zantl N, Hacker G. Chemotherapeutic drugs sensitize human renal cell carcinoma cells to ABT-737 by a mechanism involving the Noxa-dependent inactivation of Mcl-1 or A1. Mol Cancer. 2010;9:164. doi: 10.1186/1476-4598-9-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller LA, Goldstein NB, Johannes WU, Walton CH, Fujita M, Norris DA, et al. BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. J Invest Dermatol. 2009;129:964–71. doi: 10.1038/jid.2008.327. [DOI] [PubMed] [Google Scholar]
- 19.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 20.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–59. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owellen RJ, Root MA, Hains FO. Pharmacokinetics of vindesine and vincristine in humans. Cancer Res. 1977;37:2603–7. [PubMed] [Google Scholar]
- 23.Owellen RJ, Hartke CA, Hains FO. Pharmacokinetics and metabolism of vinblastine in humans. Cancer Res. 1977;37:2597–602. [PubMed] [Google Scholar]
- 24.Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Yao Y, Mason KD, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114:663–6. doi: 10.1182/blood-2009-01-200345. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–51. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 26.Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91:2387–96. [PubMed] [Google Scholar]
- 27.Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114:4441–50. doi: 10.1182/blood-2009-07-233718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balakrishnan K, Burger JA, Wierda WG, Gandhi V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood. 2009;113:149–53. doi: 10.1182/blood-2008-02-138560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamilton E, Pearce L, Morgan L, Robinson S, Ware V, Brennan P, et al. Mimicking the tumour microenvironment: three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br J Haematol. 2012;158:589–99. doi: 10.1111/j.1365-2141.2012.09191.x. [DOI] [PubMed] [Google Scholar]
- 30.Neron S, Pelletier A, Chevrier MC, Monier G, Lemieux R, Darveau A. Induction of LFA-1 independent human B cell proliferation and differentiation by binding of CD40 with its ligand. Immunol Invest. 1996;25:79–89. doi: 10.3109/08820139609059292. [DOI] [PubMed] [Google Scholar]
- 31.Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–13. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- 32.Kater AP, Evers LM, Remmerswaal EB, Jaspers A, Oosterwijk MF, van Lier RA, et al. CD40 stimulation of B-cell chronic lymphocytic leukaemia cells enhances the anti-apoptotic profile, but also Bid expression and cells remain susceptible to autologous cytotoxic T-lymphocyte attack. Br J Haematol. 2004;127:404–15. doi: 10.1111/j.1365-2141.2004.05225.x. [DOI] [PubMed] [Google Scholar]
- 33.Smit LA, Hallaert DY, Spijker R, de Goeij B, Jaspers A, Kater AP, et al. Differential Noxa/Mcl-1 balance in peripheral versus lymph node chronic lymphocytic leukemia cells correlates with survival capacity. Blood. 2007;109:1660–8. doi: 10.1182/blood-2006-05-021683. [DOI] [PubMed] [Google Scholar]
- 34.Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P, et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ. 2008;15:820–30. doi: 10.1038/cdd.2008.25. [DOI] [PubMed] [Google Scholar]
- 35.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chauhan D, Velankar M, Brahmandam M, Hideshima T, Podar K, Richardson P, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26:2374–80. doi: 10.1038/sj.onc.1210028. [DOI] [PubMed] [Google Scholar]
- 37.Ugarenko M, Nudelman A, Rephaeli A, Kimura K, Phillips DR, Cutts SM. ABT-737 overcomes Bcl-2 mediated resistance to doxorubicin-DNA adducts. Biochem Pharmacol. 2010;79:339–49. doi: 10.1016/j.bcp.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 38.High LM, Szymanska B, Wilczynska-Kalak U, Barber N, O'Brien R, Khaw SL, et al. The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs. Mol Pharmacol. 2010;77:483–94. doi: 10.1124/mol.109.060780. [DOI] [PubMed] [Google Scholar]
- 39.Mason KD, Khaw SL, Rayeroux KC, Chew E, Lee EF, Fairlie WD, et al. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia. 2009;23:2034–41. doi: 10.1038/leu.2009.151. [DOI] [PubMed] [Google Scholar]
- 40.Paoluzzi L, Gonen M, Bhagat G, Furman RR, Gardner JR, Scotto L, et al. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood. 2008;112:2906–16. doi: 10.1182/blood-2007-12-130781. [DOI] [PubMed] [Google Scholar]
- 41.Whitecross KF, Alsop AE, Cluse LA, Wiegmans A, Banks KM, Coomans C, et al. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood. 2009;113:1982–91. doi: 10.1182/blood-2008-05-156851. [DOI] [PubMed] [Google Scholar]
- 42.Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100:1795–801. [PubMed] [Google Scholar]
- 43.Kitada S, Zapata JM, Andreeff M, Reed JC. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;106:995–1004. doi: 10.1046/j.1365-2141.1999.01642.x. [DOI] [PubMed] [Google Scholar]
- 44.Willimott S, Baou M, Naresh K, Wagner SD. CD154 induces a switch in pro-survival Bcl-2 family members in chronic lymphocytic leukaemia. Br J Haematol. 2007;138:721–32. doi: 10.1111/j.1365-2141.2007.06717.x. [DOI] [PubMed] [Google Scholar]
- 45.Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119:5807–16. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than BclxL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis. 2012;3:e366. doi: 10.1038/cddis.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Lopez H, George NM, Liu X, Pang X, Luo X. Selective involvement of BH3-only proteins and differential targets of Noxa in diverse apoptotic pathways. Cell Death Differ. 2011;18:864–73. doi: 10.1038/cdd.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez H, Zhang L, George NM, Liu X, Pang X, Evans JJ, et al. Perturbation of the Bcl-2 network and an induced Noxa/Bcl-xL interaction trigger mitochondrial dysfunction after DNA damage. J Biol Chem. 2010;285:15016–26. doi: 10.1074/jbc.M109.086231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hagenbuchner J, Ausserlechner MJ, Porto V, David R, Meister B, Bodner M, et al. The anti-apoptotic protein BCL2L1/Bcl-xL is neutralized by pro-apoptotic PMAIP1/Noxa in neuroblastoma, thereby determining bortezomib sensitivity independent of prosurvival MCL1 expression. J Biol Chem. 2010;285:6904–12. doi: 10.1074/jbc.M109.038331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tromp JM, Geest CR, Breij EC, Elias JA, van Laar J, Luijks DM, et al. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18:487–98. doi: 10.1158/1078-0432.CCR-11-1440. [DOI] [PubMed] [Google Scholar]
- 51.Kang MH, Kang YH, Szymanska B, Wilczynska-Kalak U, Sheard MA, Harned TM, et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood. 2007;110:2057–66. doi: 10.1182/blood-2007-03-080325. [DOI] [PubMed] [Google Scholar]
- 52.Tagscherer KE, Fassl A, Campos B, Farhadi M, Kraemer A, Bock BC, et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene. 2008;27:6646–56. doi: 10.1038/onc.2008.259. [DOI] [PubMed] [Google Scholar]
- 53.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.