

Abstract
Our recent study showed critical roles of Dmp1 as a sensor of oncogenic Ras, HER2/neu signaling and activation of the Arf-p53 pathway. To elucidate the role of human DMP1 (hDMP1) in breast cancer, one hundred and ten pairs of human breast cancer specimen were studied for the alterations of the hDMP1-ARF-Hdm2-p53 pathway with follow up of clinical outcomes. Loss of heterozygosity (LOH) of the hDMP1 locus was found in 42% of human breast carcinomas, while that of INK4a/ARF and p53 were found in 20% and 34%, respectively. Hdm2 amplification was found in 13% of the same sample, which was found independently of LOH for hDMP1. Conversely, LOH for hDMP1 was found in mutually exclusive fashion with that of INK4a/ARF and p53, and was associated with low Ki67 index and diploid karyotype. Consistently, LOH for hDMP1 was associated with luminal A category and longer relapse-free survival, while that of p53 was associated with non-luminal A and shorter survival. Thus, loss of hDMP1 could define a new disease category associated with prognosis of breast cancer patients. Human breast epithelial cells/cancer cells with wild-type p53 were sensitive to growth inhibition by activated Dmp1:ER while those that delete p14ARF or p53, and/or Hdm2 amplification showed partial or nearly complete resistance, indicating that p53 is a critical target for hDMP1 to exhibit its biological activity.
Keywords: Dmp1 (Dmtf1), breast cancer, loss of heterozygosity, relapse-free survival, Ki67, prognostic marker
Introduction
Breast cancer is the most common malignancy in women and remains significant health issue in industrialized countries (1-3). Strong evidence supports the idea that breast cancer is initiated by defined genomic alterations, many of which are currently used as therapeutic targets or biomarkers (4). However, it is still unclear which and how genomic alterations in human breast cancer contribute to its biology. Furthermore, it is unknown whether they drive progression of the disease, response to therapy, or if they could be used as prognostic/predictive markers for better patient stratification and molecular subtyping. Recently, the potential of DNA copy number aberrations for molecular subtyping of breast cancer has been re-evaluated. It suggests that specific DNA deletions and/or amplifications may be independent predictors of patient outcomes apart from analysis of other macromolecules, and warrants future clinical implementation (5).
Dmp1, a cyclin D binding myb-like protein 1 (also called Dmtf1), was originally isolated in a yeast two-hybrid screen of a murine T-lymphocyte library with cyclin D2 as bait (6, 7). Dmp1 shows its activity as a tumor suppressor by directly binding to the Arf promoter to activate its gene expression and, thereby, induces Arf- and p53-dependent cell cycle arrest (8). The activity of the Arf-53 pathway is significantly attenuated in Dmp1-deficient cells since those cells can easily give rise to immortalized cell lines that retain wild-type p19Arf and functional p53 and are transformed by oncogenic Ras alone (9, 10). The murine Dmp1 promoter is efficiently activated by oncogenic Ras, as well as by constitutively active MEK1/2 and/or ERK1/2 in primary culture cells (11). Thus, Dmp1 is a key mediator between Ras-Raf-MEK-ERK mitogenic signaling and the Arf-p53 tumor suppressor pathway.
Dmp1-deficient mice are prone to tumor development. Tumors induced by the Eμ-Myc or K-Ras transgene were greatly accelerated in both Dmp1+/− and Dmp1−/− backgrounds with no differences between groups lacking one or two Dmp1 alleles (9, 10, 12). Indeed, nearly all tumors from Dmp1+/− mice retained and expressed the wild-type Dmp1 allele, and most expressed wild-type Dmp1 mRNA and protein, suggesting typical haploid-insufficiency of Dmp1 in tumor suppression (10, 12, 13-15).
We recently characterized the signaling pathway between HER2/neu and Dmp1 using MMTV-neu mice as a model (16). Both Dmp1 and p53 were induced in pre-malignant hyperplastic lesions from MMTV-neu mice, and mammary carcinogenesis was significantly accelerated in both Dmp1+/− and Dmp1−/− mice (16). We also observed selective deletion of Dmp1 in >50 % of wild-type HER2/neu carcinomas, while the involvement of Arf, Mdm2, or p53 was rare. Tumors from Dmp1-deficient mice showed significant downregulation of Arf and p21Cip1, showing p53 inactivity and more aggressive phenotypes than tumors without Dmp1 deletion (16). Thus, our study shows the pivotal roles of Dmp1 in HER2/neu-p53 signaling and breast cancer development.
The human DMP1 (hDMP1; hDMTF1) gene is located on chromosome 7q21, a region often deleted in human breast/lung cancers and hematopoietic malignancies (17-19). We recently analyzed 51 human non-small cell lung carcinoma (NSCLC) samples and found that loss of heterozygosity (LOH) of hDMP1 was present in ~35 % of lung cancers (12) in a mutually exclusive fashion with that of INK4a/ARF and/or p53 in the same samples. This raised the possibility that hemizygous hDMP1 deletion might define a new disease entity with different response to therapy (12, 15). The current study was conducted to demonstrate the frequency and pattern of genes involved in the hDMP1-ARF-Hdm2-TP53 pathway in human breast cancer. We analyzed 110 pairs of normal and cancer tissues from breast cancer for LOH of hDMP1, INK4a/ARF, p53 and gene amplification of Hdm2 (20, 21), and correlated the results of LOH/gene amplification with disease-free survival and known prognostic markers for human breast cancer (reviewed in 22, 23).
Results
The human DMP1 gene (hDMP1; hDMTF1) is often deleted in human breast cancer
To determine the frequency and patterns of inactivation of the hDMP1-ARF-Hdm2-p53 pathway in human breast cancers, we extracted DNA from 110 pairs of clinical samples and conducted LOH analyses for hDMP1, INK4a/ARF, p53, and gene copy number assay for Hdm2 (exon 4). Representative patterns for LOH-positive cases for each locus are shown in Figure 1. The results from a total of 110 patients are summarized in Table 1 (66 cases with promoter methylation assays for hDMP1) and Supplementary Table S1 (the other 44 cases). LOH for hDMP1 was found in 27 samples with the 5′ probe (#92465, 24.5 %), 30 cases (#198004, 27.3 %) with the 3′ probe, and 46 of 110 cases (41.8 %) with either the 5′ or 3′ probes. None of the 61 samples we studied showed methylation of the hDMP1 promoter (Table 1, the 4th column). None of the 15 randomly chosen breast cancer samples showed mutation(s) for the hDMP1 gene except for the polymorphisms at codon 91 (data not shown). Detailed mapping of the genomic fragment deleted in breast cancer showed that gene deletion was limited to the hDMP1 locus (from #69164 to #251945) (12) in 30 of 32 cases of LOH (93.8 %) (Supplementary Figure S1), a higher percentage than hDMP1 deletion in human NSCLC (78.9 %) (12). In one case, the hDMP1 deletion was not detectable by the regular LOH assays since the gene deletion was limited to the exons 8 -20 (case #2005-930) (Table 1).
Figure 1. Representative patterns of LOH for hDMP1, INK4a/ARF, and p53 in human breast carcinoma.
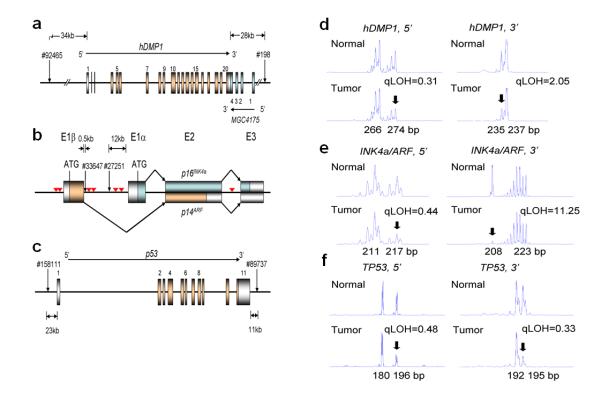
Genomic DNA was extracted from paired normal and malignant breast cancer specimen and PCR was conducted with 6-FAM-labeled primers that amplify the dinucleotide repeats within (or close to) each locus (12, 15). The area peaks of the PCR products were quantitated by ABI 3730xl DNA analyzer. The qLOH values were determined through the following equation: qLOH = Area Peak 1/Area Peak 2 (normal tissue) divided by Area Peak 1′/Area Peak 2′ (tumor tissue). The arrows indicate the peak that was lost in tumor cells. The sample was considered to have LOH when the value was >2.0 or <0.5.
a, genomic locus of the hDMP1 gene. The two different primer sets were designed to amplify the dinucleotide repeat sequences located on the 5′ and 3′ end of the hDMP1 gene. The noncoding exons were colored silver and the coding exons were colored gold.
b, genomic structure of the human INK4a/ARF locus. The two sets of PCR primers were designed to detect the dinucleotide repeats within 500 bps of Exon 1β (#33647) and those between Exon 1β and Exon 1α (#27251). The inverted triangles shown in red indicate the location of high-affinity hDMP1-binding sites.
c, genomic structure of the human p53 gene and the location of the PCR primers used for LOH analyses.
d, LOH analysis of breast cancer with hDMP1 primer sets. 5′:#2006-1202, qLOH = 0.31; 3′: #2004-817, qLOH = 2.05
e, LOH analysis with INK4a/ARF primer sets. 5′: #2008-1476, qLOH = 0.44; 3′: #1999-84, qLOH = 11.25
f, LOH analysis with p53 primer sets. 5′: #2008-1272, qLOH = 0.48; 3′: #2008-26, qLOH = 0.33
Table 1. LOH analyses of 66 pairs of human breast carcinomas for the hDMP1, INK4a/ARF, and p53 loci.
Positive results for LOH (qLOH >2.0 or <0.5) are shown in bold red type. When one of the two markers (5′ or 3′) showed qLOH value >2.0 or <0.5, the sample was considered positive for LOH for the tumor suppressor locus. Cases of mutually exclusive inactivation of hDMP1 and INK4a/ARF or hDMP1 and p53 are shown “yes” in bold blue type. Exclusive of hDMP1: LOH of INK4a/ARF (or p53) or amplification of Hdm2 is not overlapping with LOH of the hDMP1 locus in the same sample. Light brown shading indicates cases with LOH for hDMP1. Detailed analysis by real-time PCR showed that case #2005-930 had an internal deletion for hDMP1 that (darker brown shading). The hDMP1 gene was sequenced in samples with #. The p53 gene was sequences in samples with * (no mutation), and ** (with mutation). Abbreviations. H.D.: hemizygous deletion as determined by real-time PCR; No del: no deletion by real-time PCR; Un: unmethylated. Homo Del: homozygous deletion; single, LOH was not evaluated due to a single peak result; n.d.: not determined.
| hDMP1 LOH | INK4a/ARF LOH | p53 LOH | Hdm2 Amplification | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| Patient ID |
hDMP1 #92465 5′ |
hDMP1 #198004 3′ |
hDMP1 MSP |
INK4a/ARF #33647 5′ |
INK4a/ARF #27251 3′ |
p14 MSP |
p16 MSP |
Exclusive to hDMP1 LOH |
p53 #15811 5′ |
p53 #89737 3′ |
Exclusive to hDMP1 LOH |
Exclusive to INK4a/ARF LOH |
Hdm2 Ampl. |
Exclusive to hDMP1 LOH |
| 1999-12 | 1.21 | 2.57 | Un | 0.97 | 1.17 | Un | Un | yes | 0.92 | 0.49** | no | yes | No | yes |
| 1999-13 | 0.36 | 1.04 | Un | 0.95 | 0.90 | Un | Un | yes | 0.89 | 1.53* | yes | No | yes | |
| 2000-210 | 13.82 | 0.88 | n.d. | 0.95 | single | n.d. | n.d. | yes | 1.34 | 0.82 | yes | No | yes | |
| 2002-105 | 0.15 | 1.27 | Un | 0.90 | single | Un | Un | yes | single | 7.68** | no | yes | No | yes |
| 2002-358 | 1.17 | 3.19 | Un | 1.16 | 1.06 | Un | Un | yes | 0.63 | 0.95 | yes | No | yes | |
| 2002-378 | 0.42 | 2.05 | Un | single | 1.25 | Un | Un | yes | 1.68 | 1.51 | yes | no | No | yes |
| 2002-386 | 0.15 | 0.49 | Un | 1.40 | 0.67 | Un | Un | yes | 0.04 | 8.64 | no | yes | No | yes |
| 2003-424 | >10 | 4.81 | Un | single | 1.52 | Un | Un | yes | 0.89 | 1.01 | yes | No | yes | |
| 2003-452 | 2.72 | 1.34 | Un | 1.04 | 0.71 | Un | Un | yes | 0.84 | 1.10 | yes | No | yes | |
| 2004-516 | 8.11 | 2.02 | Un | 0.86 | 1.15 | Un | Un | yes | 0.83 | 1.13 | yes | No | yes | |
| 2004-720 | 1.16 | >10 | Un | 1.03 | 0.90 | Un | Un | yes | 0.98 | single | yes | No | yes | |
| 2004-753 | 0.96 | 2.08 | Un | 1.17 | 0.72 | Un | Un | yes | 1.03 | 1.14 | yes | 4.66 | no | |
| 2004-780 | 1.31 | 0.32 | Un | 0.73 | 0.94 | Un | Un | yes | 1.18 | 0.98 | yes | No | yes | |
| 2004-817 | 0.97 | 0.49 | Un | 1.13 | 1.00 | Un | Met | yes | 1.13 | 1.23 | yes | No | yes | |
| 2004-850 | 2.53 | No del | Un | 1.05 | 1.02 | Un | Un | yes | 0.75 | 1.75 | yes | No | yes | |
| 2004-857 | 2.03 | 1.25 | Un | 0.86 | 0.96 | Un | n.d. | yes | 0.70 | 1.25 | yes | No | yes | |
| 2005-78 | 2.06 | 1.52# | Un | 1.17 | 1.19 | Un | Un | yes | 0.78 | 0.97 | yes | No | yes | |
| 2005-483 | 0.92 | 2.04# | Un | 7.03 | 0.36 | Un | Un | no | 0.40 | 0.49* | no | no | No | yes |
| 2005-686 | No del | 2.94 | Un | 1.34 | 0.92 | n.d. | Un | yes | 1.56 | 0.97 | yes | No | yes | |
| 2005-823 | 1.01 | H.D. | n.d. | 1.10 | 1.01 | Un | Un | yes | 1.06 | 0.97 | yes | 3.09 | no | |
| 2005-930 | 1.20 | 1.03# | Un | 0.84 | 1.80 | Un | Un | yes | 0.93 | 1.02 | yes | 3.21 | no | |
| 2005-958 | 2.18 | H.D. | Un | 0.64 | 1.01 | Un | Un | yes | 1.57 | 0.56 | yes | No | yes | |
| 2005-972 | 2.85 | H.D. | Un | 0.92 | 0.65 | Un | Un | yes | 0.93 | 0.93 | yes | No | yes | |
| 2006-501 | 0.47 | H.D.# | Un | 1.34 | 0.70 | Un | Un | yes | 0.94 | 1.08 | yes | No | yes | |
| 2006-545 | 0.37 | No del | Un | 0.88 | 1.02 | Un | n.d. | yes | 0.79 | 0.95 | yes | No | yes | |
| 2006-819 | 3.08 | No del | Un | 1.08 | 0.77 | Un | Un | yes | 1.03 | 0.89 | yes | No | yes | |
| 2006-1169 | H.D. | 0.96# | Un | 1.20 | 1.03 | Un | Un | yes | 2.17 | 1.15 | no | yes | 11.58 | no |
| 2007-88 | 1.31 | H.D. | Un | 1.04 | 1.01 | partial | Un | yes | 0.98 | 1.10 | yes | No | yes | |
| 2007-91 | 0.48 | H.D. | Un | 0.98 | 0.95 | Un | Un | yes | 1.10 | 1.73 | yes | No | yes | |
| 2007-127 | >10 | No del | Un | 0.14 | 6.02 | Un | Un | no | 0.24 | 0.92 | no | no | No | yes |
| 2007-245 | 1.01 | H.D. | Un | 1.16 | 0.98 | Un | Un | yes | 1.01 | 0.95 | yes | No | yes | |
| 2007-285 | Del | No del | Un | 0.98 | 1.00 | Un | Un | yes | 1.57 | 1.84 | yes | No | yes | |
| 2007-404 | 0.46 | No del# | Un | 1.10 | 1.06 | Un | Un | yes | 0.93 | 1.12 | yes | No | yes | |
| 2007-425 | 1.32 | 2.12 | Un | 0.60 | 1.59 | Un | Un | yes | 2.87 | 2.70* | no | yes | No | yes |
| 1999-84 | 0.64 | No del | Un | 1.30 | 11.25 | Un | Met | yes | 0.59 | 4.08** | yes | no | No | |
| 2002-135 | 1.05 | 0.72 | Un | 1.07 | 0.79 | Un | Un | 1.49 | 0.43 | yes | yes | No | ||
| 2002-239 | 0.52 | 1.19 | Un | 0.83 | 0.21 | Un | Un | yes | 0.20 | single | yes | no | No | |
| 2002-272 | 1.01 | No del | Un | 1.75 | 0.69 | Un | Un | 0.98 | 7.14 | yes | yes | No | ||
| 2002-325 | 1.05 | No del | Un | 0.87 | 0.85 | Un | Un | 1.55 | single | 67.75 | yes | |||
| 2003-226 | 0.64 | 0.57 | Un | 1.12 | Homo Del | Un | Un | yes | 1.39 | 0.61 | yes | 4.37 | yes | |
| 2004-132 | 0.88 | 0.68 | Un | 4.66 | 2.77 | Met | Un | yes | 3.37 | 0.23* | yes | no | 3.01 | yes |
| 2004-738 | 0.89 | No del | Un | 1.06 | 0.95 | Un | Met | 1.03 | 0.93* | No | ||||
| 2004-851 | 0.72 | No del | Un | 1.38 | 0.75 | n.d. | Un | 1.77 | 0.33 | No | ||||
| 2004-1001 | 1.70 | No del# | Un | 0.23 | 1.16 | n.d. | Un | yes | 2.93 | 2.76 | yes | no | No | |
| 2004-1033 | 1.11 | No del# | Un | 0.78 | 1.05 | Un | Un | 0.61 | 1.45 | No | ||||
| 2005-173 | 0.84 | No del | Un | 0.23 | 2.01 | partial | Un | yes | 6.23 | 0.30 | yes | no | No | |
| 2005-448 | 1.14 | No del | Un | 0.68 | 1.18 | Un | Un | 0.84 | single* | No | ||||
| 2005-546 | 0.77 | No del# | n.d. | 0.94 | 1.15 | Un | Un | 0.92 | 0.92 | No | ||||
| 2005-627 | 0.80 | No del# | Un | 2.75 | 0.70 | Un | Un | yes | 5.86 | 0.12 | yes | no | No | |
| 2005-694 | 0.94 | No del# | Un | 1.03 | 1.06 | Un | Un | 0.89 | 0.73* | 3.02 | yes | |||
| 2005-702 | 1.17 | No del | Un | 1.51 | 1.08 | Un | Un | 0.79 | 0.57 | No | ||||
| 2005-705 | 1.59 | 1.16# | Un | 0.42 | 1.49 | Un | Un | yes | 1.11 | 0.92 | yes | 4.12 | yes | |
| 2005-787 | 1.05 | No del# | Un | 1.03 | 0.99 | Un | Un | 0.97 | 1.14 | No | ||||
| 2005-831 | 1.12 | 1.00# | Un | 2.11 | 1.22 | Un | Un | yes | 1.05 | 0.42 | yes | no | No | |
| 2005-876 | 0.90 | 0.83# | Un | 2.01 | 1.73 | Un | Un | yes | 0.73 | 0.47 | yes | no | No | |
| 2006-438 | 0.88 | No del | Un | 1.01 | 0.89 | n.d. | Un | 1.15 | 2.13 | yes | yes | No | ||
| 2006-633 | 0.97 | 1.09 | Un | 1.00 | 1.05 | Un | Un | 0.92 | 0.09 | yes | yes | No | ||
| 2006-777 | 0.93 | No del | Un | 1.00 | 1.03 | n.d. | Un | 0.98 | 0.99 | 3.79 | yes | |||
| 2006-843 | 0.96 | 0.57# | Un | 1.09 | 0.79 | Un | Un | 0.97 | 1.01 | No | ||||
| 2006-1091 | 0.78 | 0.99 | Un | 3.75 | 0.29 | Un | Un | yes | 0.99 | 1.06 | yes | No | ||
| 2007-24 | 1.23 | 0.78 | Un | 0.98 | 1.01 | Un | Un | 1.01 | 0.89* | No | ||||
| 2007-38 | 1.06 | No del | Un | 1.12 | 1.11 | Un | Un | 1.08 | 0.34* | yes | yes | No | ||
| 2007-486 | No del | No del | Un | 0.99 | 1.00 | Un | Un | 1.02 | 0.68* | No | ||||
| 2007-537 | 1.07 | No del | Un | 1.09 | 1.07 | Un | Un | 1.01 | 0.71 | No | ||||
| 2007-692 | 1.40 | No del | n.d. | 0.98 | 0.91 | partial | Un | 0.40 | 2.34* | yes | yes | No | ||
| 2007-729 | 1.03 | No del | n.d. | single | 0.79 | n.d. | Un | 0.46 | 2.35** | yes | yes | No | ||
|
| ||||||||||||||
| All samples including Table S1 | ||||||||||||||
| n=110 95% CI |
41.8% | 20.0% | 95.4% 89.8-100 Exclusine p=0.0027 |
33.6% | 86.3% 78.4-94.2 Exclusive p=0.025 |
68.9% 55.4-82.4 Non-exclusive p=0.0009 |
12.7% | 93.0% 87.1-98.9 Exclusive p=0.282 |
||||||
With INK4a/ARF probes, LOH (including homozygous deletion in #2003-226) was detectable in 19 cases with the 5′ probe #33647 (17.3 %), 10 cases (9.1 %) with the 3′ probe #27251, and 22 of 110 (20.0 %) with either the 5′ or 3′ probe. Likewise, LOH for the TP53 locus was detectable in 22 cases (20.0 %) with the 5′ probe #15811, 30 with the 3′ probe #89737 (27.3 %), and 37 of 110 (33.6 %) with either the 5′ or 3′ probes. This percentage was higher that the reported percentage of p53 mutations in sporadic breast cancers (20%, 24). We then sequenced the DNA-binding domain of the p53 gene in 10 p53 LOH (+) samples and found that the remaining p53 allele was mutated in 4 of 10 p53 LOH (+) cases (Table 1, Supplementary Figure S2). We then stained tissue blocks from breast cancer (13 p53 LOH [+] cases and 8 p53 LOH [−] cases) with a specific antibody to p53 (DO-1) and found overexpression of p53 in 6 of 13 p53 LOH(+) cases (46.2%), but not in any of the 8 cases with p53 LOH(−) breast cancers (Supplementary Figure S2). Importantly, all breast cancers with p53 mutation as demonstrated by sequencing showed overexpression of the p53 protein (Supplementary Figure S2). Conversely, none of the 8 samples without LOH for p53 showed high expression of p53 as studied by immunohistochemistry. These results are consistent with the previous report that showed frequent association of p53 mutations with loss of the other p53 allele in breast cancer (25). Thus, the hDMP1 locus was more frequently deleted in our breast cancer samples than the INK4a/ARF or p53 locus.
LOH for hDMP1 and INK4a/ARF was found to be mutually exclusive in 62 of 65 cases (95.4 %, p = 0.0027, χ2 = 8.977; 95 % confidence interval, 89.8-100 %) (Table 1 and Supplementary Table S1). Likewise, LOH for hDMP1 and p53 was also mutually exclusive in 63 of 73 cases (86.3%, p = 0.025, χ2 = 5.013; 95% confidence interval, 78.4-94.2 %). On the other hand, LOH for INK4a/ARF and p53 was exclusive only in 31 of 45 cases (68.9 %, p = 0.0009, χ2 = 11.088 against mutually exclusive hypothesis; 95 % confidence interval, 55.4-82.4%). The Hdm2 gene amplification (more than 6 copies) was found in 14 of 110 samples (12.7%, Table 1 and Supplementary Table S1). The Hdm2 gene amplification and LOH for hDMP1 appeared to occur independently of the other locus (93.0%, p = 0.282, χ2 = 1.157, not exclusive; 95 % confidence interval, 87.1-98.9%). Thus, our data demonstrate that 1) LOH for hDMP1 is typically found in human breast cancers with wild-type INK4a/ARF and p53 genomic loci, 2) LOH for INK4a/ARF and p53 occur simultaneously, and 3) LOH for hDMP1 and Hdm2 amplification occur at random with respect to one another.
We next studied the correlation between LOH for hDMP1 and known prognostic factors for breast cancer: HER2, estrogen receptor (ER), progesterone receptor (PR), Ki67, DNA ploidy, clinical stage, and age (data not shown). Setting the cut off level at 20%, we found significantly more cases with low Ki67 expression (i.e., Ki67+ < or = 20%) in the hDMP1 LOH (+) group in comparison to the LOH (−) group (p = 0.0266, χ2 = 4.92). Conversely, breast cancers with LOH for p53 were associated with high Ki67 (> 20%) (p = 0.0153, χ2 = 5.88) while LOH for INK4a/ARF or Hdm2 amplification was not associated with this proliferation marker (p = 0.196 and p = 0.522 respectively). We also found that breast cancers with LOH for hDMP1 more often had diploid DNA content than LOH (−) cases (p = 0.0463, χ2 = 3.97). On the other hand, LOH for INK4a/ARF or p53 was associated with aneuploidy of DNA (p= 0.0217, χ2 = 5.08; p = 0.0141, χ2 = 6.03, respectively). Conversely, Hdm2 amplification was not associated with ploidy of tumor DNA (p = 0.701). HER2 protein overexpression (2+ - 3+) was found in both hDMP1 LOH (+) (10/41, 24.4 %) and (−) (25/59, 42.4 %) without a statistically significant difference (p = 0.064). This finding is in agreement with the fact that MMTV-neu tumor development was accelerated in both Dmp1-null (16) and p53-mutant (26) backgrounds. There was no statistically significant difference in ER, PR, clinical stage, patients’ age and LOH for hDMP1.
We then classified all the breast cancer cases based on the data from histochemical studies for ER, PR, HER2, Ki67, cytokeratin, and morphology of tumor cells as proposed from the Komen Website http://ww5.komen.org/BreastCancer/SubtypesofBreastCancer.html into luminal A, luminal B, HER2, triple-negative, and unclassified/normal-type (27). The Ki67 positivity ratio of 14% was used to differentiate luminal A and luminal B subtypes, and breast cancers with HER2 (+++) was categorized into HER2 subtype. According to these criteria, 30.8% (32 of 104) of total cases were classified into luminal A, 23.1% (24 cases) were luminal B, 19.2% (20 cases) were HER2 type, 17.3% (18 cases) were triple-negative/basal-type, and 9.6% (10 cases) were unclassified/normal-type (Table 2), which were close to those that had been shown in the literature (27). Six of 110 cases could not be classified due to lack of paraffin sections. We then conducted statistical analyses and found that hDMP1 LOH (+) breast cancers were significantly associated with luminal A group of breast cancers (p = 0.0085; χ2 = 6.924) while p53 LOH(+) breast cancers were significantly associated with non-luminal A subtype (p = 0.0234; χ2= 5.141) (Table 2). Since LOH for hDMP1 is associated with low Ki67 index, higher incidence of a diploid karyotype, and luminal A subcategory, it was expected that deletion of hDMP1 will be a favorable prognostic factor for breast cancer patients.
Table 2. Subclassification of breast cancers studied and relationship with hDMP1 and p53 LOH.
All the breast cancer cases (n=104, enough information was not available in 6 cases) have been subclassified into luminal A, luminal B, HER2, triple-negative/basal type, and unclassified/normal-type based on the data from histochemical studies for ER, PR, HER2, Ki67, cytokeratin, and morphology of tumor cells as described in the Materials and Methods. The percentage of our breast cancer samples in each category was very close to those reported in the literature. hDMP1 LOH(+) breast cancers were significantly associated with luminal A category while p53 LOH(+) breast cancers were associated with non-luminal A subtype.
| hDMP1 LOH(+) | hDMP1 LOH(−) | Pecentage | p values | |
|---|---|---|---|---|
| Luminal A | 19 | 13 | 45.2 | 0.0085 |
| Luminal B | 8 | 15 | 19.0 | 0.5350 |
| HER2 | 5 | 17 | 21.1 | 0.0573 |
| Triple-negative | 6 | 12 | 11.9 | 0.5026 |
| Normal/unclassified | 4 | 5 | 9.5 | 0.7951 |
| Not evaluated | 4 | 2 | ||
| total | 46 | 64 | ||
| p53 LOH(+) | p53 LOH(−) | Pecentage | p values | |
| Luminal A | 6 | 26 | 16.7 | 0.0234 |
| Luminal B | 8 | 15 | 22.2 | 0.9848 |
| HER2 | 10 | 12 | 27.8 | 0.2288 |
| Triple-negative | 8 | 10 | 22.2 | 0.3342 |
| Normal/unclassified | 4 | 5 | 11.1 | 0.5546 |
| Not evaluated | 2 | 4 | ||
| total | 38 | 72 | ||
| All cases | Pecentage | Reported percentage | ||
| Luminal A | 32 | 29.1 | 28 | |
| Luminal B | 23 | 20.9 | 19 | |
| HER2 | 22 | 20.0 | 17 | |
| Triple-negative | 18 | 16.4 | 27 | |
| Normal/unclassified | 9 | 8.2 | 8 | |
| unknown | 6 | 5.4 | ||
| total | 110 |
Correlation of DMP1 protein expression with hDMP1 LOH and HER2 status in human breast cancer
We then studied whether LOH for hDMP1 affects protein expression in breast cancer samples by immunohistochemistry with specific antibodies (28, 29). The nuclear hDMP1 expression levels were categorized into four grades, 0 to 3++ (Figure 2a). Breast cancer samples without LOH for hDMP1 showed more intense nuclear staining for hDMP1 (mostly grades 2-3) while tumors with LOH showed weaker staining (mostly grades 0-1) (p = 0.0006, Figure 2). Normal breast epithelial cells also showed weak (1+) hDMP1 staining (data not shown). We found a significant increase in hDMP1 staining in breast carcinomas that showed HER2 overexpression (2+ or 3+) (p = 0.0038, Figure 2b), regardless of LOH for hDMP1. Together, our data show that: 1) hDMP1 protein is downregulated in clinical samples that showed LOH for hDMP1 and 2) HER2 and hDMP1 expression levels are positively correlated.
Figure 2. Histological grading of hDMP1 in human breast carcinoma.

a, human breast cancer tissues were stained with Dmp1-specifc RAX antibody (28) and the intensity of the nuclear staining was graded from 3(++), 2(+), 1(+/−), and 0 (negative). The scale bar is 100 μm.
b, correlation between LOH for hDMP1 and immunohistochemical grading of breast cancers. Breast cancer samples without LOH for hDMP1 showed significantly stronger nuclear signals for hDMP1. The hDMP1 signals were significantly higher in HER2 3+ or 2+ samples than in HER2 1+ or negative samples indicating the presence of the signaling pathway between HER2 and hDMP1 in breast cancers. Two different intensity values for hDMP1 indicate that the staining pattern for hDMP1 was mosaic; the average values (DMP1 scores) were used for statistical analyses.
Impact of LOH for hDMP1, INK4a/ARF, p53, and Hdm2 amplification on breast cancer survival
We then studied the impact of LOH for hDMP1, INK4a/ARF, p53, and Hdm2 amplification in stage I to III patients (n = 108; 2 cases of stage IV patients were eliminated from the survival study, Figure 3). Breast cancers with LOH for DMP1 had longer relapse-free survival than those without LOH (p = 0.0092, χ2 = 6.79; 70% survival 1,987 vs. 1,036 days) (Figure 3a). LOH for INK4a/ARF had no impact on patients’ survival (p = 0.591, χ2 = 0.289; 70% survival 1,121 vs. 1,830 days) (Figure 3b). Conversely, breast cancer with Hdm2 amplification showed significantly shorter survival than those without gene amplification (p = 0.0217, χ2 = 5.27; 70% survival 499 vs. 1,830 days) (Figure 3c). Likewise, LOH for p53 had significantly negative impact on patients’ disease-free survival (p = 0.0211, χ2 = 5.41; 70% survival 1,036 vs. 1,932 days) (Figure 3d) consistent with the finding that ~50% of p53 LOH cases showed simultaneous mutation of the remaining p53 allele (Supplementary Figure S2). The survival of breast cancer patients without LOH for the three loci and absence of Hdm2 amplification was not significantly different from those with involvement of the pathway (Supplementary Figure S3). Together, our data indicate that the more downstream the molecule is localized in DMP1-ARF-Hdm2-p53 signaling, the more negative impact the marker shows on breast cancer.
Figure 3. Relapse-free survival of 108 cases of human breast carcinoma dependent on LOH for hDMP1, INK4a/ARF, p53 or Hdm2 amplification.

Kaplan-Meier analyses have been conducted to study the impact for the impact of loss or gain of each locus on breast cancer patients’ disease-free survival up to 3,000 days. Only patients with stage I to III disease have been analyzed. Positive cases for gene deletion or amplification are indicated in solid lines and negative cases are shown in discontinuous lines. LOH for hDMP1 (a) has significantly positive impact (i.e. fair prognosis) on patient’s relapse-free survival while Hdm2 amplification (c) or LOH for p53 (d) had significantly negative impact. LOH for INK4a/ARF (b) had little influence on breast cancer patients’ long-term survival.
Growth inhibition of human breast epithelial cells by Dmp1:ER
Finally, we studied whether conditional activation of Dmp1:ER affects the growth of human breast epithelial and cancer cells. Non-transformed human mammary epithelial cells (MCF10A, human mammary epithelial cells [HMEC]) and breast carcinoma cell lines (MCF7, MDA-MB-175VII, ZR-75-1, BT-549, and HCC1569) were infected with Dmp1:ER or empty vector virus, and puromycin-resistance cells were cultured under the presence of 2 μM 4-hydroxytamoxifen (4-HT) (8, 12). The genomic statuses for p14ARF, Hdm2, p53, p16INK4a, and HER2 for human breast epithelial or cancer cell lines are summarized in Supplementary Table S2. Cell growth was completely inhibited by expressing Dmp1:ER in both MCF10A and tert-immortalized HMEC (Figure 4a, b). Significant inhibition of cell growth by Dmp1:ER was also observed in ZR-75-1 (Figure 4e) and MDA-MB-175VII (data not shown) breast cancer cells with wild-type ARF and p53 although the effect was significantly weaker in breast cancer cells than in HMEC or MCF10A. Western blotting (and real-time PCR in HMEC) analyses showed significant accumulation of p14ARF, p53, p21CIP1, and Hdm2 in response to activation of Dmp1:ER in HMEC and ZR-75-1 cells (Figure 5a, e; Supplementary Figure S4). In MCF10A cells, significant accumulation of p53 and p21CIP1 was observed at 12-36 hours in response to Dmp1:ER (Figure 5b) although p14ARF did not accumulate due to gene deletion. This data is consistent with our recent findings that Dmp1 physically interacts with p53 to neutralize the activities of Mdm2 in ARF-null cells (30). β-gal staining showed that ~40% of MCF10A cells underwent senescence by Dmp1 while ~70% of HMEC became senescent suggesting mixed growth inhibitory response (Supplementary Figure S5). The growth of MCF7 cells (ARF-null, p53 wild-type) was partially inhibited by Dmp1:ER (Figures 4c). Conversely HCC1569 cells with p53 deletion or BT-549 cells with p53 mutation did not slow down their growth by Dmp1:ER (Figure 4d, f). Indeed lysate analyses showed consistently high levels of p14ARF and undetectable p53 targets p21CIP1 or Hdm2 in these cells (Figure 5d, f).
Figure 4. Proliferation assay of non-transformed human breast epithelial cells and breast carcinoma cell lines that overexpress Dmp1:ER.

(a) HMEC (human mammary epithelial cells); HER2low, ARF+, p53+
(b) MCF10A; HER2low, ARF−, p53+
(c) MCF7; HER2low, ARF−, p53+
(d) HCC1569; HER2++, ARF+, p53del
(e) ZR-75-1; HER2high, ARF+, p53+
(f) BT-549; HER2low, ARF+, p53mut
solid lines show the growth curves of Dmp1:ER virus-infected cells treated with 2 μM 4-HT, discontinuous lines show those of mock-infected cells with 4-HT.
Figure 5. Western blotting analyses of (breast epithelial or cancer cells expressing activated Dmp1:ER or empty vector.

Lysate analyses were conducted by Western blotting with specific antibodies to Dmp1, p14ARF, p53, Hdm2, and p21CIP1. (a) HMEC, (b) MCF10A, (c) MCF7, (d) HCC1569, (e) ZR-75-1, and (f) BT-549 cells. Bottom axis shows hours after addition of 2 μM 4-HT.
We studied the growth of breast epithelial/cancer cells depleted of DMP1 by shRNA (12). Western analyses showed more than 90% downregulation of the hDMP1 protein in all of these three breast cancer cells and inactivation of the p53 pathway in MCF10A (Supplementary Figure S6). Depletion of hDMP1 by shRNA accelerated the growth of MCF7 cells (Supplementary Figure S7), but not T47D or MDA-MB-361 (wild-type ARF, mutant p53), suggesting that endogenous DMP1 is inhibiting the growth of p53 wild-type cells, but not in cells with mutant p53. The growth of p53 mutant cells by shRNA were retarded, possibly because shRNA to hDMP1 affected the function of other splicing variants (31) or hDMP1 interacts with mutant p53 for stabilization.
Then we conducted cell invasion assay using MCF7 cells with or without depletion for hDMP1 (see Supplementary Materials and Methods). Our results show 3.31 +/− 0.603 MCF7 cells with hDMP1 downregulation invaded from upper to lower chamber while only 1.57 +/− 0.970 cells migrated to the lower chamber in mock infected cells (p = 0.048). Conversely there was no significant effect of DMP1 expression in invasion assay with p53 mutant BT549 cells (55.2 +/− 9.25 vs. 64.5 +/− 14.1). Together, our data indicate that 1) both non-transformed human mammary epithelial cells and breast cancer cells with wild-type ARF and/or p53 (HMEC, MCF10A, MDA-MB-175VII, and ZR-75-1) are sensitive to growth inhibition/senescence by Dmp1 while breast cancer cells that delete ARF or deleted/mutant p53 show partial (MCF7) or nearly complete (HCC1569, BT-549) resistance to growth inhibitory effect by Dmp1, 2) Endogenous hDMP1 inhibits the growth of breast cancer cells with wild-type p53, and 3) DMP1-loss is associated with invasive phenotypes of breast cancer cells.
Discussion
In this study we analyzed 110 pairs of human breast cancer samples and demonstrated that hDMP1 is deleted in 42% of the cases. This percentage is even higher than the involvement of INK4a/ARF (~20%) or p53 (~35%) of the samples we analyzed, and importantly, was found in mutually exclusive fashion from LOH for INK4a/ARF or p53. On the other hand, LOH for INK4a/ARF and p53 were apparently overlapping, suggesting collaboration of these two loci, possibly through the synergism of p16INK4a loss and p53 inactivation. Deletion of hDMP1 was limited to the hDMP1 locus in 94% cases showing specificity of hDMP1 deletion in breast cancer. Importantly, deletion of the hDMP1 locus resulted in significant downregulation of the nuclear expression of the hDMP1 protein in breast cancer cells, signifying that the gene deletion significantly affected hDMP1 function and contributed to breast carcinogenesis. DMP1 protein expression was significantly higher in HER2(+) tumors than HER2(−), consistent with our recently published data showing that HER2/neu induces Dmp1 in mouse model of breast cancer and that HER2 activates hDMP1 promoter in human mammary epithelial cells (16).
Our study shows that LOH of hDMP1 is associated with relatively low Ki67 index and increased frequency of diploid DNA, both of which are indicators for favorable prognoses of breast carcinomas (23,32,33). In agreement, hDMP1 LOH(+) breast cancer was associated with luminal A subtype, and relapse-free survival was significantly longer (1,987 vs. 1,036 days) for hDMP1 LOH (+) cases than (−) patients. On the other hand, p53 LOH(+) breast cancer was associated with non-luminal A subtypes, both Hdm2 amplification and LOH for p53 were associated with shorter disease-free survival. Of note, although breast cancers with LOH for hDMP1 was associated with relatively low Ki67 index in comparison to p53 LOH samples, the former samples still showed higher Ki67 index (mean 19% in our samples) than normal breast epithelial cells (~2%; 34), indicating that loss of hDMP1 is associated with proliferation of normal breast epithelial cells, which can collaborate with other genetic alterations to develop breast cancer.
Our study shows that 35% of human breast cancers have LOH for p53 and 46% of such cases have mutation(s) of p53. This means ~16% of breast cancers have mutation(s) for p53 in our samples. Interestingly this percentage of p53 mutation is close to those that have been reported in the literature (20%) in sporadic breast cancers (24). Our data also indicate that approximately half of p53 LOH cases retain one p53 allele without p53 mutation. It has been reported that p53 heterozygous mice develop tumors at a mean latency of 70 weeks without losing or mutating the wild-type p53 allele in mice (35) suggesting that loss of one allele of p53 contributes to tumorigenesis in vivo. Although we currently do not have enough samples for survival analyses of p53+/− breast cancers, with or without p53 mutation, we continue the study to investigate the impact of single allelic p53 loss with or without p53 mutation on survival of breast cancer patients.
Since hDMP1 is a transactivator for the ARF promoter and p14ARF indirectly regulates the activity of p53 through Hdm2, there is a gradient of prognosis of breast cancer patients from (fair) hDMP1 LOH > INK4a/ARF LOH > Hdm2 amplification > or = p53 LOH (poor) possibly because : i) the closer the molecule is to p53, the more seriously p53 function will be affected, ii) LOH of p53 may be associated with a gain-of-function mutation of p53 (36), and iii) Hdm2 has multiple interacting partners other than p53 (e.g., E2F1, YY1, RB, ribosomal proteins) that explain its oncogenic potential (37). Furthermore, depending on which therapies were used to treat our cohort of patients, it is possible that loss of hDMP1 spared deletion of p53 gene, increased effectiveness of chemotherapy and radiation treatment and, thereby, extended time to relapse.
It should be noted that loss of hDMP1, INK4a/ARF, p53, or Hdm2 amplification did not exclusively correlate with currently used prognostic markers for breast cancer (ER, PR, HER2) (23). Thus, LOH studies for hDMP1, INK4a/ARF, p53, and real-time PCR assay for Hdm2 will be independent laboratory tests to predict the prognosis of breast cancer patients. Although hDMP1-loss is a favorable prognostic factor associated with longer relapse-free survival of patients than hDMP1 intact cases, 35% of breast cancer patients relapsed during the observation period of 8 years. Thus, it is likely that other genetic alteration(s) collaborate with DMP1-loss to accelerate recurrence of the disease. Further molecular genetic studies are required to clarify which molecular events collaborate with hDMP1-loss in breast cancer progression.
Our data show that shRNA to hDMP1 stimulated proliferation of breast cancer cells with wild-type p53, but inhibited cell growth of cells with mutant p53. There are two possible explanations why p53 mutant cells proliferate slower with hDMP1 knockdown. First, the shRNA used downregulates all the three DMP1 splicing variants including the tumor suppressor DMP1α. The function of other two transcripts is unknown although published study suggested the β and γ variants might be blocking the activity of hDMP1α (31). Development of splicing isoform-specific shRNA will be needed to elucidate the function of each variant on cell growth. The second possibility is that hDMP1 may directly interact with mutant p53 and hDMP1 knockdown may affect p53 gain-of-function, and thereby, reduce proliferative capacity of cells with specific p53 mutation. In support of later, patients with hDMP1 LOH(+) tumors have favorable prognosis compared to patients with hDMP LOH(−), half of which harbor p53 mutation, further suggesting that hDMP1 may promote breast cancer progression by stabilizing mutant p53. Thus, it would be of great interest to understand whether DMP1 affects function of mutant p53.
In conclusion, we have characterized the frequency and the pattern of alteration of the hDMP1-ARF-Hdm2-p53 pathway in human breast cancer. Each component in the signaling pathway can define a different disease entity associated with prognosis. Hemizygous deletion of DMP1 is found in nearly half of human breast carcinomas that often retain the wild-type p53 and INK4a/ARF loci. This finding is significant as we move closer towards personalized therapy for each breast cancer patient based on their tumor genetic alterations. Our data suggests that patients with hDMP1 LOH should be selected for current and future therapies whose efficacy is dependent on an intact p53 gene. On the other hand, patients with wild-type hDMP1 (~50% of all breast cancer patients in this study) in their tumor biopsy should be spared toxic side-effects from treatments that would be ineffective with p53 LOH. Alternatively, further research is necessary to develop small molecules that specifically activate the hDMP1 promoter or protein which will be a feasible approach to treat human breast cancer patients with DMP1 LOH since their tumors maintain a second wild-type DMP1 allele without mutation or promoter methylation.
Materials/Subjects and Methods
The protocols for LOH assay, statistical analyses, immunohistochemical studies of breast cancer samples, cell invasion assay, and real-time PCR are described in Supplementary Materials and Methods.
Human breast cancer samples and cell lines
One hundred and ten pairs of human breast carcinomas and their normal counterparts were obtained from the Tissue Procurement Core Facility of Wake Forest University. The patients’ profiles are as follows. Age: 37-89 years old, mean 57 years; stage I: 30 %, stage II: 45 %, stage III: 23 %, stage IV: 2 %; histology, ductal carcinoma (ca): 87 %, lobular ca: 6 %, metaplastic ca: 3 %, mucinous ca: 2 %, papillary ca: 2 %; HER2, 3+: 22 %, 2+: 15 %. These cases comprise a population-based cohort treated at Wake Forest Baptist Medical Center from 1999-2008. Standard of care treatments included hormone therapy (i.e., tamoxifen monotherapy), chemotherapy (anthracyclines, taxanes), no systemic therapy, and local radiation. Disease-free survival events were defined as local, regional or distant recurrence during the time interval from diagnosis to last follow-up.
Classification of human breast cancers
Breast cancer samples were classified into 5 types (luminal A, luminal B, HER2, triple-negative/basal, and normal/unclassified) based on the data from histochemical studies for ER, PR, HER2, Ki67, cytokeratin, and morphology of tumor cells as proposed from the Komen Website http://ww5.komen.org/BreastCancer/SubtypesofBreastCancer.html. These pathological examinations have been conducted at Wake Forest University Breast Cancer Center of Excellence. The Ki67 positivity ratio of 14% was used to differentiate luminal A and luminal B subtypes (27), and breast cancers with HER2 (+++) was categorized into HER2 subtype.
Western blotting
Proteins were extracted with ice-cold EBC buffer with proteinase inhibitors (7). After gel electrophoresis and transfer to nitrocellulose membranes, proteins were visualized by immunoblotting with affinity-purified polyclonal antibodies to Dmp1 (RAX) , p53 (sc-6243G, Santa Cruz), Hdm2 (ab16895 [2A10], Abcam, Cambridge, MA), p14ARF (sc-53639, 53640), p21CIP1 (sc-6246), or β-Actin (sc-1615), followed by incubation of the filters with HRP-conjugated second antibodies, and reaction with the enhanced ECL detection kit (PerkinElmer, Boston, MA).
Supplementary Material
Acknowledgements
We are grateful to R. Weinberg for HMEC cells; C. Sherr, and M. Roussel for plasmid DNAs. We thank G. Hawkins for LOH analyses of breast cancer samples. We also thank G. Kucera for providing information on human breast cancer patients’ samples, Tim Kute for helpful discussions, Guorui Deng, and Jordan Chapman for technical assistance. K. Inoue is supported by ACS RSG-07-207-01-MGO, NIH/NCI 5R01CA106314, and by Director’s Challenge Award #20595 from WFUHS. P. Taneja was supported by the Susan G. Komen Foundation postdoctoral fellowship KG080179. D. Maglic has been supported by DOD pre-doctoral fellowship BC100907.
Footnotes
Conflict of interest The authors declare no conflict of interest.
Supplementary information is available at Oncogene’s website.
References
- 1.Hortbagyi GN. Treatment of breast cancer. N Engl J Med. 1998 Oct 1;339(14):974–84. doi: 10.1056/NEJM199810013391407. Review. [DOI] [PubMed] [Google Scholar]
- 2.Ross JS, Hortobagyi GN, editors. Molecular Oncology of Breast Cancer. Jones and Bartlett Publishers; Sudbury, Massachusetts: 2005. [Google Scholar]
- 3.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. 2011 Jul-Aug;61(4):212–36. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 4.Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006 Dec;10(6):529–41. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Smeets SJ, Harjes U, van Wieringen WN, Sie D, Brakenhoff RH, Meijer GA, et al. To DNA or not to DNA/ That is the Question, When It Comes to Molecular Subtyping for the Clinic! Clin Cancer Res. 2011 Aug 1;17(15):4959–64. doi: 10.1158/1078-0432.CCR-11-0462. [DOI] [PubMed] [Google Scholar]
- 6.Hirai H, Sherr CJ. Interaction of D-type cyclins with a novel myb-like transcription factor, DMP1. Mol Cell Biol. 1996 Nov;16(11):6457–67. doi: 10.1128/mcb.16.11.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. 1998 Mar;18(3):1590–600. doi: 10.1128/mcb.18.3.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue K, Roussel MF, Sherr CJ. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci USA. 1999 Mar 30;96(7):3993–8. doi: 10.1073/pnas.96.7.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, et al. Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev. 2000 Jul 15;14(14):1797–809. [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001 Nov 15;15(22):2934–9. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K. Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol. 2005 Jan;25(1):220–32. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, Choudhary M, et al. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell. 2007 Oct;12(4):381–94. doi: 10.1016/j.ccr.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue K, Mallakin A, Frazier DP. Dmp1 and tumor suppression. Oncogene. 2007 Jun 28;26(30):4329–35. doi: 10.1038/sj.onc.1210226. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue K, Sugiyama T, Taneja P, Morgan RL, Frazier DP. Emerging roles of DMP1 in lung cancer. Cancer Res. 2008 Jun 15;68(12):4487–90. doi: 10.1158/0008-5472.CAN-07-6791. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugiyama T, Frazier DP, Taneja P, Morgan RL, Willingham MC, Inoue K. Role of DMP1 and its future in lung cancer diagnostics. Expert Rev Mol Diagn. 2008 Jul;8(4):435–47. doi: 10.1586/14737159.8.4.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taneja P, Maglic D, Kai F, Sugiyama T, Kendig RD, Frazier DP, et al. Critical role of Dmp1 in HER2/neu-p53 signaling and breast carcinogenesis. Cancer Res. 2010 Nov 15;70(22):9084–94. doi: 10.1158/0008-5472.CAN-10-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bieche I, Champeme MH, Matifas F, Hacene K, Callahan R, Lindereau R. Loss of heterozygosity on chromosome 7q and aggressive primary breast cancer. Lancet. 1992 Jan 18;339(8786):139–43. doi: 10.1016/0140-6736(92)90208-k. [DOI] [PubMed] [Google Scholar]
- 18.Kristjansson AK, Eiriksdottir G, Ragnarsson G, Sigurdsson A, Gudmundsson J, Barkardottir RB, et al. Loss of heterozygosity at chromosome 7q in human breast cancer: association with clinical variables. Anticancer Res. 1997 Jan-Feb;17(1A):93–8. [PubMed] [Google Scholar]
- 19.Bodner SM, Naeve CW, Rakestraw KM, Jones BG, Valentine VA, Valentine MB, et al. Cloning and chromosomal localization of the gene encoding human cyclin D-binding Myb-like protein (hDMP1) Gene. 1999 Mar 18;229(1-2):223–8. doi: 10.1016/s0378-1119(98)00591-5. [DOI] [PubMed] [Google Scholar]
- 20.Marchetti A, Buttitta F, Girlando S, Dalla Palma P, Pellegrini S, Fina P, et al. Mdm2 gene alterations and mdm2 protein expression in breast carcinomas. J. Pathol. 1995 Jan;175(1):31–8. doi: 10.1002/path.1711750106. [DOI] [PubMed] [Google Scholar]
- 21.Turbin DA, Cheang MC, Bajdik CD, Gelmon KA, Yorida E, De Luca A, et al. MDM2 protein expression is a negative prognostic marker in breast carcinoma. Mod Pathol. 2006 Jan;19(1):69–74. doi: 10.1038/modpathol.3800484. [DOI] [PubMed] [Google Scholar]
- 22.Masood S. Prognostic/predictive factors in breast cancer. Clin Lab Med. 2005 Dec;25(4):809–25. viii. doi: 10.1016/j.cll.2005.08.012. Review. [DOI] [PubMed] [Google Scholar]
- 23.Taneja P, Maglic D, Kai F, Zhu S, Kendig RD, Fry EA, et al. Classical and novel molecular prognostic markers for human breast cancer and their clinical significance. Clinical Medicine Insights: Oncology. 2010 Apr 20;4:15–34. doi: 10.4137/cmo.s4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gasco M, Yulug IG, Crook T. TP53 mutations in familial breast cancer: functional aspects. Hum Mutat. 2003 Mar;21(3):301–6. doi: 10.1002/humu.10173. [DOI] [PubMed] [Google Scholar]
- 25.Wasielewski M, Elstrodt F, Klijn JG, Berns EM, Schutte M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res Treat. 2006 Sep;99(1):97–101. doi: 10.1007/s10549-006-9186-z. [DOI] [PubMed] [Google Scholar]
- 26.Li B, Rosen JM, McMenamin-Balano J, Muller WJ, Perkins AS. neu/ERBB2 cooperates with p53-172H during mammary tumorigenesis in transgenic mice. Mol Cell Biol. 1997 Jun;17(6):3155–63. doi: 10.1128/mcb.17.6.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009 May 20;101(10):736–50. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mallakin A, Taneja P, Matise LA, Willingham MC, Inoue K. Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene. 2006 Dec 14;25(59):7703–13. doi: 10.1038/sj.onc.1209750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallakin A, Sugiyama T, Kai F, Taneja P, Kendig RD, Frazier DP, et al. The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of Amphiregulin, Thrombospondin-1, JunB and Egr1. Int J Cancer. 2010 Mar 15;126(6):1403–16. doi: 10.1002/ijc.24938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frazier DP, Kendig RD, Kai F, Maglic D, Sugiyama T, Morgan RL, et al. Dmp1 physically interacts with p53 and positively regulates p53’s stabilization, nuclear localization, and function. Cancer Res. 2012 Apr 1;72(7):1740–50. doi: 10.1158/0008-5472.CAN-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tschan MP, Fischer KM, Fung VS, Pirnia F, Borner MM, Fey MF, et al. Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J Biol Chem. 2003 Oct 31;278(44):42750–60. doi: 10.1074/jbc.M307067200. [DOI] [PubMed] [Google Scholar]
- 32.Ross JS, Linette GP, Stec J, Ross MS, Anwar S, Boguniewicz A. DNA ploidy and cell cycle analysis in breast cancer. Am J Clin Pathol. 2003 Dec;120(Suppl):S72–84. doi: 10.1309/QD096UGF70T5H46G. Review. [DOI] [PubMed] [Google Scholar]
- 33.Wiesner FG, Magener A, Fasching PA, Wesse J, Bani MR, Rauh C, et al. Ki-67 as a prognostic molecular marker in routine clinical use in breast cancer patients. Breast. 2009 Apr;18(2):135–41. doi: 10.1016/j.breast.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Shetty A, Loddo M, Fanshawe T, Prevost AT, Sainsbury R, Williams GH, et al. DNA replication licensing and cell cycle kinetics of normal and neoplastic breast. Br J Cancer. 2005 Nov 28;93(11):1295–300. doi: 10.1038/sj.bjc.6602829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, et al. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J. 1998 Aug 17;17(16):4657–67. doi: 10.1093/emboj/17.16.4657. [Pubmed 970425] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009 Oct;9(10):701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 37.Manfredi JJ. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010 Aug 1;24(15):1580–9. doi: 10.1101/gad.1941710. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.