

Abstract
Transposons are mobile genetic elements that are capable of self-directed excision and subsequent reintegration within the host genome. Transposase such as piggyBac, Sleeping Beauty and Tol2 catalyze these reactions and have shown potential as tools for the stable integration of transgenes when used in the binary plasmid mode. Recent modifications to the transposase and/or the terminal repeats of the transposon have increased their integration efficiency and/or specificity. We recently described the development of a piggyBac transposase system, the helper independent, single construct self-inactivating plasmid called GENIE. Here we describe the structure, safety and function of these transpositional vectors and their use in animal transgenesis and cell transfection.
Keywords: transposon, transposase, transgenesis, transgene, piggyBac, cell transfections
Since its conception in 1980, mammalian transgenesis has been the process of injecting linear DNA into the male pronucleus of fertilized oocytes by pronuclear microinjection (PNI),1 or into MII oocytes during intracytoplasmic sperm injection (ICSI) after its adherence to pretreated spermatozoa.2 The efficiency of both methods is low in terms of transgenic offspring obtained per one cell zygotes or oocytes injected, and additionally PNI and ICSI have been shown to frequently result in concatemerized transgene insertions.2,3 The introduction of an “Active Transgenesis” approach with the use of lentiviruses as vectors for the transgene opened a new avenue for germline transgenesis.4 The lentiviral approach for the production of transgenic animals is very efficient, perhaps too efficient, as many micromanipulated embryos (73%) do not reach term. Nonetheless, of the animals born during this procedure approximately 80% are transgenic. To our knowledge, no mechanisms potentially responsible for the high mortality rate of the micromanipulated embryos has been identified, however lethal mutations by over-insertions of the transgene may contribute to the observed phenomenon. There are several other factors that have prevented the lentiviral system from taking over as the preferred transgenesis method, the details of which are beyond the scope of this report.5,6 Transposons are mobile genetic elements that are capable of self-directed excision and subsequent reintegration within the host genome. Several transposases catalyzing this cut-and paste mobilization have been described among them piggyBac, Sleeping Beauty and Tol2. Specifically, transposases recognize and bind to the inverted repeat elements flanking transposons, cut this DNA segment from the donor and reinsert it to the recipient genome. These properties have been proven to be an invaluable tool for gene delivery in applications such as transgenesis, mutagenesis for cancer research or gene therapy.7-11
We have evaluated several binary, plasmid-based, class II transposase/transposon systems, where the donor plasmid contains the transgene within the transposon, while the helper plasmid encodes the transposase. In our hands the piggyBac (pB) transposase isolated from the moth Trichoplusia ni proved the most efficient during the transfection of several mammalian cell lines.12 We have employed pB in an alternative non-viral “Active Transgenesis” approach that makes use of this system for the delivery of transgenes to the genome of gametes. In our first ICSI transgenesis experiments using the binary pB system with fresh unfrozen spermatozoa we obtained very few transgenic animals. We attempted to avoid freeze-thawing of spermatozoa, used during classical ICSI transgenesis (ICSI-Tr) to adhere DNA to spermatozoa as it often results in chromosomal damage of the treated spermatozoa.13,14 We therefore generated several single plasmid, self-inactivating pB based vectors containing the transposase and transposon in a single construct.9 We recently described a novel variant of these self-inactivating single vectors, containing the mammalian codon biased hyperactive pB transposase (pmhyGENIE-3) and an antibiotic-free selection cassette in the plasmid backbone and demonstrated highly efficient mouse transgenesis in combination with the transposase-enhanced pronuclear microinjection procedure (te-PNI).8
As plasmid constructs are injected into the male pronucleus of the one cell embryo during te-PNI this method permits the use of binary plasmid constructs, which in turn enables researches to bypass the complications of producing a helper independent self-inactivating single GENIE construct. However, there are some drawbacks in using binary vectors. We and others have noted that a small percentage of transposon bearing donor plasmids are inserted into the host cells genome when7,12,15 donor only transfection are performed in control experiments.7,12,13 This is perhaps due to nicked plasmids generated during the plasmid preparation and purification procedure. Cytosolic nucleases might also linearize plasmids, facilitating plasmid fragment integrations into the host genome via the non-homologous recombination machinery.3 If a donor plasmid is nicked or cut in its backbone region where often the origin of replication and the bacterial antibiotic selection gene are located, the likelihood of an intact transposon getting inserted into the genome is highly probable. Given that numerous plasmids are introduced into cells during cell transfection, the generation of nicked plasmid in the above scenario is always possible. We hypothesized that if nicking and linearization occurs for donor plasmids it is very likely to happen to helper plasmid as well, where an active transposase gene will be likely introduced into the cells genome. In our analysis of cells transfected with the binary pB system we discovered that in some instances the incorporation of an active pB gene derived from the helper plasmid into the cells genome9 as had been previously reported by the research team who tested the pB transposase in mammalian cells and animals using the binary vector system.7 We therefore developed the single plasmid, self-inactivating GENIE vectors which provided us a better opportunity to avoid genotoxic events at the rare occasions where an active transposase is incorporated into the host genome (Fig. 1).9
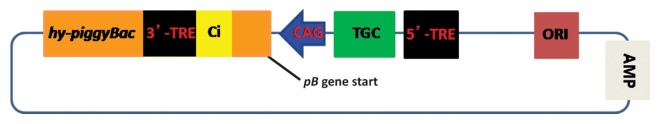
Figure 1. Schematic representation of pmhyGENIE-3. The transposon cassette for genomic integration contains the transgene cassette (TGC) and is delimited by the 3′- and 5′-TREs. Ci represents the chimeric intron, CAG the CAG promoter.
Safety of GENIE plasmids
In our previous reports8,9 we have described the safety mechanisms of pmGENIE-3 we engineered into the constructs to prevent the piggyBac-driving promoter (Fig. 1) from maintaining its activity after excision of the transposon from the plasmid. Specifically, this CAG promoter located in the transposon should not be able to contribute to aberrant gene activation, as the remaining fragment of the pB transposase gene and the chimeric intron contain two stop codons (UAA) engineered into the transposon construct. However, concerns have been raised that the CAG promoter once integrated into the host genome may prove problematic16 as it could influence expression of host genes located nearby. To address these concerns, we have performed an experiment where we designed a transposon to which we added another outward directed CAG promoter-chimeric intron construct to the 5′-TRE end of the pmhyGENIE-3 transposon. Hereafter this construct will be referred to as double reporter construct (DRC) (Fig. 2C). We placed the EGFP or DsRED Express2 genes respectively on either side of the transposon. The 5′- and 3′-TTAA sequences abate a Kozak consensus sequence, which will initiate transcription from the chromophore genes. These genes contain a polyA sequence downstream of their termination codon. The EGFP gene is situated immediately downstream of the CAG promoter, a fragment containing the start of the hyperactive pB gene, the chimeric intron which contains the 5′-TRE and the TTAA pB recognition sequence. Integration of the transposon into the TTAA target sequence within the host genome introduced 8 in frame stop codons within the region spanning from the end of the truncated pB gene to the start of the EGFP gene. The DsRed Express2 gene is situated at the opposite side of the transposon and is preceded by a similar arrangement of a CAG promoter, TRE and chimeric intron. However, this time the chimeric intron contains the 3′-TRE of pB terminating with the TTAA sequence of transpositional insertion. Here there are 10 in frame stop codons within the region spanning from the end of the truncated pB gene to the beginning of the DsRed Express2 gene. As the transposon can be inserted into DNA in one of three possible codon frames, the number of stop codons between the truncated end of the pB gene and the chromophores will vary, however there will always be at least two or more, depending on the site of insertion.

Figure 2. CAG promoter influence on neighboring genes. To assess the potential of the piggyBac-driving CAG promoter to influence expression of host genes located nearby after it's integration into the host genome we used several different vectors. (A) pmhyGENIE-3-EGFP as positive control for EGFP expression. (B) pmhyGENIE-3-DsRed as positive control for DsRed Express2 expression. (C) The DRC plasmid contains a transposon to which we added another outward directed CAG promoter-chimeric intron construct to the 5′-TRE end of the pmhyGENIE-3 transposon and EGFP or DsRED Express2 genes on either side of the transposon. (D) To assess the influence of TREs within this construct, we removed the CAG promoters from the DRC plasmid (DRC noCAG). (E) Finally, we also removed the TREs from the DRC noCAG construct to generate the DRC noCAG noTRE plasmid to evaluate promoterless and TRE-free expression.
HEK293 cells were transfected with equimolar concentrations of this DRC construct. pmhyGENIE-3 with either an EGFP (pmhyGENIE-3 EGFP) or DsRed Express2 (pmhyGENIE-3 DsRed) as a transgene served as controls. Chromophore expression was evaluated at four different time points (24, 48, 72 or 144 h). The expression of the positive controls increased over time and was at a maximum at 144 h (Fig. 3). To accommodate the strong EGFP signal at 144 h, we reduced the EGFP exposure time by 10-fold at this time point. We noticed a marked reduction in chromophore expression for the DRC plasmid when compared with the positive control, with DsRed expression slightly higher than EGFP. To assess the influence of the CAG promoter on this residual expression we removed this promoter from the DRC plasmid (DCR noCAG) (Fig. 2D). Transfection with DRC noCAG did not result in DsRed expression after 144 h, while EGFP remained at levels of DRC plasmid transfections. As a final modification we also removed the TREs from the DRC noCAG construct to generate the DRC noCAG noTRE plasmid (Fig. 2E). Here we did not observe any expression from the chromophores 144 h post transfection. It is not evident to us why we observed minor DsRed expression for the DRC construct and EGFP expression from the DRC noCAG plasmid, however it is possible that it originates from undefined low level transcriptional activity from the plasmid backbone. It is also possible that the observed EGFP expression of the CAG promoterless plasmid was due to outward-directed promoter activity of the 5′-TRE. Inward directed promoter activity has previously been reported for both the 5′- and the 3′-TREs of pB.17,18 However, outward directed promoter activity for other transposon inverted repeat elements have been described.19,20 We would like to note that this observation of a potential outward-directed promoter activity from pB TREs was not the emphasis of our experimental setup. Larger scale, more focused investigations will be necessary to elucidate this observation. In summary, our results clearly show that there is a drastic reduction in chromophore expression as a consequence of the vector design, more specifically the stop codons situated between the CAG promoter and the beginning of chromophore genes.

Figure 3. Analysis of DRC plasmid-mediated chromophore expression by fluorescent microscopy. 0.5 × 105 HEK293T cells were transfected with 400 ng of pmhyGENIE-3 containing either EGFP (G3 EGFP) or DsRed Express2 (G3 DsRed) in its transgene cassette, DRC, DRC noCAG or DRC noCAG noTRE. Images of EGFP expression followed by DsRed expression were taken for each construct at 24, 48, 72 and 144 h. *Exposure time for EGFP of the pmhyGENIE-3 EGFP at 144 h was reduced to 10% due to overexposure at original exposure time.
Finally, an often-overlooked but crucial parameter that influences the success and the safety of transposition for transgenesis, transfection and gene therapy is the integrity and purity of plasmids. This is of particular concern for transposase-mediated delivery of large DNA fragments as it has been shown to be possible with pB (up to 100 kb).21 For optimal plasmid manipulation procedures the best commercially available gravity-flow purification kits must be used to obtain only covalently closed circular (supercoiled-ccc configuration) plasmids. The desired effects after gentle lysis of the host bacterial cells are the elimination of intracellular macromolecules and the enrichment and purification of plasmid DNA. Even with these kits, care must be taken to assess each batch by gel electrophoresis for both purity in terms of contaminating bacterial DNA and ccc-configuration. Once the correct configuration plasmids as proven by electrophoresis are purified, they can then be confirmed for functionality by testing them in easy to transfect cell lines such as HEK293. Additionally, we resuspend plasmids used for transgenesis research in Sigma Water for Embryo Transfer (Sigma-Aldrich) and store 10 µL aliquots in a −20°C defrost free freezer. In our experience, all of these parameters, if not taken care of deliberately can significantly bias the efficiency and safety of transposition.
Transgenesis techniques with the pmhyGENIE plasmids
Transgenesis by microinjection is a technically challenging procedure, which needs specialist for its implementation. In addition to the technical difficulties of the process, care must be taken not to shear the transgene DNA during the microinjection process. This becomes of even greater importance when large circular plasmids are used during Active Transgenesis. pmGENIE plasmids, in addition to the transposon with the transgene in between its terminal repeat elements (TREs), the GENIE plasmid DNA also contains a transposase enzyme with all its regulatory elements. The transposase has to be first transcribed once the plasmid DNA reaches the nucleus, then processed into mRNA and finally translated into protein. The nuclear localization signal of the pB transposase must facilitate its translocation back into the nucleus. Once in the nucleus, the transposase locates the TREs of the transposon and initiates formation of the synaptic-complex. After excision of the transposon from the plasmid DNA via the synaptic-complex, the resulting circular structure with two transposase molecules holding it together at the TREs is referred to as the transposome. It is these transposase molecules in the transposome that select the target TTAA tetranucleotide sequence for transposition and finally insert the transposon into the host’s genome. As can be inferred from the above description there are several distinct regions of the plasmid DNA that must retain their structural integrity. Nicked or linearized plasmids may impede the successful completion of the transposition process and must therefore be avoided at all costs.
To accommodate this requirement for only using ccc-configuration plasmid, we have modified the traditional PNI procedure and created the new te-PNI method.8 During te-PNI a 2 µm internal diameter blunt end microinjection pipette is used. This pipette has to be well lubricated with 15% polyvinyl pyrrolidone (PVP) in order to render it non-sticky. The diameter size of the pipette allows the unimpeded transfer of precise amounts of plasmid DNA into the pronucleus without shearing it. However, the blunt ended pipette creates a difficulty for the penetration of the one-cell zygotes protective layers (zona pellucida and plasma membrane), which can be overcome by the use of a Piezo actuator (Prime Tech, Japan). The Piezo actuators ability to act as a small pneumatic drill facilitates the traversing of the protective layers of the embryo without manual force being applied to the zygote. This prevents the constriction of the embryo and allows smooth passage through the membranes of the one cell, two pronuclear zygote. Additionally, it confers control in regards to the area of the male pronucleus to be penetrated by the injection. Once the plasmid has been delivered to the pronucleus, care must be exercised not to extract a nucleolus while retracting the pipette. Therefore, the less sticky the microinjection pipette by PVP lubrication, the better this outcome. The flow regulation from the pipettes during the screw-controlled injection process is mediated by the small amount of mercury back-loaded into the pipettes. The mercury is subsequently positioned near the thin tip of the micropipette with an injection syringe. An air-filled space separates the mercury from the mineral oil filled plastic tube leading to the screw-controlled syringe. Clockwise turns move the mineral oil forward, applying pressure to the air and in turn to the mercury. Because of the very high surface tension of mercury, large displacements of the mineral oil volume brought about by the turn of the screw-controlled syringe, result in very small displacements of the volume in front of the mercury therefore allowing precise control of the injection process.22 The plasmid solution is drawn into the pipette by turning the screw-controlled syringe in a counter-clockwise orientation. This DNA solution is then injected into the pronucleus, by turning the screw-controlled syringe in a clockwise orientation.8 Precise quantitation of the amount of plasmid injected can be calculated with the following equation: π r2 length. An injection length of 65 μm equals 2.04 × 10−7 μL in volume. At a concentration of 10 ng/μL for plasmid solution (pmhyGENIE-3-RNA-OUT, 14,542 bp), the total amount of DNA injected is 2.04 femtograms (fg) or about 130 copies of plasmid per pronucleus.8 Obviously, not all the plasmid molecules deposited into the pronucleus integrate into the genome, as we consistently find 1, 2, 3 or 4 monomeric transgenes integrated into the genome of the host mouse by Southern blots.8,9 Similar infrequent transposition observations for the Sleeping Beauty mechanism were made in a recent review, where all the necessary transposition components were present in excess.16 In fact, during hydrodynamic delivery via tail vein injection, only about one in 10,000 Sleeping Beauty transposons delivered to the liver actually transpose into chromatin.23 In summary, under ideal conditions with ccc-configuration plasmids, the efficiency of transposition relies in several parameters working optimally for successful transposon integration.16
pmhyGENIE-4
The pB transposase, unlike Sleeping Beauty when expressed at high levels is not subject to overexpression inhibition.8,9,24 We have sought to overexpress the pB transposase in HEK293 cells by co-transfecting a helper plasmid for the transposase together with a mouse codon bias based pmGENIE-3 plasmid containing a hygromycin resistance gene in its transposon. We intended to assess if a larger amount of transposase will indeed increase transfection efficiency. After three weeks of hygromycin selection we assessed the number of stably transfected HEK293 colonies and noted that, there were 30.6% more colonies formed from the helper plasmid boosted pmGENIE-3 plasmid transfection, than the one where only pmGENIE-3 was used. Due to these findings we proceeded to construct the pmhyGENIE-4 plasmid, which contains two independent self-inactivating hyperactive pB transposase genes in its structure, together with an antibiotic gene free backbone for transgenesis experiments (Fig. 4). The antibiotic free backbone construct was achieved by replacing the ampicillin resistance gene and pUC ori in the backbone of the original construct for pmhyGENIE-4, as previously described for pmhyGENIE-3-RNA-OUT-MSC.8

Figure 4. Schematic representation of pmhyGENIE-4. This plasmid construct contains two independent self-inactivating hyperactive pB transposase genes in its structure, together with an antibiotic gene free backbone for transgenesis experiments. The transposon cassette for genomic integration contains the transgene cassette TGC and is delimited by the 3′- and 5′-TREs. Ci represents the chimeric intron, CAG the CAG promoter, and RNA OUT the antibiotic gene free selection cassette.
A comparison of transfection efficiencies of pmhyGENIE-4 and pmhyGENIE-3 in HEK293 cells transfection is depicted in Figure 5. Both versions of pmhyGENIE-4 (either containing an antibiotic gene in the backbone or RNA-OUT) were considerably more effective in transfecting HEK293 cells than pmhyGENIE-3. lt is of note that in these preliminary experiments we used the same plasmid concentrations for each transfection. As the pmhyGENIE-4-RNA-OUT-EGFP construct is larger than pmhyGENIE-3-EGFP by 3,185 bp, a lower number of pmhyGENIE-4-RNA-OUT-EGFP molecules was transfected and we therefore expect the efficiency of the pmhyGENIE-4 plasmids to be even higher when equimolar concentrations of plasmids are used.
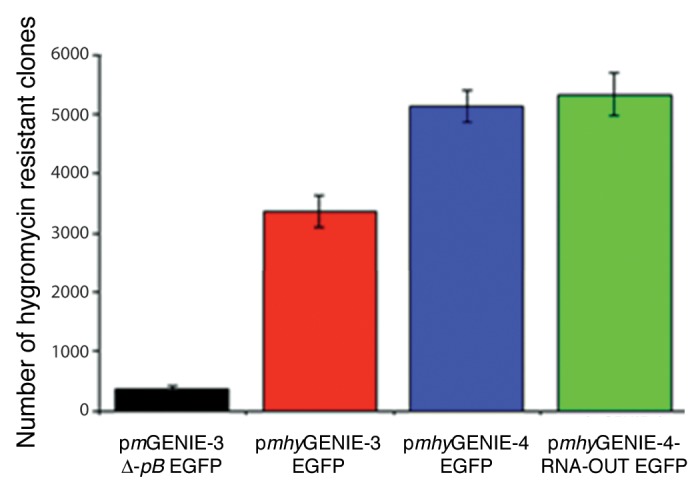
Figure 5. Analysis pmGENIE-4 mediated transposition. 0.5 × 105 HEK293T cells were transfected with 400 ng of pmhyGENIE-3 EGFP, pmhyGENIE-4 EGFP, pmhyGENIE-4-RNA-OUT EGFP or pmGENIE-3 ∆-pB. Transposition activity was measured by counting methylene blue stained, hygromycin-resistant colonies after a three-week selection period. Data are shown as mean values with SD (n = 3).
Transgenesis with pmhyGENIE-4
We previously demonstrated that te-PNI is very effective for the production of transgenic mice. The efficiencies for transgenic mice production with this method, in terms of embryos microinjected, were improved from an average of 4.6% for PNI to an average of 25.6% for te-PNI.8 In preliminary experiments, we have now produced transgenic mice with pmhyGENIE-4. We injected 13 embryos by te-PNI with a plasmid solution of 3 ng/ul. All 13 embryos developed to the 2-cell stage and where then transferred to a single surrogate mother. Eleven pups were born (85% of embryos injected), five of which were transgenic (representing 38.5% of embryos injected or 45.5% of animals born). These transgenic rates represent an increase of 50.3% over the rate of pmhyGENIE-3 te-PNI demonstrating the potential of pmhyGENIE-4 vectors. Of note, in contrast to previous te-PNI experiments,8 these results were achieved at a significantly lower plasmids concentration (10 ng/ul of pmhyGENIE-3 vs. 3 ng/ul of pmhyGENIE-4) injected into the pronucleus.
In conclusion, our results lead us to suggest that our pmhyGENIE plasmids are effective, efficient and safe to use in a wide array of application such as cell transfections, gene therapy and transgenesis.
Acknowledgments
We thank Scott Campbell, Joel Marh, Marlee Elston, Ilko Stoytchev and Zoia Stoytcheva, for help with the manuscript. The Sanger Institute, Cambridge, England for the mouse codon-optimized hyperactive piggyBac plasmid. This work was supported by National Institutes of Health Grants 5P20RR024206 and R01 GM083158-01A1 to S.M.
Glossary
Abbreviations:
- PNI
pronuclear microinjection
- ICSI
intracytoplasmic sperm injection
- pB
piggyBac
- ICSI-Tr
ICSI transgenesis
- te-PNI
transposase-enhanced pronuclear microinjection procedure
- TREs
terminal repeat elements
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mge/article/25167
References
- 1.Gordon JW, Scangos GA, Plotkin DJ, Barbosa JA, Ruddle FH. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc Natl Acad Sci U S A. 1980;77:7380–4. doi: 10.1073/pnas.77.12.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perry AC, Wakayama T, Kishikawa H, Kasai T, Okabe M, Toyoda Y, et al. Mammalian transgenesis by intracytoplasmic sperm injection. Science. 1999;284:1180–3. doi: 10.1126/science.284.5417.1180. [DOI] [PubMed] [Google Scholar]
- 3.Smith K. Theoretical mechanisms in targeted and random integration of transgene DNA. Reprod Nutr Dev. 2001;41:465–85. doi: 10.1051/rnd:2001102. [DOI] [PubMed] [Google Scholar]
- 4.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 5.Ellis J, Yao S. Retrovirus silencing and vector design: relevance to normal and cancer stem cells? Curr Gene Ther. 2005;5:367–73. doi: 10.2174/1566523054546233. [DOI] [PubMed] [Google Scholar]
- 6.Park F. Lentiviral vectors: are they the future of animal transgenesis? Physiol Genomics. 2007;31:159–73. doi: 10.1152/physiolgenomics.00069.2007. [DOI] [PubMed] [Google Scholar]
- 7.Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–83. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Marh J, Stoytcheva Z, Urschitz J, Sugawara A, Yamashiro H, Owens JB, et al. Hyperactive self-inactivating piggyBac for transposase-enhanced pronuclear microinjection transgenesis. Proc Natl Acad Sci U S A. 2012;109:19184–9. doi: 10.1073/pnas.1216473109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urschitz J, Kawasumi M, Owens J, Morozumi K, Yamashiro H, Stoytchev I, et al. Helper-independent piggyBac plasmids for gene delivery approaches: strategies for avoiding potential genotoxic effects. Proc Natl Acad Sci U S A. 2010;107:8117–22. doi: 10.1073/pnas.1003674107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rad R, Rad L, Wang W, Cadinanos J, Vassiliou G, Rice S, et al. PiggyBac transposon mutagenesis: a tool for cancer gene discovery in mice. Science. 2010;330:1104–7. doi: 10.1126/science.1193004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilber A, Ulloa Montoya F, Hammer L, Moriarity BS, Geurts AM, Largaespada DA, et al. Efficient non-viral integration and stable gene expression in multipotent adult progenitor cells. Stem Cells Int. 2011;2011:717069. doi: 10.4061/2011/717069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, et al. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A. 2006;103:15008–13. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szczygiel MA, Moisyadi S, Ward WS. Expression of foreign DNA is associated with paternal chromosome degradation in intracytoplasmic sperm injection-mediated transgenesis in the mouse. Biol Reprod. 2003;68:1903–10. doi: 10.1095/biolreprod.102.012377. [DOI] [PubMed] [Google Scholar]
- 14.Yamauchi Y, Riel JM, Ward MA. Paternal DNA damage resulting from various sperm treatments persists after fertilization and is similar before and after DNA replication. J Androl. 2012;33:229–38. doi: 10.2164/jandrol.111.013532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–10. doi: 10.1016/S0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 16.Hackett PB, Largaespada DA, Switzer KC, Cooper LJ. Evaluating risks of insertional mutagenesis by DNA transposons in gene therapy. Transl Res. 2013;161:265–83. doi: 10.1016/j.trsl.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadiñanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007;35:e87. doi: 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi X, Harrison RL, Hollister JR, Mohammed A, Fraser MJ, Jr., Jarvis DL. Construction and characterization of new piggyBac vectors for constitutive or inducible expression of heterologous gene pairs and the identification of a previously unrecognized activator sequence in piggyBac. BMC Biotechnol. 2007;7:5. doi: 10.1186/1472-6750-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moldt B, Yant SR, Andersen PR, Kay MA, Mikkelsen JG. Cis-acting gene regulatory activities in the terminal regions of sleeping beauty DNA transposon-based vectors. Hum Gene Ther. 2007;18:1193–204. doi: 10.1089/hum.2007.099. [DOI] [PubMed] [Google Scholar]
- 20.Rappleye CA, Roth JRA. A Tn10 derivative (T-POP) for isolation of insertions with conditional (tetracycline-dependent) phenotypes. J Bacteriol. 1997;179:5827–34. doi: 10.1128/jb.179.18.5827-5834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li MA, Turner DJ, Ning Z, Yusa K, Liang Q, Eckert S, et al. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res. 2011;39:e148. doi: 10.1093/nar/gkr764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffe LA, Terasaki M. Quantitative microinjection of oocytes, eggs, and embryos. Methods Cell Biol. 2004;74:219–42. doi: 10.1016/S0091-679X(04)74010-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hackett CS, Geurts AM, Hackett PB. Predicting preferential DNA vector insertion sites: implications for functional genomics and gene therapy. Genome Biol. 2007;8(Suppl 1):S12. doi: 10.1186/gb-2007-8-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Lin C, Lu D, Ning Z, Cox T, Melvin D, et al. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:9290–5. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]