

Abstract
Dicer-substrate siRNAs equipped with CpG oligodeoxyribonucleotides overcome the major hurdle in cell-specific siRNA delivery. The CpG-siRNA molecules are actively internalized by TLR9+ cells, without the need for transfection reagents, leading to RNA interference both in vitro and in vivo. Here, we elucidate the molecular mechanisms of CpG-siRNA processing in target cells. We show that shortly after uptake into early endosomes (EE), CpG and siRNA parts of the conjugate are uncoupled in the presence of Dicer endonuclease. Diced siRNA molecules are translocated from endosomes to endoplasmic reticulum, where they can interact with the RNA interference machinery. We previously observed that even though TLR9 is not involved in CpG-siRNA uptake, it is indispensable for induction of gene silencing. To explain the role of TLR9 in intracellular processing of CpG-siRNA, we used primary macrophages derived from wild-type and Tlr9-deficient mice. Macrophages lacking TLR9 showed extended endosomal colocalization of CpG and siRNA parts of the conjugate. However, Tlr9 ablation did not interfere with the interaction of CpG-siRNA with Dicer as shown by in situ proximity ligation assay. Using CpG-siRNA labeled with pH-sensitive dye, we finally identified that lack of TLR9 in macrophages resulted in significant retention of the siRNA in endosomes. Thus, TLR9 facilitates the critical step following CpG-siRNA uncoupling, which is cytoplasmic release of the diced siRNA. These findings suggest that the class of immunostimulatory siRNAs may benefit from activation of certain endosomal immune receptors, such as TLR9, in augmented gene silencing and therapeutic efficacy.
Keywords: siRNA, TLR9, CpG, Dicer, cytoplasmic release, endosomal escape
1. Introduction
Since its discovery by Fire and Mellow in 1998, siRNA has proven to be a powerful tool for modulating the expression of almost any gene in various species. Several studies demonstrated the feasibility of in vivo siRNA delivery, leading to therapeutic effects not only in mice [1–5] but also in non-human-primates [6, 7]. More recently, gene silencing after systemic siRNA administration was demonstrated in humans [8]. However, there are still severe hurdles to wide therapeutic application of this technology, such as cell-specific siRNA delivery and efficient release into cytoplasm [9]. There have been several innovative attempts to solve these problems using modular design of siRNA conjugated to cell-specific antibodies [1] or RNA aptamers [4, 5]. Others used combinations of siRNA with lipid reagents or cholesterol to enable siRNA uptake without the need for molecule encapsulation using potentially toxic lipid reagents [9]. Nevertheless, most of delivery methods result in endosomal rather than cytoplasmic siRNA uptake [10].
Due to rapid progress of this field, the rational design of siRNA reagents is hampered by gaps in our understanding of their intracellular processing. For many delivery strategies the mechanisms of siRNA release into cytoplasm are still unclear. In case of modified siRNAs that undergo rapid protonation in endosomes, the suggested release mechanism is swelling and burst of endosomes called a “proton sponge effect” [10]. This process does not explain the cytoplasmic release of siRNA conjugates linked to antibodies or RNA aptamers which is usually limited due to their large size [10, 11]. Timely siRNA release is critical because loaded early endosomes (EE) undergo a series of transformational events called maturation, resulting in significant decrease of endosomal pH and recruitment of proteases and RNAases [12]. The conversion of early into late endosomes may take as little as 20 min, and additional 20 min are required to fuse with lysosomes leading to the degradation of the vesicle content [12]. In addition, location of various downstream components of siRNA processing machinery is not well defined. Dicer endonuclease was originally found in the cytoplasm [13], but later studies indicated that it might be associated with the endoplasmic reticulum (ER) [14]. More recent reports found that Ago2 endonuclease, a major component of the RNA-induced silencing complex (RISC), likely resides on multi-vesicular bodies (MVB), which are intermediates between early and late endosomes [15]. These findings agree well with one of two major hypotheses of RISC complex assembly, which identifies Ago2 on the surface of EE and MVBs [10]. A slight variation of this assumption is that RISC is located in P-bodies, which in turn are associated with GW-bodies located on the cytoplasmic side of MVB membranes [10, 15]. In contrast, others have shown that RISC colocalizes with large subunits of ribosomes anchored to the rough ER, thus blocking translation of target mRNA [16].
We previously developed an immunostimulatory siRNA molecule by conjugating Dicer-substrate siRNA [17] to a CpG oligodeoxyribonucleotide (ODN), which acts as a targeting moiety for the intracellular toll-like receptor 9 (TLR9). CpG-siRNAs were successfully used for targeting various genes in normal and malignant TLR9-positive cells in vitro and in vivo. CpG-Stat3 siRNA was shown to generate potent systemic antitumor immunity [18], and prevented tumor metastasis in mouse models [19, 20]. Very recently, we optimized CpG-siRNA design to target human immune and cancer cells. Blocking of STAT3 or BCL-XL survival signaling inhibited in vivo growth in several xenotransplanted blood cancer models [21]. In addition, the conjugate retained its immunostimulatory properties and activated various populations of human DCs and B cells [21]. An unexpected finding of our recent studies was that both TLR9 expression and activation were required for the target gene silencing but not for the conjugate uptake. In vitro studies using CpG-siRNA in Tlr9−/− cells failed to detect RNA interference (RNAi) in target cells. Here, we delineate the critical initial steps and the role of TLR9 in CpG-siRNA processing. We believe that identifying biomarkers for target selection and molecules indispensable for the therapeutic effect will accelerate the clinical translation of the CpG-siRNA strategy for cancer therapy. Finally, we anticipate that this information will support and accelerate the rational design of other types of immunostimulatory siRNAs.
2. Materials and methods
2.1. Oligonucleotide design and synthesis
The phosphorothioated ODN and antisense strands (AS) of siRNAs were linked using 5 units of C3 carbon chain linker, (CH2)3 from Glen Research (Sterling, VA). The resulting constructs were hybridized with complementary sense strands (SS) of siRNAs to create chimeric ODN-siRNA constructs (deoxyribonucleotides are underlined). Sequences of single stranded constructs are listed below; the siRNA sequence is specific for mouse Stat3 gene (NM_213659, bases 1898–1922). CpG1668-mouse Stat3 siRNA(AS): 5′TCCATGACGTTCCTGATGCT-linker-UUAGCCCAUGUGAUCUGACACCCUGAA 3′. Mouse Stat3 siRNA (SS): 5′CAGGGUGUCAGAUCACAUGGGCUAA 3′. Constructs were fluorescently labeled using fluorescein (FITC), Cy3 (Lumiprobe, Hallandale Beach, FL) or pHrodo (Life Technologies, Carlsbad, CA) at sites indicated in the text. For FRET experiments we generated the following constructs of tripartite design using complementary 2′fluoro-modified “stick” adapters (Q = 2′fluoro-U; Z = 2′fluoro-C): CpG1668-linkerFITC-stick:5′-TCCATGACGTTCCTGATGCT-linkerFITC-AGGQGZGZZAZQQZZU-3′. Mouse Stat3 siRNA(SS)-linker-stick: 5′-CAGGGUGUCAGAUCACAUGGGCUAA -linker- AGGAAGQGGZGZAZZU-3′. Mouse Stat3 siRNA Cy3(AS): 5′Cy3- linker- UUAGCCCAUGUGAUCUGACACCCUGAA-3′. We have confirmed that modification of CpG-siRNA conjugates with various fluorochromes does not alter their internalization by target cells or gene silencing effect (Supplementary Fig. 9). The sequence of firefly luciferase-specific 25/27mer siRNA (Luc1 R 25D/27) used for the CpG(1668)-Luc siRNA conjugate molecule was published elsewhere [22]. The formation of siRNA duplex was confirmed by electrophoresis in 15% polyacrylamide/7.5M urea gel.
2.2. Cells
Mouse B16 melanoma, L929 fibroblasts, and RAW 264.7 macrophage cell lines were purchased from ATCC. B16 were cultured in RPMI medium with Glutamax (Life Technologies) and 5% fetal bovine serum (FBS, CellGro, Manassas, VA); L929 and RAW 264.7 were cultured in DMEM with Glutamax and 10% FBS. Bone marrow-derived Macrophages (BMDM) were differentiated from bone marrow cells of WT and Tlr9−/− C57BL/6 mice as described elsewhere [23]. Differentiation status and CpG-siRNA uptake efficiency were confirmed by flow cytometry with CD11b and F4/80 antibodies.
2.3. Fluorescent confocal microscopy and fluorescence resonance energy transfer (FRET)
BMDM or RAW 264.7 cells were treated using 500 nM CpG-siRNA for 30 min in a culture medium with or without 5% FBS, respectively. The medium was then replaced and incubations were continued for indicated times. Cells were fixed with 2% paraformadehyde for 20 min, permeabilized in PBS containing 0.1% Triton X-100, 1 mM MgCl2, and 1 mM CaCl2 (PBST) for 10 min, and quenched in 50 mM NH4Cl/PBST for 5 min before blocking in 1% BSA/PBST for 1 h. Samples were stained with the antibodies specific to EEA1 (H-300), Dicer (H-212, C-20) (Santa Cruz, Dallas, TX), Rab7 (D95F2 XP) (Cell Signaling, Boston, MA), Calnexin (GenScript, Piscataway, NJ), TLR9 (IMG-431) (Imgenex, San Diego, CA), FITC (Life Technologies), Ago2 (11A9) (Sigma, St. Louis, MO), and then detected with secondary Alexa 488-, 555-, 633-, and 647-coupled antibodies (Life Technologies). Slides were mounted in DAPI-containing mounting medium (Vector Labs, Burlingame, CA). Confocal imaging was carried out using C-Apochromat 40x/1.2 water-, C-Apochromat 63x/1.2 water-, or Plan-Apochromat 63x/1.4 oil-immersed objectives on cLSM510-Meta inverted confocal microscope (Zeiss, Thornwood, NY). We used LSM software v.4.2 SP1 for image acquisition, and LSM Image Browser v.4,2,0,121 for post-acquisition analysis (Zeiss).
For time-lapse confocal microscopy in living cells, cells were cultured on 35/14 mm #1.5 glass bottom tissue culture dishes (MatTek, Ashland, MA). After washing cells twice in phenol red-free media, cells were treated using CpG-siRNA for 20–30 min. Images were acquired using C-Apochromat 40x/1.2 water immersed objective, in 20–30 min time increments, in Z stacks. Microscope chamber for environmental control (37 °C, 5% CO2) was used in all experiments with living cells. For quantification purposes, Z-stacks were collapsed post-acquisition, and signal intensity in selected regions of interest (ROI) was determined using LSM software (Zeiss). In experiments with pHrodo-labeled CpG-siRNA each ROI encompassed a single cell, and ROI settings remained constant throughout the experiment. Reverse correlation between the intensity of pHrodo succinimidyl ester emission and pH was verified in a series of 50 mM citric buffers with pH values 4.5–8.0 (Supplementary Fig. 7A). Experiment was carried out in quadruplicates, and intensity of emission was analyzed using SpectraMax fluorescence plate reader (Molecular Devices, Sunnyvale, CA) using 544 nm excitation and 590 nm emission filters. FRET was carried out in living RAW 264.7 macrophages with tripartite CpG-Stat3 siRNA constructs labeled with FITC within the carbon linker between CpG and a “stick” adapter, and Cy3 on 5′ end of siRNA (AS). This design allowed for more control over the distance between two fluorophores for inducing the FRET effect. Cell culturing and image acquisition were carried out as described above for living cells: in time series and Z-stack mode. 2-FRET intensity was analyzed in a series of Z-stack projections using LSM Macro software (Zeiss) with N-FRET (Xia) algorithm.
For staining of ER in living cells, we used Green ER Tracker (Life Technologies) in a concentration of 1.0 μM. Staining was carried out in Hanks Balanced Salt Solution supplemented with calcium chloride, magnesium sulfate, and Glutamax (Life Technologies) for 30 min at 37 °C.
2.5. Transmission electron microscopy with immunostaining
RAW 264.7 cells were treated with CpG-siRNA5′SS-FITC, then pelleted and cryo-fixed in Leica EM PACT2 high pressure freezer (Leica, Buffalo Grove, IL). Specimens were then freeze-substituted in anhydrous acetone containing 0.5% glutaraldehyde in automated freezer substitution system (Leica). The temperature progression was 49 h at −90 °C, −90 °C to −25 °C at 5 °C/h, −25 °C for 12 h. At −25 °C, the specimen was rinsed with acetone 3 times, and infiltrated with 50% Lowicryl H2O in acetone for 1 h, 67% for 1 h, and 100% overnight. Infiltrated specimen was polymerized in Lowicryl H2O for 50 h at −25 °C under a UV lamp, and then at 25 °C for another 50 h. Ultra-thin sections (ca. 70 nm thick) were cut using Ultra Cut UCT microtome (Leica) and picked up on 200 mesh nickel grids. Post embedding labeling was carried out using rabbit anti-FITC antibody. Secondary IgG antibody was conjugated to 10 nm colloidal gold. Grids were stained with 2% uranyl acetate in 70% ethanol for 1 min, followed by staining with Reynold’s lead citrate for 1 min. Electron microscopy (EM) was done on FEI Tecnai 12 transmisison electron microscope equipped with a CCD camera (FEI, Hillsboro, OR).
2.6. In situ proximity ligation assay (PLA)
Cells were plated onto Nunc Lab-Tek 16-well chamber slides (Thermo Fisher Scientific, Marietta, OH) and incubated overnight prior to the treatment with CpG-siRNA. Duolink™ in situ PLA was carried according to a manufacturer’s protocol (Olink, Uppsala, Sweden) using WT and Tlr9−/− BMDM. We used goat anti-Dicer and rabbit anti-FITC primary antibodies, PLA probes anti-goat Plus and anti-rabbit Minus, and far red detection reagents (Olink) to detect the colocalization events in intact cells. Slides were later mounted and scanned on confocal microscope as described in p.2.3. Z-stacks of images were collapsed to allow for quantification of total PLA events within a cell. All samples were duplicated and we analyzed at least 3 scan areas per sample for quantification purposes.
2.7. Quantitative real-time PCR
Total RNA isolation and cDNA synthesis were carried out as described previously [21]. Samples were then analyzed using pairs of primers specific for Stat3 and 18S cDNA (Qiagen, Germantown, MD). Sequence-specific amplification was detected by fluorescent signal of SYBR Green (Qiagen) by using CFX96 Real-time PCR Detector (Bio-Rad, Hercules, CA).
2.8. In vivo experiments
Mouse care and experimental procedures were performed under pathogen-free conditions in accordance with the approved protocols from the IACUC of the City of Hope. For subcutaneous tumor challenge, we injected 5×105 B16 tumor cells into 7–8 week old C57BL/6 WT or Tlr9−/− mice kindly provided by Dr. Don Diamond (City of Hope). After tumors had reached average size of ca. 5 mm, mice were injected peritumorally with 0.78 nmole (1 mg/kg) of CpG-siRNA. Tumor growth was monitored every other day using electronic caliper. For the analysis of Stat3 gene silencing, mice were euthanized after 5 days of treatment, tumor draining lymph nodes were collected, and CD11b+ cells were magnetically enriched using EasySep kit (Stem Cell Technologies, Vancouver, Canada) as described previously [18].
2.9. Flow cytometry and scavenger receptor inhibition assays
Single cell suspensions of cultured or lymph nodes cells were prepared by mechanic tissue disruption and collagenase D/DNase I treatment as described [24], and stained with 7AAD (BD Biosciences, San Jose, CA). To analyze conjugate internalization, RAW 264.7 cells were pre-treated using 50 μg/ml dextran sulfate or chondroitin sulfate (Sigma) for 1 h prior to treatment with 500 nM CpG-siRNA for 1 h. For assessment of uptake at +4 °C, cells were placed on ice 10 min before stimulation, then stimulated with 500 nM of CpG-Stat3 siRNA for 30 min, and immediately analyzed by flow cytometry. Fluorescence data were collected on BD Accuri C6 cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR).
2.10. Statistical analysis
Unpaired two-tailed t-test was used to calculate two-tailed P value to estimate statistical significance of differences between two treatment groups in the whole study. Statistically significant P values were indicated in figures and/or legends and labeled as follows: ***; P < 0.001; **, P < 0.01 and *, P < 0.05. Data were analyzed using Prism v.5.0 software (GraphPad, La Jolla, CA).
3. Results
3.1. Target immune cells actively internalize CpG-siRNA by scavenger receptor-mediated mechanism
Our previous studies demonstrated that immunostimulatory CpG-Stat3 siRNA conjugates are potent antitumor therapeutics in both mouse and human systems [18, 21]. Even though TLR9 did not participate in the CpG-siRNA uptake, it was critical for silencing of various target genes in vitro in mouse and human cells. Altogether, these results prompted us to elucidate the role of TLR9 in facilitating CpG-siRNA-mediated gene silencing effect. First, we assessed the process of CpG-siRNA internalization by target immune cells. We have previously demonstrated that CpG-siRNAs can rapidly enter target cells through endocytosis. CpGFITC-Stat3 siRNA5′SS-Cy3 was clearly detectable in endosomes within less than 30 min of incubation, as shown by transmission electron microscopy (Fig 1A) in agreement with our previous study [18]. The CpG-siRNA was actively internalized by target immune cells under normal physiological conditions and the process was abrogated at 4°C (Supplementary Fig. 1). The internalization of CpG ODNs is reportedly mediated by surface receptors from scavenger family [25, 26]. Due to functional redundancy of scavenger receptors (SRs), we decided to assess the effect of dextran sulfate, a general competitive SR inhibitor [27], on CpG-siRNA uptake, as described before [25, 26]. As shown in Fig. 1B, pre-incubation of RAW 264.7 cells with dextran sulfate blocked the internalization of CpG-siRNA5′SS-Cy3 as measured using flow cytometry. In contrast, the control chondroitin sulfate, which does not bind SRs [27], failed to affect the conjugate internalization.
Fig. 1.
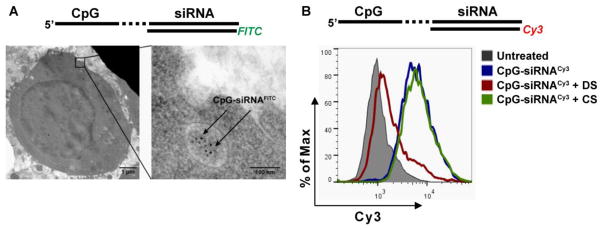
CpG-siRNA is actively internalized by scavenger receptor-mediated endocytosis in RAW 264.7 macrophages. (A) Transmission electron microscopy on fixed cells treated for 30 min using CpG-Stat3 siRNA5′SS-FITC. Sections of flash-frozen cell pellets were incubated with FITC-specific antibodies and then detected using secondary antibodies coupled to 10 nm gold nanoparticles. Intracellular localization of CpG-siRNA is shown with arrowheads. Scale bars: 1 μm (left) and 100 nm (right). (B) CpG-siRNA uptake is mediated by scavenger receptors. Cells were pre-treated with dextran sulfate (DS, scavenger receptor ligand) or chondroitin sulfate (CS, negative control) for 2 h, then incubated with 500 nM CpG-Stat3 siRNA5′SS-Cy3 for 1 h and then analyzed by flow cytometry. Shown are representative results of one of three independent experiments.
3.3. Rapid uncoupling of CpG and siRNA moieties in the presence of Dicer
The design of CpG-siRNA utilizes Dicer-substrate 25/27-mer siRNA sequence to enable cleavage of the molecule releasing 21-mer siRNA from the receptor-bound CpG fragment of the conjugate [18]. To assess the timing and efficiency of CpG-siRNA cleavage, we developed an in situ confocal imaging approach based on fluorescence resonance energy transfer (FRET) between both parts of the conjugate. CpG-siRNA molecules were equipped with FITC label (donor) in the linker region and with Cy3 label (acceptor) added via another linker to the 5′ end of the siRNA antisense strand. Flexibility of the linker regions permits close proximity of both fluorochromes required for the energy transfer from the excited donor to the acceptor, which results in emission in Cy3 spectrum (max. at 570 nm). The donor-acceptor pair would be separated after the conjugate cleavage by Dicer endonuclease into 3 parts: CpGFITC-linker-stick, 21-mer siRNA and short Cy3-labeled terminal fragment of AS siRNA. To verify that changes in FRET intensities result from processing of the siRNA part of the conjugate, we compared emission spectra of dual FITC/Cy3-labeled CpG-Stat3 siRNA without or with nuclease resistant 2′O-methyl (2′OMe) modified siRNA. As shown in Fig. 2A and 2B, in RAW 264.7 cells the FRET effect induced by the unmodified conjugate was transient, decreasing by half within 2.5 h. In contrast, the FRET signal induced by 2′OMe CpG-siRNA remained elevated and even slightly increased at 2.5 h. These results suggested that CpG-siRNA processing occurs early after internalization and requires cleavage of the siRNA part of the molecule.
Fig. 2.

CpG-siRNA conjugates are uncoupled in the presence of Dicer endonuclease shortly after internalization into RAW 264.7 cells. (A) Separation of the CpG and siRNA parts the CpGFITC-Stat3 siRNA5′AS-Cy3 conjugate was measured by decreasing FRET effect using time-lapse confocal microscopy. The nuclease-resistant conjugate with completely 2′OMe-modified both strands of the siRNA was used as a negative control. Shown are representative results from one of three independent experiments. Scale bars: 5 μm. (B) Quantification of average FRET intensities from three independent experiments using unmodified and 2′OMe-modified conjugates. Shown are means ± SEM. (C) CpG-siRNA5′SS-Cy3 colocalizes with Dicer within 0.5 h after uptake as assessed by confocal microscopy on fixed RAW 264.7 macrophages. (D, E) Dicer transiently interacts with CpG-siRNA shortly after endocytosis of the conjugate. Cells were incubated with CpGFITC-siRNA for indicated times, fixed, permeabilized and the interaction between CpGFITC-siRNA and Dicer was detected by in situ PLA using FITC- and Dicer-specific antibodies. (D) Shown are confocal microscopy images (Z stack projections; Scale bars: 10 μm) and (E) the average counts of PLA events per cell for indicated times, means ± SEM.
Next, we investigated whether kinetics of CpG-siRNA interaction with Dicer coincide with the rapid uncoupling of the conjugate. First, we confirmed using confocal microscopy that already 0.5 h after CpG-siRNA5′SS-Cy3 uptake, most of the conjugate was colocalized with Dicer in endosomal vesicles (Fig. 2C). In fact, both Dicer and TLR9 were found in similar vesicular localization in RAW 264.7 cells even before treatment (Supplementary Fig. 2). Thus, CpG-siRNA uncoupling can occur right after internalization into TLR9/Dicer containing endosomes. For direct proof of interaction between Dicer and its substrate, CpG-siRNA, we used the in situ proximity ligation assay (PLA) which enables visualization and quantification of the interaction between two molecules in intact fixed cells [28]. Briefly, the molecules examined are identified by a pair of unconjugated primary antibodies, and IgG secondary antibodies equipped with DNA primers which form circular template only when in close proximity. The circular DNA becomes a template for rolling circle DNA polymerase, which amplifies DNA repeats. Further, those repeats are recognized by complimentary oligonucleotides labeled with a fluorophore, and a fluorescent signal is detected using confocal microscopy. Using antibodies specific to Dicer and fluorescein in CpGFITC-siRNA, we found that both molecules colocolized in mouse macrophages as early as 0.5 h after treatment, reaching maximum at 1 h and decreasing thereafter (Fig. 2DE). These results demonstrate that Dicer interaction with CpG-siRNA coincides with siRNA uncoupling as observed by FRET assays, thus underscoring the role of Dicer for initial CpG-siRNA processing.
3.4. The siRNA released from the conjugate is relocated to the endoplasmic reticulum
To induce gene silencing, the 21/21mer siRNA uncoupled from CpG-siRNA conjugate must leave the endosome and bind to Ago2 protein as a critical component of a RISC complex. Our initial confocal microscopy studies on mouse macrophages showed that Ago2 is detectable on a subset of EE under basal conditions (Supplementary Fig. 3), and also associated with the ER but only after CpG-siRNA treatment (Fig. 3A). To follow further trafficking of fluorescently labeled siRNA molecules in RAW 264.7 cells, we used a dual-labeled CpGFITC-Stat3 siRNA5′SS-Cy3, which allowed us to visualize both parts of the conjugate before and after cleavage. We also included 2′OMe CpG-siRNA as a negative control, similar to as shown in Fig. 2A. Confocal microscopy analysis showed that initially colocalized CpGFITC and siRNA5′SS-Cy3 parts of the unmodified but not 2′OMe-modified conjugates were clearly separated at 3 h (Fig. 3B, top row). The siRNA5′SS-Cy3 molecules were found both in vesicles and within the endoplasmic reticulum as confirmed by staining for the ER maker, calnexin (Fig. 3B). In contrast, the CpGFITC was contained in a distinct vesicular compartment (Fig. 3B). Using staining with Rab 7, the late endosomal marker, we confirmed that the CpG fragment in fact remains in maturing endosomes likely bound to TLR9 (Supplementary Fig. 4). To confirm that siRNA can efficiently escape from early rather than maturing endosomes, we used chloroquine, a commonly used inhibitor of endosomal acidification that causes bursting of maturing endosomes [29]. We compared levels of Stat3 silencing induced in RAW 264.7 cells by CpG-Stat3 siRNA or control non-targeting CpG-Luc siRNA in the presence or absence of chloroquine. As shown in Fig. 3C, chloroquine did not affect Stat3 gene silencing confirming that siRNA molecules are efficiently uncoupled and released without significant endosomal retention. Finally, we verified whether the effect of 2′OMe-modified oligonucleotides in our experiments is not a result of their ability to act as potent TLR7 antagonists [30]. Our control experiments did not detect any difference in the siRNA transport to ER in the presence or absence of 2′OMe-modifed CpG-siRNA (Supplementary Fig. 8).
Fig. 3.

Diced siRNA escapes from EE to endoplasmic reticulum. (A) Treatment with CpG-siRNA induces partial translocation of Ago2 to the ER. Shown are confocal microscopy images acquired from primary mouse bone-marrow derived macrophages. Scale bars: 10 μm. (B) The intracellular localization of both parts of CpG-siRNA after endosomal processing. Unmodified (top row) and 2′OMe-modified (bottom row) CpGFITC–Stat3 siRNA5′SS-Cy3 were added to cultured RAW 264.7 cells. After 3 h, cells were fixed, stained with anti-calnexin antibody (ER marker) plus DAPI (nuclear staining; shown in white) and analyzed by confocal microscopy. Scale bars: 5 μm. (C) Disruption of late endosomes by chloroquine does not affect target gene silencing by CpG-Stat3 siRNA. Stat3 gene silencing was measured using qPCR in 264.7 cells treated with CpG-Stat3 siRNA and CpG-Luc-siRNA (negative control) with or without chloroquine (100 μM), inhibitor of endosomal maturation, as indicated. Shown are representative results from one of two independent experiments; mean ± SEM.
3.5. TLR9 is necessary for endosomal release of diced siRNA
We have previously shown that TLR9 activity is required for target gene silencing but not for the conjugate uptake. To assess whether TLR9 plays a role in the endosomal release of the diced siRNA, we used primary bone marrow-derived macrophages (BMDM) from WT and Tlr9−/− mice. First, we addressed a question of whether TLR9 expression is required for the CpG-siRNA interaction with Dicer. The PLA assay in macrophages treated with CpGFITC-Stat3 siRNA indicated that Dicer endonuclease colocalizes with CpG-siRNA in both WT and Tlr9−/− cells, independently from TLR status (Supplementary Fig. 5). However, this transient interaction was significantly extended in Tlr9−/− cells, which suggested impaired CpG-siRNA processing or endosomal release. To explain this observation, we incubated primary WT and Tlr9−/− macrophages with dual-labeled CpGFITC-Stat3 siRNA5′SS-Cy3 and assessed the intracellular conjugate processing by confocal microscopy. The release of diced siRNA5′SS-Cy3 from EE was delayed in Tlr9−/− cells and the colocalization of both fluorescent signals was still detectable after 2.5 h of treatment in contrast to WT cells (Supplementary Fig. 6).
For the direct proof of siRNA endosomal release, we generated CpG-Stat3 siRNA labeled with a pH-sensitive fluorescent dye, pHrodo. The fluorescent intensity of emission for pHrodo inversely correlates with the pH of the environment, thus permitting the detection of changes in pH due to transition from EE (pH 5.9–6.0) either to cytoplasm/ER (pH 7.0–7.2) or to late endosomes (pH 5.0–6.0) (Supplementary Fig. 7A) [31]. Using time-lapse imaging, we recorded and digitally quantified changes in the pHrodo intensity in primary WT and Tlr9−/− macrophages treated with CpG-siRNA5′SS-pHrodo. As shown in Fig. 4AB, the intensity of fluorescent signal from pHrodo in WT cells decreased rapidly by more than 60% between 1–2 h of incubation and was barely detectable after 4 h. In contrast, the signal intensity in Tlr9−/− cells dropped initially by about 30% but then remained relatively unchanged for up to 4.7 h. To verify that observed changes in pHrodo intensity in fact resulted from the conjugate cleavage and cytoplasmic release of the siRNA5′SS-pHrodo, we repeated the experiment using 2′OMe-modified CpG-siRNA similar to as in Fig. 3 (and Supplementary Fig. 7BC). Confirming our earlier observation (Fig. 2AB), only a small fraction of the unprocessed conjugate was released from endosomes in WT cells. In Tlr9−/− cells the release of intact conjugate was negligible, resulting in increase of the pHrodo signal with progressing endosome acidification (Supplementary Fig. 7BC).
Fig. 4.

TLR9 is required for efficient release of the diced siRNA from EE. (A) Representative images from time-lapse confocal microscopy in WT and Tlr9−/− primary bone-marrow derived macrophages treated using CpG-siRNA labeled with pH-sensitive fluorescent dye (pHrodo) for 0.5 h. Shown are single confocal Z layers, scale bars: 10 μm (A), and the relative average intensity of pHrodo emission as a function of time (B). Results are averaged from 2 to 3 individual cells; mean ± SEM. Shown are the results from one of two (WT) or three (Tlr9−/−) independent experiments. (C) TLR9 is required for efficient translocation of diced siRNA from endosomes to the ER. Confocal microscopy in live WT and Tlr9−/ primary BMDM treated using CpG–siRNA5′SS-Cy3 for indicated times with intravital staining using green ER Tracker. Single Z layers, scale bars: 5 μm.
Next, we verified whether TLR9 activity is also involved in the translocation of diced siRNA to the ER. We incubated WT and Tlr9−/− BMDM in the presence of fluorescently labeled CpG-siRNA5′SS-Cy3 for 1 h and 3 h and then analyzed colocalization of the siRNA5′SS-Cy3 with ER using confocal microscopy in live cells. As shown in Fig. 4C, most of the siRNA signal was detectable within the ER in WT cells starting from 1 h and increasing at 3 h of incubation. In contrast, Tlr9−/− macrophages showed only weak colocalization of the siRNA with ER even at 3 h, thereby indicating that the role of TLR9 in this process. The summary of our findings can be found in the Graphical Abstract and in the Supplementary Fig. 10.
4. Discussion
We previously developed a CpG-siRNA strategy that allows for targeted gene silencing in various TLR9-positive cells in vitro and in vivo. Both in mouse and in human target cells, TLR9 was essential for the induction of RNAi [18, 21]. Intriguingly, we found that Tlr9 deficiency does not affect CpG-siRNA uptake. In the current study, we used various imaging approaches, from confocal to transmission electron microscopy, to elucidate the role of intracellular TLR9 receptor in permitting the CpG-siRNA effect. Our studies in established mouse macrophages as well as in primary immune cells demonstrated that shortly after Dicer-mediated cleavage of the conjugate, TLR9 facilitates release of uncoupled siRNA from EE.
Mature and fully functional TLR9 is located in EE [29], thus it is not directly responsible for ligand internalization. Our results identified that CpG-siRNA undergoes endocytosis by dextran sulfate-sensitive receptors of scavenger family. This observation agrees well with previous reports that suggested role of several scavenger receptors in CpG ODN internalization, including SR-B1, CXCL16 and others due to their functional redundancy [25, 26]. In addition, binding to cofactors can also affect oligonucleotide delivery especially in vivo as shown for granulin-bound CpG ODNs, with drastically enhances delivery to endosomes [32]. Thus, CpG-siRNAs can be internalized also by TLR9-negative but SR-positive target cells. However, SR are known to be expressed mostly in immune cells with ability to internalize macromolecules, such as dendritic cells, macrophages and B cells, which also express TLR9. Thus, CpG-siRNA uptake by mouse or human target cells usually correlates with constitutive or inducible TLR9 expression, as in differentiating monocytes [21]. In all scenarios, CpG ODN is quickly internalized into EE and binds to TLR9 present in the same location as quickly as in 5 min after treatment [33]. We found similar rapid kinetics of internalization also in case of CpG-siRNA conjugates. Interestingly, our FRET studies indicated that endonuclease-dependent uncoupling of the siRNA from the CpG part occurs early after uptake of the conjugate, while still in the EE. The extended 25/27mer siRNA molecules are known to be targets of Dicer, which trims these molecules to 21/21-mer siRNA [17]. We have previously shown in vitro that Dicer cleaves CpG-siRNA in a similar manner, releasing 21/21mer siRNA [18]. Our current study verifies these results in both living and fixed cells using FRET and confocal imaging. We demonstrate for the first time that Dicer colocalizes with TLR9 in the endosomal compartment and transiently interacts with the target oligonucleotide in EE as early as 30 min after the conjugate uptake. It remains to be determined whether Dicer operates inside the endosome or rather on the endosomal membrane, where other components of the RNAi machinery are located [15].
Rapid processing of CpG-siRNA is critical for an efficient induction of RNAi. Our experiments with chloroquine, an inhibitor of endosomal maturation, confirmed that siRNA release is very effective and likely occurs before conversion of EE to MVB and late endosomes. The uncoupled siRNA molecules seem to escape from EE before they undergo acidification, fuse to lysosomes, and start degrading their content [34]. Following the endosomal escape, siRNA molecules must enter into RISC in order to initiate the RNAi. As suggested by recent studies, major RISC components, such as Ago2, are present on a cytoplasmic side of membranes of both endosomes and ER [10, 16]. Our results show that Ago2 is constitutively present on some EEs as well as partly associated with ER but only in cells treated with the CpG-siRNA conjugate. We also found that after uncoupling from the CpG-siRNA conjugate, diced siRNA is translocated to ER. According to multiple references, siRNA processed by Dicer is immediately passed over to Ago2 through the TRBP protein, a part of the RISC loading complex, which binds to both Dicer and Ago2 [11, 34, 35]. Binding to Ago2 effectively protects the AS (guide) strand of siRNA from hazardous nucleases throughout the RNAi process [36]. We anticipate that the presence of Ago2 on EE and on the ER can facilitate the transfer of siRNA from one location to another, as suggested by our results indicating relocation of newly formed endosomes from the cellular membrane towards the perinuclear cisterns of the ER. Such transport mechanism implemented by motor proteins dynein and kinesin would eliminate the need for release of naked siRNA into the cytoplasm, where viscosity and concentration of nucleases can be high [10, 37].
The fusion of newly formed endosomes with ER was previously shown in macrophages [38], and TLR9 is known to induce formation of similar tubular phagosomes [33]. These processes are related to antigen cross-presentation, which allows for delivery of phagosomal antigens to MHC class I complexes in various immune cells from DCs and macrophages to B cells [39]. In antigen cross-presentation, phagosomes fuse with the ER to acquire a number of ER-specific proteins, which are critical for transmembrane transport of various molecules [40]. TLR9 activation is known to stimulate phagocytosis and cross-presentation of antigens in the cytosol [41–43]. Specifically, TLR9 can regulate microtubule-assisted transport of EE toward the ER, and the early release of endosomal content before acidification [10, 41, 42]. Our results suggest that a similar TLR9-regulated mechanism may be employed for the release of siRNA diced from the CpG-siRNA conjugate. TLR9 does not seem to be required for recruitment of Dicer, which is already present in TLR9-positive EE. However, our experiments in Tlr9−/− macrophages provide evidence that TLR9 does facilitate endosomal release of uncoupled siRNA. Our further studies should establish whether a canonical downstream TLR9 signaling regulates the release of siRNA uncoupled from CpG-siRNA conjugates. In addition, the involvement of other endosomal TLRs, such as TLR3 and TLR7, in conjugate processing and siRNA release cannot be excluded and will be addressed by experiments in specific TLR-deficient cell models.
CpG-siRNA strategy shows promise as a cancer immunotherapeutic targeting immunosuppressive regulators such as STAT3 [18, 21]. We have also demonstrated that this approach is suitable for direct elimination of human TLR9-positive cancer cells, such as certain blood cancers [18–21]. Intriguingly, our studies identified differences in the efficiency of target gene silencing in human cells using siRNA conjugated to A- and B-types of CpG ODN (Kortylewski, unpublished data). Since various types of CpG ODNs differ in timing and type of downstream TLR9 signaling, it is likely that these events can be critical determinants of CpG-siRNA efficacy. Our findings imply that TLR9 triggering might also enhance the endosomal release of siRNA delivered by other than CpG ODN strategies, including RNA aptamers or monoclonal antibodies. We believe that understanding the requirements for effective induction of RNAi in target cells will facilitate further optimization of not only CpG-siRNA but potentially also other siRNA delivery approaches for human cancer therapy.
Supplementary Material
Acknowledgments
We are grateful to Zhuo Li, Ricardo Zerda, and Marcia Miller (Electron Microscopy Core Facility) for assistance and technical support with the TEM experiments, to Andreas Herrmann (Department of Cancer Immunotherapeutics and Tumor Immunology) for helpful comments on the design of confocal microscopy experiments, and to the staff of the Animal Research Center at City of Hope for support with in vivo experiments. This work was funded by the National Cancer Institute of the National Institutes of Health award number R01CA155367 (to M.K.), H.N. and Frances C. Berger Foundation and Parsons Foundation fellowships (to S.N), and by the Brazil National Counsel of Technological and Scientific Development, CNPq fellowship (to D.M.).
Appendix A. Supplementary data
Supplementary data to this article can be found online.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23(6):709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- 2.McNamara JO, 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol. 2006;24(8):1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 3.Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, Kirschnek S, Gaffal E, Landsberg J, Hellmuth J, Schmidt A, Anz D, Bscheider M, Schwerd T, Berking C, Bourquin C, Kalinke U, Kremmer E, Kato H, Akira S, Meyers R, Hacker G, Neuenhahn M, Busch D, Ruland J, Rothenfusser S, Prinz M, Hornung V, Endres S, Tuting T, Hartmann G. 5′-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14(11):1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- 4.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, 2nd, Giangrande PH. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27(9):839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature. 2010;465(7295):227–230. doi: 10.1038/nature08999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li BJ, Tang Q, Cheng D, Qin C, Xie FY, Wei Q, Xu J, Liu Y, Zheng BJ, Woodle MC, Zhong N, Lu PY. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat Med. 2005;11(9):944–951. doi: 10.1038/nm1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, Judge AD, Lam K, McClintock K, Nechev LV, Palmer LR, Racie T, Rohl I, Seiffert S, Shanmugam S, Sood V, Soutschek J, Toudjarska I, Wheat AJ, Yaworski E, Zedalis W, Koteliansky V, Manoharan M, Vornlocher HP, MacLachlan I. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441(7089):111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 8.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464(7291):1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457(7228):426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen J, Szoka FC. Nucleic acid delivery: the missing pieces of the puzzle? Acc Chem Res. 2012;45(7):1153–1162. doi: 10.1021/ar3000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther. 2012;20(3):483–512. doi: 10.1038/mt.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J. 2002;366(Pt 3):689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billy E, Brondani V, Zhang H, Muller U, Filipowicz W. Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc Natl Acad Sci U S A. 2001;98(25):14428–14433. doi: 10.1073/pnas.261562698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002;21(21):5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbings D, Voinnet O. Control of RNA silencing and localization by endolysosomes. Trends Cell Biol. 2010;20(8):491–501. doi: 10.1016/j.tcb.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Chendrimada TP, Finn KJ, Ji X, Baillat D, Gregory RI, Liebhaber SA, Pasquinelli AE, Shiekhattar R. MicroRNA silencing through RISC recruitment of eIF6. Nature. 2007;447(7146):823–828. doi: 10.1038/nature05841. [DOI] [PubMed] [Google Scholar]
- 17.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23(2):222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 18.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, Lee H, Scuto A, Liu Y, Yang C, Deng J, Soifer HS, Raubitschek A, Forman S, Rossi JJ, Pardoll DM, Jove R, Yu H. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27(10):925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, Jove R, Yu H. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16(12):1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, Raubitschek A, Hoon DS, Forman S, Figlin RA, Liu J, Jove R, Yu H. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21(5):642–654. doi: 10.1016/j.ccr.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Q, Hossain DM, Nechaev S, Kozlowska A, Zhang W, Liu Y, Kowolik CM, Swiderski P, Rossi JJ, Forman S, Pal S, Bhatia R, Raubitschek A, Yu H, Kortylewski M. TLR9-mediated siRNA delivery for targeting of normal and malignant human hematopoietic cells in vivo. Blood. 2013 doi: 10.1182/blood-2012-07-442590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, Rossi JJ, Behlke MA. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33(13):4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marim FM, Silveira TN, Lima DS, Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One. 2010;5(12):e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 25.Gursel M, Gursel I, Mostowski HS, Klinman DM. CXCL16 influences the nature and specificity of CpG-induced immune activation. J Immunol. 2006;177(3):1575–1580. doi: 10.4049/jimmunol.177.3.1575. [DOI] [PubMed] [Google Scholar]
- 26.Zhu P, Liu X, Treml LS, Cancro MP, Freedman BD. Mechanism and regulatory function of CpG signaling via scavenger receptor B1 in primary B cells. The Journal of biological chemistry. 2009;284(34):22878–22887. doi: 10.1074/jbc.M109.018580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acton S, Resnick D, Freeman M, Ekkel Y, Ashkenas J, Krieger M. The collagenous domains of macrophage scavenger receptors and complement component C1q mediate their similar, but not identical, binding specificities for polyanionic ligands. The Journal of biological chemistry. 1993;268(5):3530–3537. [PubMed] [Google Scholar]
- 28.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 29.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456(7222):658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robbins M, Judge A, Liang L, McClintock K, Yaworski E, MacLachlan I. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol Ther. 2007;15(9):1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 31.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5(2):121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 32.Park B, Buti L, Lee S, Matsuwaki T, Spooner E, Brinkmann MM, Nishihara M, Ploegh HL. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity. 2011;34(4):505–513. doi: 10.1016/j.immuni.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5(2):190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 34.Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123(Pt 8):1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 35.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436(7051):740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Zhu D, Huang L, Zhang J, Bian Z, Chen X, Liu Y, Zhang CY, Zen K. Argonaute 2 complexes selectively protect the circulating microRNAs in cell-secreted microvesicles. PLoS One. 2012;7(10):e46957. doi: 10.1371/journal.pone.0046957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welte MA. Bidirectional transport along microtubules. Curr Biol. 2004;14(13):R525–537. doi: 10.1016/j.cub.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 38.Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110(1):119–131. doi: 10.1016/s0092-8674(02)00797-3. [DOI] [PubMed] [Google Scholar]
- 39.Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10(6):403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 40.Kasturi SP, Pulendran B. Cross-presentation: avoiding trafficking chaos? Nat Immunol. 2008;9(5):461–463. doi: 10.1038/ni0508-461. [DOI] [PubMed] [Google Scholar]
- 41.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7(10):1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 42.Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, DiDonato J, Dziarski R, Akira S, Schoenberger SP, Raz E. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol. 2003;170(8):4102–4110. doi: 10.4049/jimmunol.170.8.4102. [DOI] [PubMed] [Google Scholar]
- 43.Mouries J, Moron G, Schlecht G, Escriou N, Dadaglio G, Leclerc C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood. 2008;112(9):3713–3722. doi: 10.1182/blood-2008-03-146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.