

Abstract
As an obligatory intracellular pathogen, human immunodeficiency virus type-1 (HIV) is dependent upon its ability to exploit host cell machinery for replication and dissemination, and to circumvent cellular processes that prevent its growth. One such intracellular process is autophagy, a component of the host defense against HIV with roles in innate immune signaling, adaptive immunity and intracellular degradation of HIV. During permissive infection, HIV down-modulates autophagy, promoting its own replication. Inducers of autophagy can overcome this suppression and inhibit HIV. This review summarizes recent advances in understanding the antiviral and replicative roles of autophagy during HIV infection. Dissecting the molecular mechanisms by which HIV utilizes autophagy may lead to the identification of novel drug candidates to treat and potentially eradicate HIV infection.
INTRODUCTION
Following an acute primary infection, human immunodeficiency virus type-1 (HIV) establishes a reservoir of infected long-lived cells while CD4+ T cells are progressively lost leading to AIDS. Whether HIV establishes true latency or a persistent infection with continued low-level viral replication during periods of prolonged viral suppression remains controversial. However, increasing data examining occult reservoirs support some continued viral replication in most persons. As an obligatory intracellular parasite, HIV survival is dependent upon its ability to exploit host cell machinery for replication and dissemination and to evade intrinsic cellular processes and defenses that may limit viral replication and pathogenesis including autophagy – a cell-autonomous innate immune defense whereby cells eliminate intracellular pathogens by capture into autophagosomes with subsequent killing through macroautophagy (a process sometimes referred to as xenophagy) [1].
Macroautophagy (hereafter referred to as autophagy) is a degradation pathway whereby cytosolic double membrane-bound compartments termed autophagosomes engulf cytoplasmic constituents such as sub-cellular organelles and microbial pathogens, including viruses, and target them for lysosomal degradation. During autophagy, the physical interaction of the phosphatidylinositol 3-kinase, catalytic subunit type 3 (PIK3C3) with Beclin-1 (BECN1) forms the PIK3C3 kinase complex producing phosphatidylinositol-3-phosphate (PI3P), which is essential for the induction of the nascent phagophore by recruiting PI3P binding proteins such as RAB5A and ATG14. To form the autophagosome, two intertwined conjugation systems play a role. In the first, ATG12 is activated by ATG7, an E1-like ubiquitin activating enzyme, transferred to ATG10, an E2-like ubiquitin conjugating enzyme which covalently conjugates it to ATG12. ATG12–ATG5 non-covalently binds ATG16L1 and further elongates the phagophore membrane. In the second, the cytosolic microtubule-associated protein 1 light chain 3 beta (LC3B)-I, encoded by MAP1LC3B, is converted to LC3B-II by a ubiquitin-like system that involves ATG7 and ATG3. The ATG12–ATG5-ATG16L1 complex functions as an E3-like ubiquitin ligase, ligating LC3B-II to the nascent autophagosome membrane through phosphatidylethanolamine with the LC3B-II associated with the inner membrane degraded after fusion of the autophagosome with lysosomes.
Although the first reports of autophagy during viral infections appeared in the 1960s [2], the potential significance of this is only recently being appreciated. An expanding literature indicates that autophagy, in a virus-specific manner, may function to both degrade viruses and promote viral replication. Moreover, autophagy is not only an important component of the innate immune system, but also helps to modulate the adaptive immune system through MHC class I and class II antigen presentation (Figure 1).
Figure 1. Modulation of autophagy.
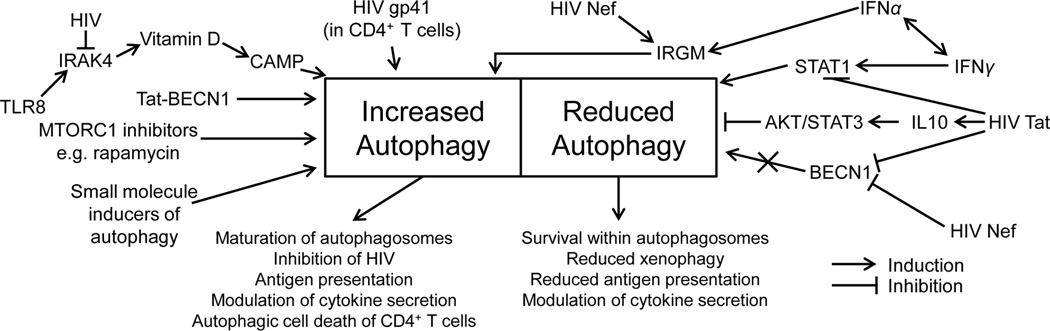
Autophagy can be pharmacologically induced in macrophages by various inducers of autophagy such as MTORC1 inhibitors, the Tat-Beclin peptide, drugs acting via inositol 1,4,5-triphosphate, by TLR8 agonists through the induction of the vitamin D pathway, by vitamin D directly through the induction of CAMP, and by cytokines such as IFNγ, which is dependent upon IRGM. The HIV protein Tat inhibits autophagy through the inihibition of STAT1 phosphorylation and by increasing the secretion of IL10. HIV Nef both increases autophagy through interactions with IRGM and inhibits autophagy through Beclin-1 sequestration. HIV infection of CD4+ T cells is associated with the initial induction of autophagy associated with the fusion activity of the HIV gp41 protein.
The role of autophagy in HIV pathogenesis has, until recently, been under appreciated. However, over the past five years an increasing number of laboratories have described the interaction of HIV and autophagy. As often occurs in new areas of investigation, there are contradictory findings. Of note, it is important to differentiate studies that are performed in continuous transformed cell lines or with pseudotyped virus from those performed using primary cells and infectious HIV isolates. Often, transformed cells are under different autophagic homeostasis than primary cells and may lead to findings that are not corroborated when evaluated in primary cells. Similarly, single-round replication deficient HIV may not provide a complete picture of cells that are infected with multiple-round replication competent virus, although they can help to dissect molecular processes. In this review, we have summarized many of the key studies that have been performed, to date, regarding HIV and autophagy.
HIV subverts autophagy to promote its own replication
Of the more than 35 human autophagy-associated genes currently known to be involved in autophagy, ten have been linked with HIV replication. Using a large-scale small RNAi screen to identify host factors required by replication competent HIV in a HeLa-derived cell line, Brass et al. [3] identified more than 250 HIV-dependency factors. These included four genes involved in the nucleation and elongation of autophagosomes (ATG7, MAP1LC3B, ATG12, ATG16L2) and two involved in lysosomal function (CLN3 and LAPTM5) that were essential for HIV replication. In another cell-line model, Eekels et al. [4] demonstrated that RNAi of PIK3R4, ATG4A, ATG5 or ATG16 resulted in the inhibition of replication competent CXCR4-tropic HIV in SupT1 cells. An additional two groups have demonstrated that HIV replication in primary cells is inhibited upon autophagy-associated gene silencing: BECN1 and ATG5 in macrophages [5–7], and BECN1 and ATG7 in monocytes [8].
In agreement with these RNAi studies, the current data suggest that the early steps of autophagy are important in HIV replication in cells of the monocyte/macrophage lineage. Espert et al. [9] demonstrated that autophagosome accumulation occurs in productively infected primary macrophages infected through exposure to chronically infected MOLT-4 effector cells. They found that virions were only detectable in cells with moderate, but not high levels of accumulated autophagosomes suggesting that HIV elicits the induction of autophagy but blocks its late proteolytic stage. Consistent with these findings, HIV assembly is thought to occur on endocytic membranes that intersect with recycling endosomes [10,11]. HIV Gag-derived proteins colocalize and interact with LC3B in macrophages, and are present at LC3B-II enriched membranes suggesting that autophagy may be involved in Gag processing and the production of nascent virions [8]. Moreover, the HIV accessory protein Nef interacts with immunity-associated GTPase family M (IRGM), which itself interacts with ATG5, ATG10, LC3B and SH3GLB1, inducing autophagosome formation and enhancing HIV replication [12]. Finally, in HIV-infected macrophages where the nucleation and formation of autophagosomes was inhibited using small-molecule chemical inhibitors or RNAi, the production of infectious virions was significantly decreased [7–9] (Figure 1).
Although HIV may upregulate autophagy during the initial stages of primary infection, HIV must control the antiviral proteolytic and degradative late stages of autophagy to avoid its degradation (Figure 2). The current data suggest that HIV has developed mechanisms to inhibit autophagic degradation in CD4+ T cells [13] and cells of the monocyte/macrophage lineage [8]. Kyei et al. [8] demonstrated that in HIV-infected macrophages, HIV Nef plays a major role in the inhibition of the proteolytic stages of autophagy by binding BECN1. Interestingly, the authors demonstrated that the Nef 174DD motif previously shown to be required for CD4 downregulation and interactions with the V1 domain of the vacuolar H+-ATPase [14] was required for Nef to act as an anti-autophagic maturation factor. Moreover, Nef-deficient HIV was unable to overcome autophagic degradation and thus replicated less efficiently. It is unlikely that Nef influences H+-ATPase assembly or activity thereby inhibiting autophagosome acidification or autophago-lysosome fusion as the inhibition of endosome acidification was previously shown to be independent of Nef [15]. Thus, the protein complex containing Nef and BECN1 may act through a mechanism other than acidification, possibly through the redistribution of membrane-associated PIK3C3 [8]. A recent study demonstrated that Nef binds to amino acids 267–284 in the BECN1 evolutionarily conserved domain [16]. This is the same region that is necessary and sufficient for BECN1 to bind GLIPR2 (GAPR-1) [16], a protein that associates with lipid rafts at the cytosolic leaflet of the Golgi membrane [17] and that negatively regulates autophagy by sequestering BECN1 to the Golgi complex (where it is inactive in autophagy) [16]. Whereas Nef may not alter autophagosome acidification, recent findings from our laboratory suggest that during permissive infection HIV prevents the fusion of autophagosomes with lysosomes through the inhibition of the hepatocyte growth factor required tyrosine kinase substrate (HGS) by a yet unidentified mechanism.
Figure 2. Schematic representation of modulation of autophagy by HIV.
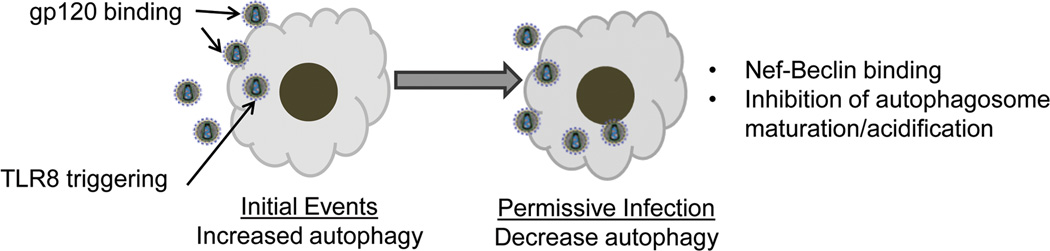
During the initial stages of infection, HIV or gp120 binding and TLR8 signaling induce autophagy to promote viral replication. However, during permissive infection autophagy is down-regulated through Nef-Beclin interaction and inhibition of autophagosome acidification promoting viral persistence and cell survival. However, as shown in Figure 1, inducers of autophagy promote increased autolysosome formation with degradation of HIV.
HIV infection of CD4+ T cells is associated with the initial induction of autophagy associated with the fusion activity of the HIV gp41 protein [18] followed by the inhibition of autophagy and reduced autophagosome accumulation during productive infection [9,13,19]. It is important to note that during permissive infection of primary cells, HIV down modulates autophagy. Nevertheless, autophagy is never completely abrogated since it is required for cell survival. However, inducers of autophagy, including amino acid starvation and rapamycin, overcome the Nef-imposed phagosome maturation block leading to inhibition of viral replication [13] (Figure 1).
HIV-induced autophagy in bystander cells
HIV infection and the progression to AIDS are characterized by the depletion of CD4+ T cells through apoptosis of the uninfected bystander cells and the direct killing of HIV-infected cells. More recently, HIV has been shown to influence autophagy in HIV-uninfected bystander cells. HIV Tat inhibits interferon-γ-induced autophagy induction in uninfected macrophages through the inhibition of STAT1 phosphorylation [20]. Tat has also been shown to inhibit autophagy in uninfected macrophages through binding CXCR4, VEGFR1 and β-integrins, and through the induction of AKT1, SRC and IL10 production [21]. As autophagy is an immune effector mechanism for the targeted destruction of several intracellular pathogens, the inhibition of autophagy in bystander macrophages may contribute to the susceptibility of HIV-1-infected patients to tuberculosis and toxoplasmosis amongst others. Moreover, as autophagy promotes antigen presentation, the inhibition of autophagy may promote pathogen immune evasion.
In contrast to the effect of Tat on uninfected macrophages, HIV Env protein induces apoptosis in uninfected CD4+ T cells [9,22] and neurons [23] through a mechanism involving the accumulation of Beclin-1 and the induction of autophagy. Within CD4+ T cells, this is dependent upon the fusion activity of the HIV gp41 protein [18] regardless of the co-receptor used for HIV entry [9]. How the fusogenic function of gp41 induces autophagy in CD4+ T cells and why gp41 induces autophagy in CD4+ T cells but not in macrophages is unknown [9]. Within the central nervous system, HIV gp120 induces autophagy in neuroblastoma cells [23]. Postmortem brains from HIV encephalitis (HIVE) patients exhibit increased markers of autophagy compared with brains from HIV infected individuals without HIVE or HIV uninfected controls [23,24]. However, it remains unknown whether autophagy proceeds to completion in these brains or if there is an accumulation of autophagic proteins without autophagy-related degradation. These findings, however, provide support for the hypothesis that the dysregulation of autophagy during HIV infection is important in the pathogenesis of neuroAIDS [25].
Utilizing autophagy to control HIV pathogenesis
Cells can use autophagy in a number of different antiviral roles. These include xenophagy, the presentation of endogenous viral antigens to the major histocompatibilty complexes and subsequent activation of the adaptive immune system, and as a mechanism to detect intracellular pathogens through delivery of viral products to endosomal pattern-recognition receptors (PRRs), which recognize signature molecules of pathogens termed pathogen-associated molecular patterns (PAMPs).
Recent research has focused on the role of autophagy in the innate and adaptive immune systems with pharmacological inducers, enhancers and inhibitors of autophagy currently being examined for the treatment of cancer [26], lymphangioleiomyomatosis, [27], proteinopathies such as Huntington’s disease [28], and infectious diseases including Mycobacterium tuberculosis [5,29] with some currently in clinical trials. In contrast, despite the strong need for the development of alternative antiretrovirals due to the emergence of resistant viruses, the complexity of current treatment regimens and the toxicity of currently used antiretrovirals, few studies have examined the use of pharmacologic enhancers of autophagy in the treatment of HIV. The induction and modulation of autophagy through pharmacological means to enhance HIV treatment is attractive. Since autophagy works at the host cellular level to improve intracellular killing of both replicating and non-replicating HIV within endosomes, it has the potential to be used in combination with antiretrovirals to treat and potentially eradicate HIV. Moreover, as autophagy is a cellular process, viral resistance is less likely to develop.
Rapamycin, a specific MTOR inhibitor and inducer of autophagy inhibits HIV replication through both the downregulation of CCR5 [30] and the induction of autophagy [7]. Rapamycin has also been found to inhibit HIV infection in human peripheral blood leucocytes-SCID reconstituted mice [31]. Of interest, rapamycin also works synergistically to enhance the activity of entry inhibitors such as vicriviroc, aplaviroc and enfuvirtide [31]. However, rapamycin has immunosuppressive and other adverse effects that limit its potential usefulness in the treatment of HIV.
A cell-permeable autophagy-inducing peptide, termed Tat–Beclin-1 derived from the region of BECN1 that interacts with HIV Nef and conjugated to the basic region of HIV Tat has recently been found to markedly inhibit HIV replication in primary human macrophages through the induction of autophagy through the canonical pathway with the same magnitude as rapamycin [16]. This peptide was also able to restrict the in vitro replication of Sindbis virus, chikungunya virus and West Nile Virus, and reduced the mortality of mice infected with West Nile Virus and chikungunya virus [16].
The antimicrobial effects of vitamin D have been well documented and association studies have linked low levels of 25-hydroxycholecalciferol (25D3) and/or the active metabolite 1α,25-dihydroxycholecalciferol (1,25D3) with increased risk of or severity of infection with HIV [32,33]. Physiological concentrations of 1,25D3 induces the dose-dependent inhibition of HIV alone and in Mycobacterium tuberculosis co-infected macrophages that is dependent upon both the induction of autophagy and autophagosomal maturation [5,7]. The precise mechanism by which 1,25D3 induces autophagy has yet to be elucidated, but it is known that it is dependent upon the human cathelicidin antimicrobial peptide (CAMP). This conclusion is supported by recent studies demonstrating that CAMP silencing inhibits 1,25D3-induced autophagy and significantly lowers the 1,25D3-mediated restriction of HIV replication and antimycobacterial activities [5,34]. Endogenous CAMP has been implicated in a number of cellular functions including the regulation of inflammatory responses [35] and the formation and maturation of autophagosomes [34]. CAMP has also been shown to play an important role in the activation of mitogen activated protein kinases and CEBPB which contribute to the transcriptional activation of BECN1 and ATG5 in response to 1,25D3 [34]. Moreover, during autophagy, autophagosomes recruit CAMP through an AMP kinase, calcium and CAMKK2 dependent mechanism where it is involved in microbial killing [34]. Further work is necessary to determine the precise role of CAMP in autophagy and antiretroviral activity.
Vitamin D has also been implicated in the autophagic response to PAMPs by PRRs. There are several classes of PRRs: Toll like receptors (TLRs), retinoic acid-inducible gene-I-like receptors and nucleotide-binding oligomerization domain-like receptors. The single-stranded RNA genome of HIV contains multiple PAMPs that are recognized by endosomal TLR7 in dendritic cells, and TLR8 in macrophages [36–38]. Upon stimulation, TLR7/8 agonists activate NFKB1 via MYD88 that leads to the induction of a cascade of antiviral effector functions including the induction of autophagy [6,39] and proinflammatory cytokines [38]; thus significantly impairing the permissivity of peripheral blood mononuclear cells to HIV infection [40]. Interestingly, HIV downregulates IRAK4, which is essential for virtually all TLR signaling [41]. In macrophages, the TLR8-mediated inhibition of HIV is dependent upon both the presence and upregulation of vitamin D metabolism and the vitamin D (1,25D3) receptor which induces autophagy through a CAMP dependent mechanism [6]. In the case of plasmacytoid dendritic cells, the TLR7-mediated expression of the proinflammatory cytokine interferon-α is dependent upon the induction of autophagy [42].
Collectively, these studies demonstrate that inducers of autophagy inhibit HIV replication. Well-controlled clinical trials are needed to determine if autophagy inducers are of value as adjunctive treatment in HIV-infected persons. Dissecting the molecular mechanisms by which HIV utilizes autophagy has the potential to lead to the identification of novel drug candidates to treat and potentially to eradicate HIV infection, as well as preventing HIV-related related complications.
Highlights.
HIV inhibits autophagy during permissive infection
Modulation of autophagy alters HIV pathogenesis and development of CNS impairment
The induction of autophagy inhibits HIV
Vitamin D induces autophagy and inhibits HIV at physiologic concentrations
TLR8 inhibition of HIV requires vitamin D and induction of autophagy and cathelicidin
ACKNOWLEDGMENTS
This work was supported by the NIAID, NIH (grant AI084573), the International Maternal Perinatal Adolescent AIDS Clinical Trials (IMPAACT) Network and the California HIV/AIDS Research Program (IDEA grant ID12-SD-255). Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) [U01 AI068632], the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the National Institute of Mental Health (NIMH) [AI068632]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the CHRP. This work was supported by the Statistical and Data Analysis Center at Harvard School of Public Health, under the National Institute of Allergy and Infectious Diseases cooperative agreement #5 U01 AI41110 with the Pediatric AIDS Clinical Trials Group (PACTG) and U01 AI068616 with the IMPAACT Group. Support of the sites was provided by the National Institute of Allergy and Infectious Diseases (NIAID) and the NICHD International and Domestic Pediatric and Maternal HIV Clinical Trials Network funded by NICHD (contract number N01-DK-9-001/HHSN267200800001C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, Debnath J, Deretic V, Elazar Z, Eskelinen EL, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–1294. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dales S, Eggers HJ, Tamm I, Palade GE. Electron Microscopic Study of the Formation of Poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- 3.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 4.Eekels JJ, Sagnier S, Geerts D, Jeeninga RE, Biard-Piechaczyk M, Berkhout B. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol J. 2012;9:69. doi: 10.1186/1743-422X-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campbell GR, Spector SA. Vitamin D Inhibits Human Immunodeficiency Virus Type 1 and Mycobacterium tuberculosis Infection in Macrophages through the Induction of Autophagy. PLOS Pathog. 2012;8:e1002689. doi: 10.1371/journal.ppat.1002689. This study expands on the previous findings by Campbell and Spector [7], Kyei et al. [8] and Yuk et al. [34] to include the control of HIV and Mycobacterium tuberculosis co-infection through the induction of autophagy.
- 6. Campbell GR, Spector SA. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLOS Pathog. 2012;8:e1003017. doi: 10.1371/journal.ppat.1003017. This study points to a connection between low vitamin D3 levels in HIV-infected individuals as a contributor to diminished autophagic defenses and thus host response to viral infections.
- 7.Campbell GR, Spector SA. Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011;286:18890–18902. doi: 10.1074/jbc.M110.206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. This study was the first to directly show that HIV utilizes autophagy for its replication, but that if Nef is inactivated autophagy can degrade and eliminate the virus.
- 9.Espert L, Varbanov M, Robert-Hebmann V, Sagnier S, Robbins I, Sanchez F, Lafont V, Biard-Piechaczyk M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLOS One. 2009;4:e5787. doi: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol. 2007;177:329–341. doi: 10.1083/jcb.200609050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelchen-Matthews A, Kramer B, Marsh M. Infectious HIV-1 assembles in late endosomes in primary macrophages. J Cell Biol. 2003;162:443–455. doi: 10.1083/jcb.200304008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grégoire IP, Richetta C, Meyniel-Schicklin L, Borel S, Pradezynski F, Diaz O, Deloire A, Azocar O, Baguet J, Le Breton M, et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLOS Pathog. 2011;7:e1002422. doi: 10.1371/journal.ppat.1002422. This study demonstrates that different families of RNA viruses all use a common strategy to manipulate autophagy to improve viral infectivity in a IRGM dependent manner.
- 13.Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS. 2008;22:695–699. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roeth JF, Collins KL. Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. Microbiol Mol Biol Rev. 2006;70:548–563. doi: 10.1128/MMBR.00042-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jouve M, Sol-Foulon N, Watson S, Schwartz O, Benaroch P. HIV-1 buds and accumulates in "nonacidic" endosomes of macrophages. Cell Host Microbe. 2007;2:85–95. doi: 10.1016/j.chom.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 16. Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. This study describes a novel beclin 1-derived autophagy-inducing peptide that has the potential to be used for the prevention and treatment of a broad range of human diseases.
- 17.Eberle HB, Serrano RL, Fullekrug J, Schlosser A, Lehmann WD, Lottspeich F, Kaloyanova D, Wieland FT, Helms JB. Identification and characterization of a novel human plant pathogenesis-related protein that localizes to lipid-enriched microdomains in the Golgi complex. J Cell Sci. 2002;115:827–838. doi: 10.1242/jcs.115.4.827. [DOI] [PubMed] [Google Scholar]
- 18.Denizot M, Varbanov M, Espert L, Robert-Hebmann V, Sagnier S, Garcia E, Curriu M, Mamoun R, Blanco J, Biard-Piechaczyk M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy. 2008;4:998–1008. doi: 10.4161/auto.6880. [DOI] [PubMed] [Google Scholar]
- 19.Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, Stalder R, Garcia E, Dinkins C, Leuba F, Wu L, et al. Human Immunodeficiency Virus-1 Inhibition of Immunoamphisomes in Dendritic Cells Impairs Early Innate and Adaptive Immune Responses. Immunity. 2010;32:654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li JC, Au KY, Fang JW, Yim HC, Chow KH, Ho PL, Lau AS. HIV-1 trans-activator protein dysregulates IFN-gamma signaling and contributes to the suppression of autophagy induction. AIDS. 2011;25:15–25. doi: 10.1097/QAD.0b013e328340fd61. [DOI] [PubMed] [Google Scholar]
- 21. Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLOS One. 2010;5:e11733. doi: 10.1371/journal.pone.0011733. This study provides a potential mechanism by which HIV blocks autophagy within macrophages.
- 22.Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011;203:1647–1657. doi: 10.1093/infdis/jir163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, Ellis R, Letendre S, Patrick C, Adame A, et al. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: reversal with beclin-1 gene transfer. J Neurovirol. 2013;19:89–101. doi: 10.1007/s13365-012-0145-7. This study demonstrates that the disruption of autophagy may contribute to age-related neuropathology in HIV-infected individuals.
- 25.Spector SA, Zhou D. Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy. 2008;4:704–706. doi: 10.4161/auto.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–678. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Parkhitko A, Henske EP. Autophagy: an 'Achilles' heel of tumorigenesis in TSC and LAM. Autophagy. 2011;7:1400–1401. doi: 10.4161/auto.7.11.17652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ní Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J. Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol. 2011;164:291–300. doi: 10.1111/j.1365-2249.2011.04381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heredia A, Amoroso A, Davis C, Le N, Reardon E, Dominique JK, Klingebiel E, Gallo RC, Redfield RR. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: an approach to suppress R5 strains of HIV-1. Proc Natl Acad Sci U S A. 2003;100:10411–10416. doi: 10.1073/pnas.1834278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicoletti F, Lapenta C, Donati S, Spada M, Ranazzi A, Cacopardo B, Mangano K, Belardelli F, Perno C, Aquaro S. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin Exp Immunol. 2009;155:28–34. doi: 10.1111/j.1365-2249.2008.03780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lake JE, Adams JS. Vitamin D in HIV-Infected Patients. Curr HIV/AIDS Rep. 2011;8:133–141. doi: 10.1007/s11904-011-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viard JP, Souberbielle JC, Kirk O, Reekie J, Knysz B, Losso M, Gatell J, Pedersen C, Bogner JR, Lundgren JD, et al. Vitamin D and clinical disease progression in HIV infection: results from the EuroSIDA study. AIDS. 2011;25:1305–1315. doi: 10.1097/QAD.0b013e328347f6f7. [DOI] [PubMed] [Google Scholar]
- 34. Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim JM, Jo EK. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6:231–243. doi: 10.1016/j.chom.2009.08.004. This study demonstrates that the hormonally active form of vitamin D3 induces autophagy through a cathelicidin dependent mechanism, providing a link between autophagy and antimicrobial peptides.
- 35.Nijnik A, Hancock RE. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr Opin Hematol. 2009;16:41–47. doi: 10.1097/moh.0b013e32831ac517. [DOI] [PubMed] [Google Scholar]
- 36.Meier A, Alter G, Frahm N, Sidhu H, Li B, Bagchi A, Teigen N, Streeck H, Stellbrink HJ, Hellman J, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol. 2007;81:8180–8191. doi: 10.1128/JVI.00421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 38.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J Immunol. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 39.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schlaepfer E, Audige A, Joller H, Speck RF. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J Immunol. 2006;176:2888–2895. doi: 10.4049/jimmunol.176.5.2888. [DOI] [PubMed] [Google Scholar]
- 41.Pathak S, De Souza GA, Salte T, Wiker HG, Asjo B. HIV induces both a down-regulation of IRAK-4 that impairs TLR signalling and an up-regulation of the antibiotic peptide dermcidin in monocytic cells. Scand J Immunol. 2009;70:264–276. doi: 10.1111/j.1365-3083.2009.02299.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhou D, Kang KH, Spector SA. Production of interferon alpha by human immunodeficiency virus type 1 in human plasmacytoid dendritic cells is dependent on induction of autophagy. J Infect Dis. 2012;205:1258–1267. doi: 10.1093/infdis/jis187. [DOI] [PMC free article] [PubMed] [Google Scholar]