

Abstract
Anti-herpes simplex virus (HSV) drug acyclovir (ACV) is phosphorylated by the viral thymidine kinase (TK), but not the cellular TK. Phosphorylated ACV inhibits cellular DNA synthesis and kills the infected cells. We hypothesize that ACV monophosphate (ACVP), which is an activated metabolite of ACV, should be efficient in killing cells independent of HSV-TK. If so, ACVP should be a cytotoxic agent if properly delivered to the cancer cells. The Lipid/Calcium/Phosphate (LCP) nanoparticles (NPs) with a membrane/core structure were used to encapsulate ACVP to facilitate the targeted delivery of ACVP to the tumor. The LCP NPs showed entrapment efficiency of ~69%, the nano-scaled particle size and positive zeta potential. Moreover, ACVP-loaded LCP NPs (A-LCP NPs) exhibited concentration-dependent cytotoxicity against H460 cells and increased S-phase arrest. More importantly, a significant reduction of the tumor volume over 4 days following administration (p<0.05~0.005) of A-LCP NPs, suggests excellent in vivo efficacy. Whereas, two free drugs (ACV and ACVP) and blank LCP NPs showed little or no therapeutic effect. It was also found that the high efficacy of A-LCP NPs was associated with the ability to induce dramatic apoptosis of the tumor cells, as well as significantly inhibit tumor cell proliferation and cell cycle progression. In conclusion, with the help of LCP NPs, monophosphorylation modification of ACV can successfully modify an HSV-TK-dependent antiviral drug into an anti-tumor drug.
Keywords: Acyclovir monophosphate, Calcium phosphate, Nanoparticles, tumor therapy
Introduction
Acyclovir (ACV), 9-[(2-hydroxyethoxy) methyl] guanine, has been widely used for the treatment of herpes simplex virus (HSV) infection. Its conversion into the corresponding monophosphate (ACVP) is mediated by the viral thymidine kinase (HSV-TK) in the HSV-infected cells. ACV-P was further phosphorylated to ACV diphosphate and triphosphate by cellular kinase in the host cells, which is capable of blocking DNA synthesis through the inhibition of the viral DNA polymerase and terminating the chain elongation of the viral DNA [1,2]. Taking advantage of this characteristic, gene therapy using a combination of ACV or ganciclovir (GCV) and the HSV-TK gene has been developed for the treatment of cancers in previous studies [3]. More interestingly, the bystander effect or metabolic cooperation correlating with the occurrence of gap junctions also contributes to the regression of tumors in these therapy modalities, despite that there was only a small proportion of tumor cells expressing TK [4,5,6].
Previous studies have demonstrated the potent antitumor effects of HSV-TK/prodrug gene therapy in animal models such as malignant gliomas [7,8]. However, clinical trials of HSV-TK-based suicide gene therapy have generally been disappointing [9]; the low transfection efficiency of the HSV-TK gene, poor prodrug activation kinetics [10,11], and the poor lipophilicity of ACV or GCV which prevents penetration of the cell membrane may all contribute to the failures. These obstacles impact the extended application of this strategy in the clinic. These studies also indicate that ACV may be more appropriate for clinical application than GCV because it is more lipophilic and less toxic [8]. Targeted delivery of the active ingredient, which is ACVP in this case, should be a major emphasis if we hope to improve the antitumor activity and decrease potential toxicity of these therapies.
The calcium phosphate (CaP)-based nanoparticles have frequently been used to deliver genes because their escape of the endosomes is pH-sensitive, which contributes to the effective drug release, biocompatibility, biodegradability, and minimal toxicity [12,13]. In our previous study, we developed a novel vector, Lipid/Calcium/Phosphate (LCP) nanoparticles (NPs) [14,15]. Compared to the CaP-based formulation, which contains no lipid, the lipid coating of the LCP NPs better prevents the core from aggregation during the preparation of the nanoparticles and facilitates the formation of the outer-leaflet layer with PEG-lipid derivates. Yang, et al. (2012) verified the potential of this system for the targeted delivery of siRNA [16]. Treatment with a relatively low dose of therapeutic siRNA in LCP NPs caused a ~70–80% reduction of lung metastases. This system also significantly prolonged the mean survival time of mice without the associated toxicity.
In the present study, ACVP was synthesized, avoiding the limiting step of monophosphorylation that depends on the effective transfection of HSV-TK gene during gene therapy. The phosphorylation also provided an active group to enable ACV to bind with CaP, creating a high encapsulation efficiency. LCP PEGylated with anisamide-containing, PEG-lipid conjugates (DSPE-PEG-AA) encapsulate ACVP and bind to the tumor cells that overexpress the sigma receptor. We hypothesized that LCP would facilitate the targeted delivery of ACVP to the tumor through the synergistic mechanism of ligand-mediated, specific tumor-targeting, the enhanced permeability and retention (EPR) effect, and reduced reticuloendothelial system (RES) uptake (as shown in scheme 1). These characteristics should facilitate the transmembrane transport of ACVP, enhance its tumor accumulation, improve the antitumor effect, and also avoid the peripheral toxicity. We investigated the in vitro and in vivo activity of ACVP-loaded LCP nanoparticles (A-LCP NPs). Moreover, the potential mechanism through which ACVP and A-LCP NPs induce DNA damage was studied by cell cycle, TdT-mediated dUTP Nick-End Labeling (TUNEL), immunohistochemical and Weston blot assays.
Scheme 1.

Illustration of the structure, uptake by tumor cells and intracellular delivery of A-LCP NPs.
2. Materials and Methods
2.1. Materials
ACV was purchased from Carbosynth Limited (Compton Berkshire, UK). 1, 2-dioleoyl-3-trimethylammonium-propane chloride salt (DOTAP), cholesterol, 1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000) ammonium salt (DSPE-PEG) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). DSPE-PE-anisamide (DSPE-PEG-AA) was synthesized in our lab as described [17]. ACVP was also synthesized by following a previously reported procedure [18]. The purity of ACVP was 92.5%. Other chemicals were obtained from Sigma-Aldrich (St. Louis, MO) and were not purified further.
The H460 (H460-TK−) cells originally obtained from American Type Culture Collection (ATCC) were cultured in an RPMI 1640 cell culture medium with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). The HSV-TK-expressing H460 (H460-TK+) cells were obtained by transfection with pCDNA3.1-HSV1-TK using lipofectamine2000. Cells were seeded at 1×105 cells in a 10-cm plate and grown overnight in the growth medium (to reach 90% confluence at the time of transfection). The primary growth medium was removed and replaced with Opti-MEM®I. The transfection complex was added and allowed to incubate for 4 h. The medium was then removed and replaced with RPMI 1640 cell culture medium. After 24 h, the cells were seeded for further study. Cells were cultivated in a humidified incubator at 37°C and 5% CO2.
Mice were purchased from the National Cancer Institute (Bethesda, MD). All experiments performed on animals were in accordance with and approved by the Institutional Animal Care and Use Committees at the University of North Carolina at Chapel Hill.
2.2 Preparation and characterization of A-LCP NPs
The LCP NPs were prepared using the methods described in our previous studies with some additional improvement [14]. Three hundred μL of P phase including 3.125 mM NaHPO4 (pH=9.0) and 24 mM ACVP were dispersed in 10 mL cyclohexane/Igepal CO-520 (71/29, v/v). The Ca phase was prepared by adding 300 μL CaCl2 (2.5 mM) into a separate oil phase. Four hundred μL (20 mM) dioleoylphosphatydic acid (DOPA) in chloroform was added to the P phase. After mixing the two microemulsions for 20 min, 20 mL of absolute ethanol was added to the mixture and centrifuged at 10,000 g for 20 min to pellet the LCP cores. After being extensively washed by ethanol twice and dried under N2, the LCP core pellets were dissolved in 1 mL chloroform and stored at −20°C for further use.
A-LCP NPs were prepared by mixing 250 μL of the cores with 145 μL of 4 mM Cholesterol, 4 mM DOTAP, 2.7 mM DSPE–PEG-2000 and 0.6 mM DSPE–PEG–AA. After evaporating the chloroform, the residual lipid was dissolved in 80 μL of THF-ethanol solution (3:5, v/v). One hundred and sixty μL of distilled water was added to form the NPs, and then dialyzed against water for 1 h (dialysis membrane MWCO 20,000). The amount of ACVP in the nanoparticles was measured at 254 nm using a DU 800 spectrophotometer (Beckman Coulter Inc.). The solvent was composed of THF-1M hydrochloric acid solution (7:3, v/v). The zeta potential of A-LCP nanoparticles were determined through dynamic light scattering measurements (Malvern ZetaSizer Nano series, Westborough, MA). The shape and surface morphology of the nanoparticles were observed by using a JEOL 100CX II transmission electron microscope (TEM) (Tokyo, Japan).
2.3 In vitro cytotoxicity studies
The cytotoxicity of the A-LCP NPs was assessed using the MTT assay. The H460-TK− and H460-TK+ cells were seeded at a density of 1×105 cells per well in 96-well microtitre plates, respectively and incubated for 24 h. The cells were treated with different formulations for 48 h. After incubation, MTT solution (15 μL, 5 mg/mL in PBS) was then added to each well and the cells were incubated further for 4 h at 37°C. The media were removed and the cells were dissolved in DMSO. Absorbance at 570 nm was measured with a microplate reader. Cell viability (%) was calculated as (OD of test group/OD of control group) ×100.
2.4 Cell cycle arrest
H460 cells growing exponentially were seeded at 1×105 cells/ml in 6-well plates. Cells were treated with free ACV, free ACVP and A-LCP NPs for 48 h. The PBS solution served as the control. Ice-cold, 70% ethanol was used to fix the cells at 4°C overnight. After centrifuging to remove the supernatant, the cells were re-suspended with 1 mL staining buffer and washed once. After re-suspension, the cells were incubated with 10 μL of RNase A (10 mg/mL) at 37°C for 30 min, and were stained with 5 μL of propidium iodide (1 mg/ml) at room temperature for 30 min. The cell-cycle analysis was performed on a FACS Canto flow cytometry (BD Biosciences, USA). The data were analyzed using ModFit LT V3.3.11 software.
2.5. In vivo anti-tumor activity
In vivo anti-tumor activity of the NPs was evaluated in H460-bearing nude mice. Female nude mice (6–8 weeks) were used in all studies. Subcutaneous injections of 5×106 cells in 100 μL of PBS into the right flanks of the mice were used to establish the xenograft model. When the tumor volume reached 100–200 mm3, mice were randomly divided into five groups and were injected with normal saline (the control group), free ACV solution, free ACVP solution, A-LCP NPs solution or the drug-free LCP NPs (i.e. blank NPs). A drug dose of 20 mg/kg was used for all treatments. Therapy was continued five times at 2-day intervals using tail vein injections. Changes in tumor mass were used as an index of antitumor activity of the formulations tested. The lengths of the longest tumor axis (a, mm) and the vertical axis (b, mm) were measured with a caliper, and the tumor volume (v, mm3) was calculated using the following equation: V=0.5×a×b2. Animals were sacrificed and tumor mass was measured the day after the last administration. Furthermore, the toxicity of the formulations was determined by monitoring the animal behavior and the weight loss. Pathologic examination of the major tissues by Hematoxylin and Eosin (HE) staining was performed to investigate the potential side effect of the formulations.
2.6 TUNEL assay and immuno-histochemical staining
H460 tumor-bearing nude mice were given three daily IV injections of each formulation at a drug dose of 20 mg/Kg. Mice were sacrificed 24 h after the final injection, and tumors were fixed in 10% formalin for at least 24 h before being embedded in paraffin and sectioned at a thickness of 5 μm. In vivo tumor cell apoptosis was determined using the TUNEL assay and HE staining assay. The TUNEL staining was performed as recommended by the manufacturer (Promega) and DAPI mounting medium was dropped on the sections for nucleus staining. Images of TUNEL-stained tumor sections were taken with a fluorescence microscope (Nikon Corp., Tokyo, Japan). The TUNEL-positive cells were counted using the Image J software.
Proliferation of tumor cells after scheduled therapy was also detected using immunohistochemistry, specifically through the use of an antibody against proliferating cell nuclear antigen (PCNA) (1:200 dilution, Santa Cruz). The immunohistochemistry was performed using a mouse-specific HRP/DAB detection IHC kit as recommended by the manufacturer (Abcam, Cambridge, MA).
2.7 Western blot analysis
H460 tumor-bearing mice were sacrificed at 24 h after three daily IV injections, and the tumors excised were homogenized in RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 1% Triton, 0.5% deoxycholate, 2 mM EDTA) supplemented with a 100× protease inhibitor cocktail (Sigma-Aldrich). Forty μg of protein per lane was separated by 4–12% SDS-PAGE electrophoresis (Invitrogen) before being transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). The membranes were blocked for 1 h with 5% silk milk at room temperature and then incubated with anti-phospho-γ-H2AX pSer 139 rabbit polyclonal antibodies (1:2000 dilution, Pierce, Thermo scientific) and NF-κB p65 (F-6) mouse monoclonal antibody (1:500 dilution; Santa Cruz biotechnology, Inc.) overnight at 4°C. β-actin antibody (1:4000 dilution; Santa Cruz biotechnology, Inc.) was probed as the loading control. The membranes were washed three times and then incubated with a secondary antibody (1:4000 dilution; Santa Cruz biotechnology, Inc.) at room temperature for 1 h. Finally, the membranes were washed 4 times and developed using an enhanced chemiluminescence system according to the manufacturer’s instructions (Thermo scientific).
2.8. Statistical analysis
Data were expressed as mean ± SD. The statistical significance of group differences was analyzed using one-way unweighted mean analysis of variance (ANOVA) and a value of p< 0.05 was considered significant.
3. Results and discussion
3.1. Preparation and characterization of A-LCP NPs
Previous studies have observed that the CaP-based nanoparticles for DNA or nucleoside analogues suffer from large size, instability and non-specific uptake by the RES [19]. In this study, amphiphilic DOPA was used to stabilize the CaP core. The lipid coating also aided in the prevention of the aggregation of the particles. Moreover, the surface modification with PEGylated lipids, such as DSPE-PEG and DSPE-PEG-AA, as targeting ligands in the outer layer further benefited the stability and tumor targeted delivery of LCP via sigma-receptor-mediated endocytosis.
The morphology of the A-LCP cores and NPs was examined by TEM without negative staining (Fig. 1). Both the cores and NPs exhibited a uniform, spherical shape. The LCP cores were about 10–20 nm in diameter, and showed the typical hollow structure as observed in our previous studies [14], which might provide an opportunity to entrap soluble drugs. LCP NPs exhibited good drug loading capacity for ACVP; the entrapped efficiency was ~70 %.The particle size of A-LCP NPs were about 20–30 nm (Fig. 1B), indicating a good nano-scaled size for tumor delivery. The zeta potential of the LCP was also determined by DLS. The A-LCP NPs had a positive surface charge of 27.6±0.7 mV, which depended on the amount of DOTAP in the formulations.
Fig. 1.

TEM images of A-LCP core (A) and nanoparticles (B).
3.2 In vitro cytotoxicity studies
The potential of ACVP and A-LCP NPs to kill the tumor cells were investigated by the MTT assay. Cell survival was measured as a function of drug concentration. As shown in Fig. 2A, free ACV had no cytotoxicity against HSV-TK− H460 cells in vitro, while free ACVP showed the loss of cell viability of about 25–30% at a higher concentration (0.36 mM). Importantly, A-LCP NPs exhibited a concentration-dependent cytotoxicity against H460 cells. The IC50 of A-LCP NPs was 0.31 mM, which was 2.8 times lower than that of ACVP (0.88 mM). Additionally, blank LCP NPs had little cytotoxicity (data not shown), suggesting its good biocompatibility.
Fig. 2.
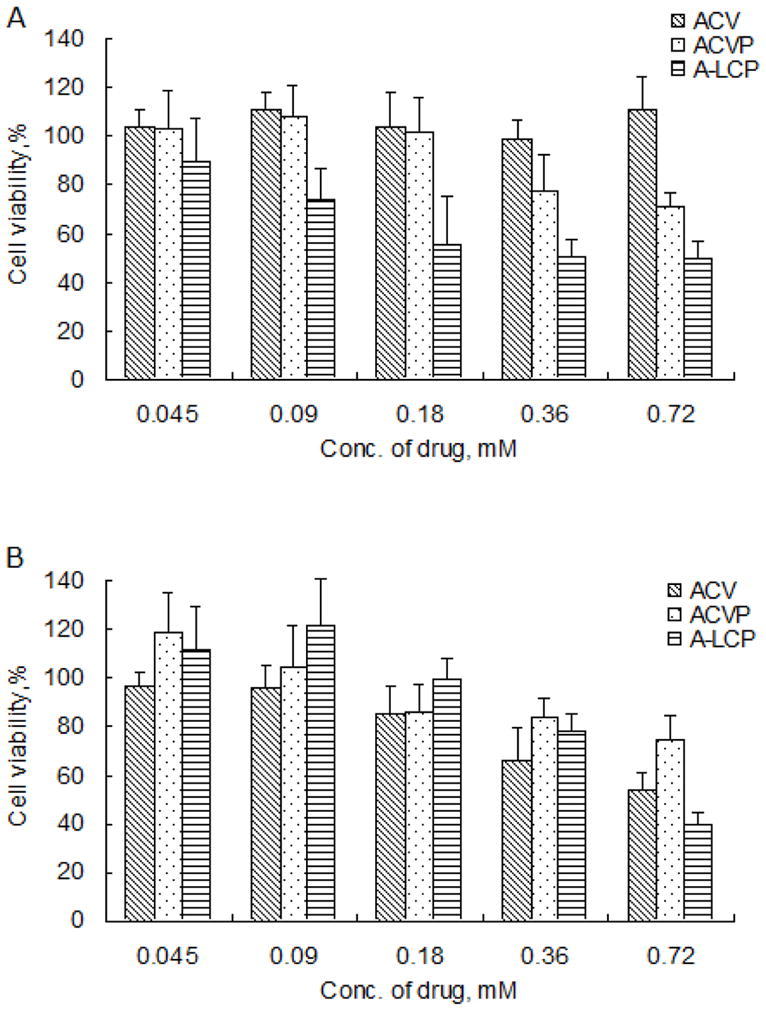
Cytotoxicity of ACV, ACVP and A-LCP NPs against HSV-TK− H460 cells (A) and HSV-TK+ H460 cells (B) in vitro. Cell survival fractions were assessed by MTT assay. (n=6, mean ± S.D.)
Next, we investigated the effect of the HSV-TK genes on the cytotoxicity of various formulations. A dramatic loss of cell viability treated with free ACV was obtained in HSV-TK+ H460 cells (in Fig. 2B) as suggested in previous studies [20–21], which was attributed to the effective phosphorylation of the ACV by HSV-TK. As for ACVP, there was no significant difference between the cytotoxicity in HSV-TK− and HSV-TK+ H460 cells. These results also indicate that chemically monophosphorylated ACV was equivalent to the combination of HSV-TK gene therapy and ACV, consistent with our working hypothesis. In addition, it was also found that the cytotoxicity of ACVP against HSV-TK+ H460 cells was not better than that of ACV, probably because it is more difficult for the relatively negatively-charged ACVP to be transported through the cell membrane compared to ACV. The negative charge of ACVP may also affect its potency of bystander effect due to the weak ionic selectivity of gap junction’s permeability [22]. The results also suggested that the use of the appropriate carrier was critical for the in vivo delivery of ACVP.
3.3 Cell cycle assay
Cell cycle checkpoints have been identified as critical molecular regulators that must be overcome in order to achieve passage programmed cell death [23]. A slowing of progression through the S and G2-M phases, are used as DNA-damage checkpoints in addition to the assessment of the G0-G1 phase. Some studies described that the nucleoside analogues GCV and ACV converted into GCV and ACV tri-phosphate, which are incorporated into nascent DNA strands during the S phase and results in chain termination and single strand breaks in the newly synthesized DNA [24]. Komindky et al. (2000) also reported that the inhibitory effects of ACV correlated with a delay/block in the S phase [25]. Therefore, in order to study the possible mechanism of action of ACVP, we investigated the cell cycle of H460 cells treated with ACVP and A-LCP NPs for 48 h.
As illustrated in Fig. 3, the percentage of cells in the S phase increased after 48 h incubation with free ACV, ACVP or A-LCP NPs, indicating the cell cycle was arrested in the S phase. There was no significant change in the mechanism of action of ACV after both phosphorylation and encapsulation in the NPs. Interestingly, compared to the free ACV, ACVP and A-LCP NPs yielded much stronger inhibition of cell cycle. The percentage of cells in the S phase after treatment with A-LCP NPs was 1.9 times of the control (49.1±2.1% v.s. 25.6±1.1%). Moreover, it was also higher than that of ACVP group (p<0.05). On the other hand, blank LCP NPs almost had no effect on the cell cycle progression of tumor cells. These results indicated that LCP NPs efficiently improved the S-phase suppression of ACVP, probably because the NPs facilitated the transmembrane delivery of the drug through sigma receptor-mediated endocytosis.
Fig. 3.

Cell cycle analysis. (A) Typical flow cytometric diagrams analyzed by ModFit LT V3.3.11 software. (B) Cell cycle distribution of the H460 cells treated with ACV, ACVP, A-LCP NPs and blank LCP NPs for 48h. The data are shown as mean ± SD (n=3).
3.4 In vivo anti-tumor activity
Although the in vitro cytotoxicity assay revealed that ACVP showed improved ability to cause the death of H460 cells over ACV, the in vivo efficacy of ACVP is still of concern because the delivery of drugs to the tumor may be affected by the complicated physiological environment such as protein binding, nonselective distribution and obstacles of the cell membrane.
As shown in Fig. 4A, there was no significant reduction of the tumor volume in mice treated with free ACV and ACVP compared to the control (saline) after a schedule of multiple doses (p>0.05). However, the mean tumor volume in mice treated with free ACVP was slightly smaller than those treated with the control. A-LCP NPs exhibited significantly more effective inhibition of tumor growth; the tumor volume in the group treated with A-LCP NPs were significantly smaller than those of other three groups (p<0.05~0.005) beginning 4 days following the first injection. A-LCP NPs also reduced tumor growth by 61.3% (p<0.05) compared to the negative control. Moreover, the A-LCP NPs induced slower tumor growth than other groups. The same phenomenon was observed in tumor weight after the termination of the therapy (in Fig. 4B), significantly decreased tumor weights of mice treated with A-LCP NPs were obtained compared to the control, ACV and ACVP groups (all of p<0.005). In addition, it was also noticed that blank LCP NPs showed no obvious suppression of tumor growth. These results indicated that the excellent in vivo efficacy of A-LCP NPs was attributed to the encapsulation of ACVP within the NPs.
Fig. 4.
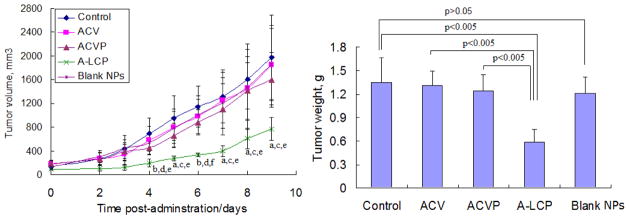
In vivo efficacy assay of A-LCP NPs. (A) Tumor growth kinetics with dosing every other day for 5 times. Data are expressed as mean ± S.D, (n=5). a p<0.05 and b p<0.01 vs. the control, c p<0.05 and d p<0.01 vs. free ACV, e p<0.05 and f p<0.005 vs. free ACVP. (B) Tumor weights of tumor-bearing mice after the treatment. Data are expressed as mean ± S.D, (n=5).
In general, free drugs exhibit rapid inactivation, excretion and non-specific distribution after systemic administration. Free ACVP and ACV also show low penetration through the cell membrane due to their poor lipophilicity. These disadvantages will most likely result in little drug accumulation in the tumor, which will affect the in vivo efficacy of the drugs. It is well known that the prolonged blood circulation is a prerequisite for EPR effect. Our previous data showed that comparing to free drug, drug loaded LCP NPs increased the area under the curve (AUC) for more than 25-fold and the mean residence time (MRT) about 7-fold [26]. In the present study, A-LCP NPs showed a dramatically improved inhibition of tumor growth, which was attributed to endocytosis mediated by the sigma receptors as well as the EPR effect [16,17,27]. Other possible mechanisms were involved in the success of the delivery system. First, the CaP-based formulation improves efficiency of the release of drugs from the endosomes because CaP can cause endosome swelling and rupture through increased osmotic pressure after they are dissolved in the low pH microenvironment of the endosome [14,16]. Second, PEG is the most widely used moiety for surface modification of the NPs. PEGylation retards the rapid uptake of NPs by the mononuclear phagocyte system [17,28]. The membrane/core structure of LCP allows a relatively high density of PEG (about 20% of total outer leaflet lipids) on the surface, which promotes the formation of a dense PEG layer. The surface modification with PEG may also minimize the interaction of the particles with circulating blood components and allows the drug to distribute into the tumor.
Side effects present one of the major obstacles for cancer chemotherapy. In this study, the toxicity of free drugs and A-LCP NPs on the major tissues, including the liver, spleen, kidney, heart and lungs, was examined using HE staining after long-term treatment. No obvious damage was observed in these tissues after their treatment with A-LCP NPs (Fig. 5). Consistently, no other side effects, for example decreased body weight and noticeable change in activity, were observed (data not shown) [16]. Thus, these findings indicate the potential of clinical application of the A-LCP NPs.
Fig. 5.
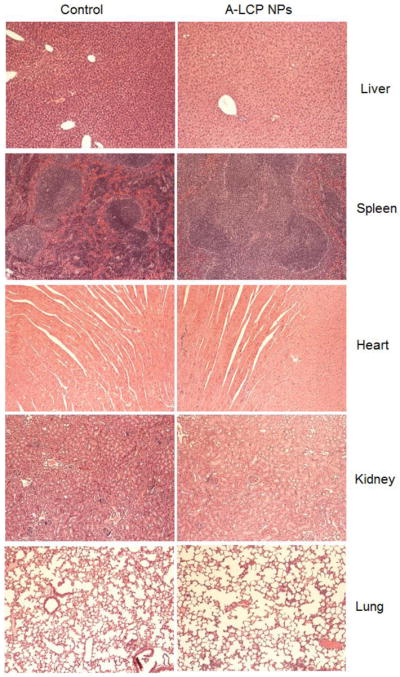
Histological analysis of major organs. HE staining of the tissue sections from H460 tumor-bearing nude mice after 5 doses.
3.5 TUNEL assay and immuno-histochemical staining
Next, we focused on the mechanism through which A-LCP NPs and ACVP caused the tumor cell death. TUNEL assay, immunohistochemistry, HE staining assay and western blot analyses were performed. To avoid the possible interference of tumor self-apoptosis due to extensive tumor growth after long-term treatment, the tumor-bearing mice were sacrificed 24 h after three daily IV injections.
As illustrated in Fig. 6, A-LCP NPs induced the most effective apoptosis of cells in H460 xenograft tumors compared with the control, free ACV and ACVP. The percentage of apoptotic cells of (7.9±4.1%) was 6.2 and 16.8 times higher than those of the ACVP and ACV, respectively. These results suggest the essential role of LCP formulation for in vivo efficacy. HE staining also indicated that A-LCP NPs caused the most severe necrosis in the tumor compared to other formulations.
Fig. 6.

In vivo tumor cell apoptosis induced by A-LCP NPs. (A) TUNEL assay of the tumor sections from H460 tumor-bearing nude mice after a schedule of 3 doses. TUNEL positive cells are shown as green dots (top) and the nuclei stained by DAPI are blue (bottom). (B) Quantification of apoptosis. *p<0.05 vs. the control (untreated), (n=8). (C) HE staining of the tumors.
The inhibition of tumor cell proliferation following different treatments was also investigated. PCNA is a very useful marker of cellular proliferation due to the specifically regulated synthesis of PCNA in only the late G1 and S phases of the cell cycle. Compared to the control (untreated group), both free ACVP and A-LCP NPs significantly decreased the number of PCNA positive cells in H460 xenograft tumors (Fig. 7). In particular, the inhibition of cell proliferation in the A-LCP NP treated group was more obvious. However, free ACV had few inhibitory effects on proliferation, probably due to the metabolism of free drugs and their lack of accumulation in the tumor site.
Fig. 7.

(A) PCNA immuno-histochemistry assay of the tumor sections from H460 tumor-bearing nude mice after a schedule of 3 doses. The PCNA positive nuclei are shown as brown dots. (B) The percentage of proliferation cells. **p<0.01 and***p<0.001 vs. the control (untreated), (n=8).
3.6 Western blot analysis
A cell cycle assay in vitro has verified the obvious enhancement of S-phase delay of the tumor cells treated with ACVP and A-LCP NPs. We also investigated if drugs or NPs had activated major responses to DNA damage. H2AX-Ser 139 phosphorylation and activation of NF-κB were monitored as an indicator of DNA-damage activation.
DNA double-strand breaks are one of the most dangerous types of DNA damage that occur within the cell and their signaling and repair is critical for all cells and organisms [29]. It is well known that H2AX phosphorylation plays a very early and important role in the cellular response to DNA double-strand breaks [30]. Western blot analysis (Fig. 8) indicated that the phosphorylation of γ-H2AX was significantly elevated after the treatment of A-LCP NPs in the tumor, and its expression level was higher than those of free ACVP and ACV. The data indicated that A-LCP NPs could induce DNA damage more effectively than ACVP and ACV. Moreover, ACVP induced higher level of DNA damage than ACV.
Fig. 8.
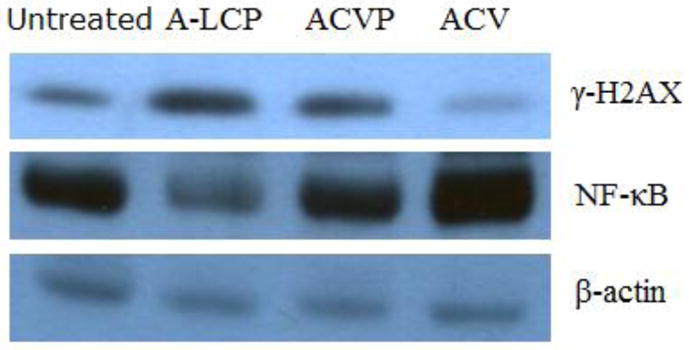
Western blot assay for γ-H2AX and NF-κB. Protein in the tumors from H460 tumor-bearing nude mice after a schedule of 3 doses was extracted and analyzed.
NF-κB is a protein complex that controls the transcription of DNA. It also has been connected with the control of apoptosis, cell cycle, differentiation and cell migration in response to DNA damage or cytokine treatment [31,32]. Inhibition of NF-κB in some cancer cell lines leads to an apoptotic response [33]. In this study, A-LCP NPs significantly down-regulated the expression of NF-κB, suggesting that the apoptosis of H460 cells by the induction of A-LCP NPs involved in blocking NF-κB activation. It was also found that free ACVP and ACV did not affect the NF-κB activation.
4. Conclusions
In this study, ACVP, a phosphorylated derivative of ACV was encapsulated by LCP NPs with the encapsulation efficiency of over 60% and the particles size of less than 50 nm. More importantly, LCP NPs facilitated the delivery of ACVP to the tumor, which contributed to the significantly enhanced inhibition of tumor growth with no obvious toxicity. This high efficacy of A-LCP NPs was associated with its ability to induce dramatic apoptosis of tumor cells as well as significantly inhibit tumor cell proliferation and cell cycle progression. In conclusion, based on the efficient delivery of LCP NPs, monophosphorylation modification of ACV by chemical method was an effective approach to turn an HSV-TK-dependent antiviral to an anti-tumor drug.
Supplementary Material
Acknowledgments
This project was supported by NIH grants CA151652 and CA149363. Dr. Jing Yao’s work in UNC was sponsored by Qing Lan Project and Jiangsu Overseas Research and Training Program for University Prominent Young and Middle-aged Teachers and Presidents. We thank Kelly Racette for editing the manuscript.
List of abbreviations
- ACV
Acyclovir
- ACVP
Acyclovir monophosphate
- A-LCP NPs
ACVP-loaded LCP nanoparticles
- CaP
Calcium phosphate
- HSV-TK
Herpes simplex virus thymidine kinase
- DOPA
Dioleoylphosphatydic acid
- DOTAP
1,2-dioleoyl-3-trimethylammonium-propane chloride salt
- DSPE-PEG
1,2-distearoryl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol-2000) ammonium salt
- DSPE-PEG-AA
DSPE-PEG-anisamide
- EPR
Enhanced permeability and retention effect
- GCV
Ganciclovir
- HE
Hematoxylin and Eosin
- LCP
Lipid/Calcium/Phosphate
- NPs
nanoparticles
- PCNA
Proliferating cell nuclear antigen
- RES
Reticuloendothelial system
- TEM
Transmission electron microscope
- TUNEL
TdT-mediated dUTP Nick-End Labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Elion GB. Mechanism of action and selectivity of acyclovir. Am J Med. 1982;73:7–13. doi: 10.1016/0002-9343(82)90055-9. [DOI] [PubMed] [Google Scholar]
- 2.Furman PA, StClair MH, Fyfe JA, Rideout JL, Keller PM, Elion GB. Inhibition of herpes simplex virus-induced DNA polymerase activity and viral DNA replication by 9-(2-hydroxyethoxymethyl) guanine and its triphosphate. J Virol. 1979;32:72–77. doi: 10.1128/jvi.32.1.72-77.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freund CT, Sutton MA, Dang T, Contant CF, Rowley D, Lerner SP. Adenovirus-mediated combination suicide and cytokine gene therapy for bladder cancer. Anticancer Res. 2000;20:1359–1365. [PubMed] [Google Scholar]
- 4.Curver KM, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer drugs for treatment of experimental brain tumor. Sciences. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 5.Ram Z, Culver KW, Walbridge S, Blaese RM, Oldfield EH. In situ restroviral-mediated gene transfer for the treatment of brain tumors in rats. Cancer res. 1993;53:83–88. [PubMed] [Google Scholar]
- 6.Qiao J, Black ME, Caruso M. Enhanced ganciclovir killing and bystander effect of human tumor cells tranduced with restroviral vector carrying a herpes simplex virus thymidine kinase gene mutant. Hum Gene Ther. 2000;22:1569–1576. doi: 10.1089/10430340050083298. [DOI] [PubMed] [Google Scholar]
- 7.Rosolen A, Frascella E, di Francesco C, Todesco A, Petrone M, Mehtali M, Zacchello F, Zanesco L, Scarpa M. In vitro and in vivo antitumor effect of retrovirus-mediated herpes simplex thymidine kinase gene-transfer in human medulloblastoma. Gene Ther. 1998;5:113–120. doi: 10.1038/sj.gt.3300559. [DOI] [PubMed] [Google Scholar]
- 8.Huang Q, Xia Z, You Y, Pu P. Wild type p53 gene sensitizes rat C6 glioma cells to HSV-TK/ACV treatment in vitro and in vivo. Pathol Oncol Res. 2010;16:509–514. doi: 10.1007/s12253-009-9240-3. [DOI] [PubMed] [Google Scholar]
- 9.Fulci G, Chiocca EA. The status of gene therapy for brain tumors. Expert Opin Biol Ther. 2007;7:197–208. doi: 10.1517/14712598.7.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ardiani A, Sanchez-Bonilla M, Black ME. Fusion enzymes containing HSV-1 thymidine kinase mutants and guanylate kinase enhance prodrug sensitivity in vitro and in vivo. Cancer Gene Ther. 2010;17:86–96. doi: 10.1038/cgt.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willmon CL, Krabbenhoft E, Black ME. A guanylate kinase/HSV-1 thymidine kinase fusion protein enhances prodrug-mediated cell killing. Gene Ther. 2006;13:1309–1312. doi: 10.1038/sj.gt.3302794. [DOI] [PubMed] [Google Scholar]
- 12.Epple M, Ganesan K, Heumann R, Klesing J, Kovtun A, Neumann S, Sokolova V. Application of calcium phosphate nanoparticles in biomedicine. J Mater Chem. 2010;20:18–23. [Google Scholar]
- 13.LeGeros RZ. Calcium phosphate-based osteoinductive materials. Chem Rev. 2008;108:4742–4753. doi: 10.1021/cr800427g. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Yang Y, Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Control Release. 2012;158:108–114. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Satterlee A, Huang L. In vivo gene delivery by nonviral vectors: overcoming hurdles? Mol Ther. 2012;20:1298–304. doi: 10.1038/mt.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Li J, Liu F, Huang L. Systemic delivery of siRNA via LCP nanoparticle efficiently inhibits lung metastasis. Molecular Ther. 2012;20:609–615. doi: 10.1038/mt.2011.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee R, Tyagi P, Li S, Huang L. Anisamide-targeted stealth liposomes: a potent carrier for targeting doxorubicin to human prostate cancer cells. Int J Cancer. 2004;112:693–700. doi: 10.1002/ijc.20452. [DOI] [PubMed] [Google Scholar]
- 18.Yoshikawa M, Kato T, Takenishi T. A novel method for phosphorylation of nucleosides to 5’-nucleotides. Tetrahedron Letters. 1967;50:5065–5068. doi: 10.1016/s0040-4039(01)89915-9. [DOI] [PubMed] [Google Scholar]
- 19.Giger EV, Puigmartí-Luis J, Schlatter R, Castagner B, Dittrich PS, Leroux JC. Gene delivery with bisphosphonate-stabilized calcium phosphate nanoparticles. J Control Release. 2011;150:87–93. doi: 10.1016/j.jconrel.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi K, Rahman SMA, Ohno H, Tanaka T, Toyooka N, Nemoto H, Hayashi T. Evaluation of scopadulcio-related moleculars for their stimulatory effect on the cytotoxicity of acyclovir and ganciclovir against herpes simplex virus type1 thymidine kinase gene-trnafected HeLa cells. Chem Pharm Bull. 2004;52:1015–1017. doi: 10.1248/cpb.52.1015. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi K, Hayashi T, Sun HD, Takeda Y. Contribution of a combination of ponicidin and acyclovir/Ganciclovir to the antitumor efficacy of the herpes simplex virus thymidine kinase gene therapy system. Hum Gene Ther. 2002;13:415–423. doi: 10.1089/10430340252792549. [DOI] [PubMed] [Google Scholar]
- 22.Veenstra RD. Size and selectivity of gap junction channels formed from different connexins. J Bioenergetics Biomembranes. 1996;28:327–337. doi: 10.1007/BF02110109. [DOI] [PubMed] [Google Scholar]
- 23.Halloran PJ, Fenton RG. Irreversible G2-M Arrest and Cytoskeletal Reorganization Induced by Cytotoxic Nucleoside Analogues. Cancer Res. 1998;58:3855–3865. [PubMed] [Google Scholar]
- 24.Reid R, Mar EC, Huang ES, Topal MD. Insertion and extension of acyclic, dideoxy, and ara nucleotides by herpesviridae, human α and human β polymerase. J Boil Chem. 1988;263:3898–3904. [PubMed] [Google Scholar]
- 25.Komindky SL, Subramaniam PS, Johnson HM, Torres BA. Inhibitory Effects of IFN-g and Acyclovir on the Glioblastoma Cell Cycle. J Interferon Cytokine Res. 2000;20:463–469. doi: 10.1089/10799900050023870. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Kim WY, Huang L. Systemic delivery of gemcitabine triphosphate via LCP nanoparticles for NSCLC and pancreatic cancer therapy. Biomaterials. 2013;34:3447–58. doi: 10.1016/j.biomaterials.2013.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechol Bioeng. 2007;96:203–209. doi: 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]
- 28.Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788:2259–2266. doi: 10.1016/j.bbamem.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohgaki T, Bohgaki M, Hakem R. DNA double-strand break signaling and human disorders. Genome Integr. 2010;1:15–29. doi: 10.1186/2041-9414-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J boil chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kB. J Clin Investi. 2001;107:241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Investi. 2001;107:135–142. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin, Baldwin AS., Jr Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.