

Abstract
Insects act as vectors for diseases of plants, animals and humans. Replacement of wild insect populations with genetically modified individuals unable to transmit disease provides a potentially self-perpetuating method of disease prevention. Population replacement requires a gene drive mechanism in order to spread linked genes mediating disease refractoriness through wild populations. We previously reported the creation of synthetic Medea selfish genetic elements able to drive population replacement in Drosophila. These elements use microRNA-mediated silencing of myd88, a maternally expressed gene required for embryonic dorso-ventral pattern formation, coupled with early zygotic expression of a rescuing transgene, to bring about gene drive. Medea elements that work through additional mechanisms are needed in order to be able to carry out cycles of population replacement and/or remove existing transgenes from the population, using second-generation elements that spread while driving first-generation elements out of the population. Here we report the synthesis and population genetic behavior of two new synthetic Medea elements that drive population replacement through manipulation of signaling pathways involved in cellular blastoderm formation or Notch signaling, demonstrating that in Drosophila Medea elements can be generated through manipulation of diverse signaling pathways. We also describe the mRNA and small RNA changes in ovaries and early embryos associated from Medea-bearing females. Finally, we use modeling to illustrate how Medea elements carrying genes that result in diapause-dependent female lethality could be used to bring about population suppression.
Keywords: Selfish genetic element, synthetic biology, maternal-effect, gene drive, mosquito, dengue, malaria
Insects act as vectors for a number of important diseases of humans, animals, and plants. Examples include malaria and West Nile (humans and birds), dengue, yellow fever, lymphatic filariasis, chikungunya, and Chagas disease (humans), Rift valley fever and trypanosomyasis (humans and livestock), and plant diseases such as Huanglongbing (citrus), Almond leaf scorch, Pierces disease (grapes), and zebra chip disease (potato) (1). Replacement of the wild population with insects engineered to be unable to transmit disease provides a method of disease prevention that complements traditional vector control methods. Population replacement is attractive as a disease prevention mechanism because it is species specific, does not involve the use chemicals, involve gross modification of the environment, or require direct contact with the disease hosts (humans, animals or plants). Perhaps most importantly, it is in principle self-sustaining, providing protection over large areas even if other vector control strategies suffer gaps in coverage.
Transgenes that mediate disease refractoriness are unlikely to confer an overall fitness benefit on insects that carry them (2, 3, 4). Therefore, an essential component of any population replacement strategy is a gene drive mechanism that will ensure the spread of linked transgenes to genotype or allele fixation in a modest number of generations following release. A number of gene drive mechanisms have been proposed (5, 6), but only one, Medea, has been engineered and shown to drive replacement in a wildtype population, in Drosophila (7). A Medea selfish genetic element consists of two chromosomally-located, tightly linked transgenes: one that encodes a toxin inherited by all progeny of Medea-bearing mothers, and a second that encodes an antidote active in the zygote. Medea spreads by causing the death of non-Medea-bearing progeny of Medea-bearing mothers (Figure 1A), thereby causing a relative increase in the population frequency of the Medea-bearing chromosome (Medea-bearing chromosomes and their non-Medea-bearing counterparts are hereafter referred to as Medea and non-Medea alleles, respectively) (8–12). In the absence of family level selection, Medea-bearing individuals and alleles experience no direct benefit from this killing, but non-Medea alleles experience Medea-dependent death (a fitness loss) in each generation that is dependent on the Medea allele frequency. Whenever the average cost associated with the Medea allele is less than that associated with the non-Medea allele, a Medea element located on an autosome spreads, with the population ultimately consisting of Medea-bearing heterozygotes and homozygotes. When Medea is located on the X chromosome in a X/Y male heterogametic species, Medea is predicted to spread to allele fixation, with wildtype alleles being completely eliminated. (12).
Figure 1. Medea genetics, molecular basis of synthetic elements, and cycles of population replacement.

Mothers carrying Medea cause the death of all progeny that fail to inherit Medea (A). Synthetic Medea elements consist of two genes. Maternally-expressed miRNAs (the toxin) silence (red line during oogenesis) the expression of a maternally-expressed transcript (grey line) that normally provides a product essential for early embryonic development. Rescue of Medea-dependent maternal-effect lethality occurs when progeny inheriting Medea express a version of the silenced maternal mRNA sufficient to rescue normal development (green line). Progeny that fail to inherit Medea die because the endogenous levels of the maternally-deposited mRNA (red line during embryogenesis) are insufficient for normal development (B). Second-generation Medea elements (n+1) carrying a new cargo, a new toxin, a new antidote, and a copy of the antidote from the previous generation (n), drive into a population at the expense of a first generation element (n), when both elements are located at the same position in the genome so as to force them to compete for germline transmission (C).
We previously generated synthetic Medea elements through maternal expression of microRNAs designed to silence the expression of a maternally-expressed transcript, myd88, whose product is required in the embryo for dorso-ventral pattern formation. Maternal expression of these small RNAs created a pre-toxic state, a loss of maternal myd88, which if left unopposed led to the death of all embryos. Zygotic rescue in this case was mediated by expression of a tightly linked transgene, encoding a microRNA-insensitive version of the myd88 transcript lacking target sites present in the endogenous transcript, driven by a transient early zygote-specific promoter (Figure 1B) (7).
There are several reasons why it is important to be able to create Medea elements that work through additional mechanisms. First, while the Toll pathway and Myd88 are likely to be required for dorso-ventral pattern formation in many if not all insects, key components may be supplied zygotically rather than maternally in some species (e.g. (13)), which would prevent maternal silencing from causing embryonic lethality, a prerequisite for Medea-dependent drive. In addition, the genes that make up synthetic Medea elements and their transgene cargo will inevitably be subject to mutation. First, the toxin encoding miRNAs, or the promoter driving their expression, can mutate to inactivity. This leads to the creation of antidote-only alleles, which are resistant to Medea-dependent killing but do not drive. If these chromosomes have a higher fitness than those carrying intact Medea elements, the former will spread at the expense of the latter, potentially resulting in the appearance of wildtype individuals capable of disease transmission (11). Second, genes mediating disease refractoriness can also mutate to inactivity. If this loss of function results in a Medea-bearing chromosome with increased fitness as compared to chromosomes carrying an intact Medea element, the former will spread at the expense of the latter, also resulting in the appearance of individuals capable of spreading disease. Finally, principles of risk management argue that it is always desirable to be able to greatly decrease the frequency of, or eliminate, a particular transgenic modification from a wild population, if desired.
One strategy that addresses the above issues, albeit not always perfectly, involves creating second-generation Medea elements that carry a new toxin, a new antidote, and if needed a new set of transgenes designed to bring about disease refractoriness. If second generation elements are located at a different position in the genome from first generation elements, the former will spread, creating a population with both elements. If instead a second generation element is located at the same position in the genome as the first generation element - and it also carries a copy of the first generation antidote - this element should spread into the wild population at the expense of the first generation element in the same way first generation element spread at the expense of wildtype (7) (Figure 1C). Thus, for an autosomal element carrying a fitness cost, the frequency of a first generation elements will be greatly decreased, but it will not eliminated (it will persist in heterozygous combination with the second generation element at a frequency that depends on the relative fitness difference between first and second generation elements) (12). In contrast, an X-linked second-generation element is predicted to completely eliminate a first generation element from the population. These manipulations do not restore the pre-transgenic state, but they do allow one to carry out multiple cycles of population replacement, with distinct cargoes, or an element with no cargo (when the goal is primarily to decrease the frequency of, or remove, a particular gene for disease refractoriness from the population) being driven into the population during each cycle. A critical prerequisite for each of these strategies is the availability of multiple, independent toxin-antidote combinations able to show Medea-based gene drive.
EXPERIMENTAL RESULTS AND DISCUSSION
Engineering novel Synthetic Medea elements
In order to generate a synthetic Medea element through zygotic rescue of a maternal loss-of-function phenotype one needs genes whose expression occurs maternally, but whose products are not required until after the initiation of zygotic transcription. Two genes with these characteristics are discontinuous actin hexagons (dah, CG6157) and O-fucosyltransferase 1 (o-fut1, also known as neurotic, CG12366). Dah is membrane-associated protein required in Drosophila for proper formation of the metaphase furrow during syncytial blastoderm stages, and cellularization during blastoderm formation (14). Maternal loss of dah results in 100% embryo lethality and embryonic death is not rescued when embryos inherit a wildtype copy of the gene from the father, indicating that dah mRNA and/or protein must normally be provided maternally. Dah is a member of the dystrotelin branch of the family of proteins that also includes dystrophins and dystrobrevins (15). Clear Dah homologs are present in all sequenced Drosophila species and in mosquitoes, but are not obvious in other sequenced insect species such as beetles (Tribolium), wasps (Nasonia) or Honeybees (Apis mellifera). More distantly related dystrotelins are found throughout the animal kingdom, though functions for many remain to be determined. O-fut1 is required for Notch signaling in Drosophila (16) (17), with maternal loss resulting in a neurogenic phenotype, in which excess neurons are produced at the expense of embryonic epidermis (16). As with dah, loss of maternally-provided O-fut1 cannot be rescued through inheritance of a wildtype copy of the gene from the father. O-fut1 fucosylates Notch, and this modification is required for many Notch-dependent signaling events. O-fut1 also plays a non-enzymatic role as an ER chaperone that promotes Notch folding (18) (19), and as an extracellular component essential for Notch endocytic trafficking. O-fut1 homologs are conserved throughout the animal kingdom, as is its role in Notch-dependent signaling (20).
We engineered synthetic Medea elements based around dah and o-fut1 using the same architecture used to generate Medeamyd88(7). In order to silence the expression of dah or o-fut1, we generated for each gene a transcription unit (the toxin) encoding an 2-mer of two synthetic miRNAs, designed to base pair with 100% complementarity to the relevant target transcript. Synthetic miRNAs were generated using the mir6.1 backbone, as described previously (7). The expression of each miRNA multimer was driven by a modified version of the maternal-specific bicoid (bcd) promoter. Adjacent to each of these transcription units we placed a second transgene (the antidote) encoding a version of either dah or o-fut1 recoded so as to be resistant to silencing by the maternally-deposited synthetic miRNAs, with expression being driven by the transient, early promoter from the bottleneck (bnk) gene (21). Elements carrying both toxin and antidote genes are known as Medeadah and Medeao-fut1. Flies carrying these constructs are characterized below.
We also generated Medea elements based around maternal silencing and zygotic rescue of a number of other genes known to have maternal-effect lethal loss-of-function phenotypes (unpublished observations). For seven of these, pelle (22), tube (23), almondex (24), bloated tubules (25), concertina (26), anillin (27) and torso (28), we did not observe maternal effect killing, presumably due to inefficient gene silencing, possibly coupled with low levels of the maternal product being sufficient to provide sufficient activity for normal development. These elements were not characterized further. For two other genes, groucho (gro), a transcriptional co-repressor required for multiple aspects of early embryonic development, including neurogenesis, sex determination and segmentation (29), and trunk (trk), a ligand for the receptor Torso, required for specification of the embryonic termini (30), we were able to create Medea elements that showed strong maternal-effect lethality, with 100% of embryos derived from matings between heterozygous mothers and wildtype fathers dying (construct details provided in the methods). Medeagro also showed zygotic rescue behavior. However, rescue was imperfect and often resulted in male-biased sex ratios. These elements did not show drive when introduced into a non-transgenic population at an allele frequency of 25%, as described previously (7). In the case of Medeatrk elements, no zygotic rescue was obtained.
The above results indicate that it can be difficult to silence maternal gene expression, and that even when good maternal silencing is achieved, it may also prove difficult to replace the needed product through zygotic expression in a way that restores normal development. In the case of gro-based Medea elements, it is likely that specific zygotic levels of gro are important, with bnk-drive expression of the rescuing transgene resulting in inappropriate levels that disrupt multiple signaling pathways (29). In the case of trk-based Medea elements the reasons for lack of rescue are unclear. Injection of trk mRNA into embryos from homozygous mutant trk mothers has been shown to rescue embryonic pattern formation, albeit at a low frequency (31). It is possible that, as with gro, timing and levels of protein expression are critical. While we cannot draw strong conclusions from this limited data set, our results may suggest that genes whose products provide a necessary condition for a process, but whose expression is not sufficient to drive the process (adaptors such as myd88 and dah), are better candidates than those whose expression drives a process in a dose-dependent manner (signal transduction effectors such as gro and trk).
Medeadah and Medeao-fut1 show maternal-effect lethal and zygotic rescue behavior
Multiple transgenic lines were generated for each of the above constructs. One line each was selected for further characterization. Matings between heterozygous Medeadah/+, or Medeao-fut1/+ males (where + indicates a wild-type chromosome) and +/+ females resulted in high levels of embryo viability, similar to those for the w118 strain used for transformation (Table 1). In addition, ~50% of adult progeny carried Medea, as expected for Mendelian segregation without dominant fitness costs. Matings among homozygotes for Medeadah or Medeao-fut1 also resulted in high levels of egg viability. In contrast, when heterozygous Medeadah/+ or Medeao-fut1/+ females were mated with homozygous +/+ males, ~50% of embryos died and all adult progeny were Medea-bearing. These observations, together with the results of several other crosses (Table 1), indicate that a single copy of each Medea toxin is sufficient to induce 100% maternal-effect lethality and a single copy of each rescue transgene is sufficient to rescue normal development of embryos derived from mothers expressing one or two copies of the toxin.
Table 1. Medeadah and Medeao-fut1 show Medea-like selfish genetic behavior.
Progeny of crosses between parents of several different genotypes are shown. M refers to the Medeadah- or Medeao-fut1-bearing chromosome; + refers to the non–Medea-bearing homolog. The maternal copy number (0 to 2) of bcd-driven miRNAs (maternal toxin) targeting the endogenous dah or o-fut1, and the zygote copy number (0 to 2), and percentage of embryos inheriting bnk-driven dah or o-fut1 (zygotic antidote) are indicated, as are the adult progeny genotypes predicted for Mendelian inheritance of Medeadah or Medeao-fut1. The percent embryo survival was normalized to that of wild-type (w1118), which was 93.80±3.19. −, not measured.
| Inherited by the | |||||||
|---|---|---|---|---|---|---|---|
| Parental Genotype | Oocyte | Embryo | Adult M progeny % | Embryo Survival % | |||
| Male | Female | Maternal Toxin | Zygotic Antidote % | Predicted | Observed (dah, o-fut1) | Predicted | Observed (dah, o-fut1) ± sd |
| M/+ | +/+ | 0 | 50(0); 50(1) | 50 | 50 (n>12,000), 50 (n>11,000) | 100 | 98.05±2.3, 96.28±2.8 |
| M/M | M/M | 2 | 100(2) | 100 | - | 100 | 98.97±6.0, 92.74±6.0 |
| +/+ | M/+ | 1 | 50(0); 50(1) | 50 | 100 (n>10,000), 100 (n>12,000) | 50 | 49.56±0.5, 47.43±2.0 |
| M/M | M/+ | 1 | 50(1); 50(2) | 100 | - | 100 | 98.05±2.3, 97.35±2.0 |
| M/+ | M/+ | 1 | 25(0); 50(1); 25(2) | 75 | - | 75 | 73.62±1.5, 73.9±1.7 |
| M/+ | M/M | 2 | 50(1); 50(2) | 100 | - | 100 | 97.7±1.0, 98.41±1.5 |
| +/+ | M/M | 2 | 100(1) | 100 | - | 100 | 99.47±1.5, 94.51±2.0 |
| M/M | +/+ | 0 | 100(1) | 100 | - | 100 | 95.22±1.1, 96.99±2.5 |
| +/+ | +/+ | 0 | 100(0) | 0 | - | 100 | 93.80±3.19 |
Medeadah and Medeao-fut1 drive population replacement
The above observations suggested that Medeadah and Medeao-fut1 should drive population replacement. To test this prediction we mated equal numbers (1:1 ratio) of hetrozygous Medeadah/+ or Medeao-fut1/+ males to WT (+/+) females in separate bottles in triplicate, giving rise to progeny populations with Medeadah or Medeao-fut1 present at an allele frequency of ~25%. Populations were followed for a total of 20 generations, counting the number of Medea-bearing adults each generation, as described previously (7). In all cages non-Medea-bearing individuals disappeared from the population between generations 12 and 16 (Figure 2).
Figure 2. Medeadah and Medeao-fut1 drive population replacement.

Fraction of the adult population that is transgenic is plotted versus number of generations for both Medeadah (thin blue lines) and Medeao-fut1 (thin green lines). Expected transgenic frequencies for a Medea with no fitness cost (s = 0) are indicated by the black line. Predicted behavior of Medea elements with fitness costs corresponding to best-fit estimates Medeadah (bold blue line) and Medeao-fut1 (bold green line) were derived from actual Medeadah and Medeao-fut1 behavior. 95% confidence intervals are indicated by light green and blue shading. s = additive fitness cost in Medea homozygotes.
The observed changes in Medea frequency trailed the predicted rate of increase for a Medea element with no fitness cost, with the delay being more extreme for Medeadah than for Medeao-fut1. The magnitude of the fitness costs were estimated by calculating the expected dynamics for a variety of fitness costs (both additive and dominant), calculating the likelihood of the observed data for each fitness cost, and selecting the fitness cost having the greatest likelihood (for more details, see the materials and methods). Confidence intervals for the fitness costs were then obtained using a Markov Chain Monte Carlo sampling procedure (32). We found that additive fitness costs provided a much better fit to the data for Medeadah, while for Medeao-fut1, both additive and dominant fitness costs performed equally well. Assuming additive fitness costs, we estimate a cost in homozygotes of 27.3% for Medeadah (95% confidence interval: 26.8–27.8%) and of 17.4% for Medeao-fut1 (95% confidence interval: 16.5–18.2%). The basis for these costs is unknown. It could reflect insertion site-dependent effects on the expression of neighboring genes. It could also reflect the effects of manipulating the developmental expression of the targeted genes (imperfect rescue, for example), or other off-target effects on unknown genes. The results of crosses tabulated in Table 1 do not identify any obvious fitness costs associated with survival of embryos or adults carrying Medeadah or Medeao-fut1. However, many aspects of life history such as response to stress, longevity, and fertility/fecundity remain to be explored. Characterization of identical Medea elements inserted at new locations should provide insight into whether the costs observed are site-specific or intrinsic to these elements.
These results, in conjunction with our earlier work, demonstrate that Medea selfish genetic behavior in Drosophila can be engineered through the manipulation of diverse signaling pathways. Each of the pathways targeted - embryonic dorso-ventral pattern formation (33), blastoderm formation (34), and Notch signaling (35), requires the products of multiple genes in order to bring about normal early embryonic development. Many of these are also provided maternally. In consequence, it may - with the caveats noted above - be possible to generate Medea elements through maternal silencing and zygotic rescue of those whose activity is not required in some other context during oogenesis. Elements generated this way, even those that target different components of the same pathway, will function as independent, non-cross-rescuing Medea elements.
Our positive results notwithstanding, it remains to be demonstrated that synthetic Medea elements can be generated using the above strategy in pest insects. In unpublished work we have tried, and thus far been unable to bring about maternal effect killing in the mosquito Aedes aegypti in response to maternal expression of miRNAs designed to silence Aedes myd88, dah or o-fut1. While transcript levels of the targeted genes are decreased in response to miRNA expression, it is possible that remaining levels of maternally deposited product are sufficient to bring about normal development. Extensive transcriptional profiling of Aedes oogenesis and early embryonic development (unpublished observations) also leaves open the possibility that early zygotic transcription of myd88, dah and o-fut1 may provide some rescuing activity, limiting our ability to kill through maternal silencing alone. These difficulties highlight the fact that the approach to Medea development described herein requires some detailed knowledge of oogenesis and embryonic development with respect to when specific transcripts/proteins are required, and how they are provided. This information exists for the genes we targeted in Drosophila, and provided the basis for our use of them, but it does not exist for other insects. An alternative approach to Medea development involves the use of proteins or RNAs that provide a toxic dominant gain of function activity that acts in the zygote. If these are generally toxic to insect embryos (though not oocytes), this strategy would appear to require less knowledge of the insect under study (primarily information relating to promoters and UTRs that provide control over RNA stability and protein translation during oogenesis), which may make the system more portable across species. In summary, while our results demonstrate that multiple synthetic Medea elements can be generated through maternal silencing and zygotic rescue, it remains unclear if this design strategy provides a straightforward path to creation of similar elements in other insects, for which more limited information is available.
Molecular characterization of Medea-dependent gene silencing
An important unanswered question with respect to the synthetic Medea elements generated here and previously (7) is the extent to which the targeted maternal transcripts are being silenced, and the levels of synthetic miRNAs associated with these levels of silencing. MicroRNA-dependent silencing can occur through translational inhibition and/or transcript degradation (36). To provide a minimum estimate of the extent to which maternal myd88, dah and o-fut1 were being silenced, we used next generation sequencing technology (RNA-seq) to measure transcript levels in both the mature oocytes (stage 14, s14) of Medea/+ mothers and early embryos (0–1 hr) derived from crosses between these mothers and +/+ males. In s14 oocytes transcript levels were decreased by 5.0-fold (myd88), 14.27-fold (dah), or 30-fold (o-fut1) with respect to the levels of the endogenous transcripts. Similar, though somewhat lower levels of silencing were also observed in the early embryo (0–1 hr): 4.1-fold for (myd88), 7.12-fold for (dah)and 5.53-fold for (o-fut1) (Figure 3A). The somewhat lower level of knockdown in the 0–1 hr early embryo versus the s14 oocyte may reflect poly A addition to previously non-polyadenylated transcripts (37). It may also reflect, in part, the initiation of zygotic transcription and expression of the rescue transgenes.
Figure 3. Molecular characterization of stage 14 oocytes and 0–1 hr embryos from mothers of specific genotypes.

Levels of myd88, o-fut-1 and dah mRNA determined using RNA-seq (expressed as Fragments per kilobase per million reads, FPKM) in wildtype stage 14 oocytes and 0–1 hr embryos from wildtype mothers and wildtype fathers (+/+), and from oocytes and embryos derived from heterozygous Medeamyd88 (green bars), Medeadah (blue bars) or Medeao-fut1 (red bars) mothers crossed to (+/+) males. Asterisks indicate the number of replicates (A). The structures of the miRNAs used to silence dah, o-fut1, and myd88 are indicated. The orange shaded region indicates the predicted miRNA guide strand; the yellow shaded region represents the miRNA* strand. Horizontal arrows indicate sites of Drosha and Dicer cleavage (B). Small RNA reads for synthetic miRNAs were expressed as reads per million reads (RPM), which normalizes read count to the size of each library. To provide a sense of scale, the expression levels of synthetic miRNAs were also compared to those of endogenous miRNAs expressed in the ovary, both in terms of absolute rank and in terms of rank percentile.
To begin to identify the characteristics of synthetic miRNAs able to bring about effective silencing of the maternal transcripts discussed above, we sequenced libraries of small RNAs isolated from s14 oocytes and 0–1 hr embryos derived from crosses between mothers heterozygous for Medeamyd88, Medeadah or Medeao-fut1 and +/+ fathers (Supplementary Table 1). We detected the expression of 154 (64.7%) of the 238 total Drosophila melanogaster annotated miRNAs in miRbase (v19), with the top 15 expressed miRNAs constituting the bulk, between 89.14 and 97.81%, of the total reads mapping to miRNAs. modENCODE does not have an equivalent data set, the only related one being small RNAs from whole ovaries, which includes all stages of ovarian development (38). Therefore, to gain a sense of reproducibility from sample to sample we calculated correlation coefficients, comparing levels of miRNAs in wildtype s14 oocytes or 0–1 hr embryos with comparable stages derived from Medeamyd88, Medeadah or Medeao-fut mothers (Supplement Tables 2 and 3). Correlation coefficients were high for DAH (s14=0.84 and 0–1 hr=0.96) and myd88 (s14=0.91 and 0–1 hr=0.97), but were significantly lower for o-fut1 (s14=0.66 and 0–1 hr=0.92). Consistent with this, the rank abundance of the top 20 miRNAs in DAH and Myd88 samples were similar (though not identical) to those from wildtype, while those from o-fut-1 samples were much less similar. The basis for the low correlation coefficients observed with o-fut1 samples requires further exploration. It could be that expression of synthetic miRNAs targeting o-fut1, and/or position effects related to the site of Medea insertion in the genome, resulted in significant changes in the patterns of small RNA gene expression. Alternatively, differences in sample preparation may be responsible.
We calculated levels of synthetic miRNAs in terms of reads per million total reads (RPM), and noted their rank percentile abundance with respect to endogenous miRNAs expressed in the ovary or embryo to provide a sense of scale (Figure 3C). For all synthetic miRNAs we only detected mature miRNAs from the guide strand (Figure 3B). For Medeao-fut1, both synthetic miRNAs were undetectable in early embryos, and only one, o-fut1-miRNA-2, was observed at low levels in the s14 oocyte (rank percentile of 34). For Medeadah, dah-miRNA-1 was not detected in the ovary, and was found only at very low levels in the embryo. In contrast, dah-miRNA-2 was found at somewhat higher levels in the oocyte and embryo (rank percentile of 25% and 20%, respectively). Finally, for Medeamyd88 we observed that one miRNA, myd88-miRNA-1, was present at relatively high levels in the ovary and embryo (rank percentile of 12% and 5%), while myd88-miRNA-2 was expressed at much lower levels.
Given the high correlation coefficients observed in myd88 and dah samples, the observed levels of synthetic miRNAs may be an accurate reflection, at least to some extent, of their endogenous levels. This cannot be said for the o-fut1 samples. There are several possible explanations for why the levels of specific miRNAs in the myd88 and dah samples might be low and/or discordant with those of other miRNAs expressed from the same transcript. First, miRNAs loaded into Ago1-containing RISC complexes, and bound to targets with perfect complementarity, are degraded through a tailing and trimming process, while those bound to Ago2-containing RISC undergo 2′-O-methylation at their 3′ ends, which prevents degradation through this pathway (39) (40). All of our miRNAs are designed to have perfect complementarity with their targets, and while we designed them using rules thought to promote loading into Ago2 (41) (42) (43), their actual loading preferences have not been determined. Thus, it is possible that Ago-loading preferences are influencing their fate. That said, if the low levels of specific synthetic miRNAs we observe in the ovary and early embryo result from their degradation subsequent to binding to fully complementary mRNA target transcripts while in association with Ago1, the fact that we still observe good maternal transcript silencing would suggest that these miRNAs have significant ability to silence prior to degradation. In previous work in S2 cells, Ago1-bound miRNAs with perfectly complementary to their targets were unable to bring about detectable silencing (44). It will be interesting to determine if Ago-1-bound miRNAs behave differently in the ovary.
Preferential processing of specific miRNAs from the polycistronic transcript may also contribute the low and/or discordant levels observed for specific miRNAs in a polycistron. For example, once processing of one of the miRNAs occurs, if processing of the second miRNA is delayed, exonuclease activity may result in degradation of the cleaved precursor before processing of the second miRNA occurs (45). Finally, active turnover may promote the degradation of specific mature miRNAs (46). Clearly more work is needed in order to understand which miRNAs are bringing about silencing, and which Ago-RISC complex they are associated with. Generation of Medea elements that drive population replacement, and that express single miRNAs, in conjunction with immunoprecipitation of Ago1- and Ago2-bound miRNAs from Medea-bearing females, should provide additional insights into the characteristics of highly active maternal miRNAs.
Using Medea to bring about environmental cue-dependent population suppression
Here we consider a second possible use of Medea: to bring about population suppression, and perhaps eradication, following population replacement with a transgene cassette that results in female-specific lethality in response to a specific environmental cue such as diapause, which allows many insects to survive long periods of adverse conditions such as cold or drought (47) (48, 49). An attractive feature of an environmental cue-dependent population reduction strategy is that, in contrast to the use of insecticides, the strategy is species specific. A cassette to be driven into the population for such a strategy might consist of a gene expressed under the control of a diapause-induced promoter, encoding a transcript that undergoes female-specific splicing to produce a toxin in females but not males (Figure 4). We use discrete-generation difference equations to theoretically explore this idea. We consider the Medea element as a single allele, which we denote as “M” and refer to the corresponding position on the wild-type chromosome as “m.” A one-locus model can then be used for the discrete-generation dynamics. We assume random mating, 100% toxin efficiency, and equal fitness costs in males and females having the element in the absence of the external cue. We also consider density-dependence. We model the effects of an external cue by modulating the fitness costs based on the presence or absence of the cue. In the absence of the cue, we consider additive fitness costs of 17.4% in homozygous males and females (i.e. sf = sm = 0.174 and hf = hm = 0.5), corresponding to fitted estimates for Medeao-fut1, which suffered smaller fitness costs than Medeadah. These fitness costs are unlikely to reflect fitness in the wild. They should perhaps be thought of as providing a minimum estimate, and are incorporated for illustrative purposes only. In the presence of the cue, we consider the case of dominant, female-specific lethality, so that male fitness costs are unchanged and female fitness costs are 100% in both heterozygotes and homozygotes (i.e. sf = 1 and hf = 1). To focus on mosquitoes as a specific example, if the female-specific toxin is under the control of a diapause-specific promoter, then there should be little or no fitness cost associated with the presence of this transgene during the ~12 generations of the rainy season, while female-specific lethality should result during the ~1 generation of the dry season in which diapause occurs. We consider a release at the beginning of the rainy season, neglecting seasonal changes in the environmental carrying capacity. Details of the model are found in the methods. Here we focus on several important conclusions.
Figure 4. Hypothetical Medea elements carrying a cargo transgene cassette able to induce cue-dependent female killing.
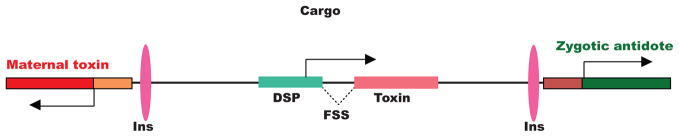
The Medea illustrated carries a cargo gene consisting of a diapause-specific promoter (DSP) driving a toxin that is only synthesized as an intact protein in response to female-specific splicing (B). Chromatin insulators (Ins), female specific splicing events (FSS), and 3′ untranslated regions (UTR) are indicated.
We consider a release of homozygous transgenic males at the beginning of the rainy season (generation 1). The rainy season lasts 12 generations, during which time the transgene spreads into the population. Then, as the dry season begins (generation 13), transgenic females suffer a lethal fitness cost such that the female population is greatly reduced by generation 14 (the beginning of the next rainy season), with a consequent drop in total population size by generation 15. This scenario is shown in Figure 5A, B. In Figure 5A 4,000 transgenic males are released into a wild-type population of 10,000. By generation 13, the transgene has almost reached transgene fixation (i.e. almost all individuals have either one or two copies of the Medea element); but because transgene fixation is not complete, a few wild-type females survive diapause and the population rebounds during the next rainy season. However, by the time of the next diapauses (generation 26), the population is entirely transgenic and so, this time, the female population collapses at generation 27 and there is a total population crash by generation 28. Figure 5B illustrates a scenario in which 6,000 transgenic males are released. In this case the population is entirely transgenic by the time of the first diapause (generation 13). As a result a permanent population crash occurs by generation 15.
Figure 5. Modeling the ability of Medea elements carrying genes that cause cue-dependent female lethality to bring about population eradication.

4,000 males homozygous for a Medea carrying a transgene cassette that induces diapause-dependent female killing are released into a total wild population of 10,000. The fate of the total population (red line), transgenics (blue line), and females (green line) are followed for 30 generations. Diapause occurs in generation 13 and generation 26 (A). 6,000 transgenic males are released into the wild population as above (B).
To summarize, the key result from our modeling is the finding that that an environmental cue can be used to bring about a population crash, provided that Medea is given time to spread to transgene fixation before the cue appears. Appearance of the cue prior to transgene fixation may lead to transient population suppression, but the continued presence of wildtype females allows the population to ultimately rebound. However, because Medea-bearing males are not killed by the cue, population replacement continues, and cue-dependent suppression can be initiated the next season. While the numbers of Medea-bearing males that need to be released in order for population replacement to happen within one season is high (~1:1 with respect to wildtype males), these numbers are still significantly lower than those associated with sterile male release programs designed to bring about population suppression, and do not constitute a fundamental limitation of the technology (50).
Can the components required to bring about Medea-dependent population suppression be created? In the above model the encoded toxin would only be active in females as a result of a female-specific splicing event. Genes with these splicing characteristics have been created to bring about female-specific killing for other vector-control strategies (51) (52) (53), suggesting such a toxin can be generated. However, diapause-specific genes and promoters have not yet been identified. In addition, because the cue-dependent transgene causes lethality, mutants that lack this activity, or the Medea toxin, which is required for drive, will be strongly selected for. Multiple copies of the cue-dependent lethality cassette, and the gene encoding the Medea toxin (the maternal promoter and its associated poly-miRNA encoding transcript, each copy of which may include multiple miRNAs, targeting different regions of the target transcript), can delay the effects of mutation and selection. However, they cannot prevent them. Therefore, use of such a technology to bring about a population crash would likely be most effective in contexts such as islands in which population size is limited and migration of wildtypes into the population is negligible. More detailed ecological models will be required to identify specific species and environments in which such a strategy for population suppression might be successful.
Methods
Construction of a miRNA’s
The Drosophila miRNA mir6.1 stem-loop was modified to target dah, o-fut1, gro or trk. To generate a mir6.1 stem-loop backbone that generates a mature miRNA complementary to one or the other of these target sites we annealed pairs of primers. For example, to make a miRNA that targets dah site 1, primers dah-1-f (5′-CTT AAT CAC AGC CTT TAA TGT AGG GAA ATA TAT AAC AAT ACA CTA AGT TAA TAT ACC ATA TCT-3′) and DAH-1-r (5′-ATG TTA GGC ACT TTA GGT ACA GGG AAA TAT ATA ACA ATA AAC TAG ATA TGG TAT ATT AAC TTA G-3′) were annealed; to target dah site 2, primers dah-2-f (5′-TTA AAC TTA ATC ACA GCC TTT AAT GTA ACC AGG ATG CGA ACT ATA CAC TAA GTT AAT ATA CCA TAT CTA G-3′) and dah-2-r (5′-AAT GAT GTT AGG CAC TTT AGG TAC AAC CAG GAT GCG AAC TAT AAA CTA GAT ATG GTA TAT TAA CTT AG-3′) were annealed; to target o-fut1 target site 1, primers o-fut1-1-f (5′-AAA CTT AAT CAC AGC CTT TAA TGT AGT TTT ATT ACA TTG ATT ACG CTA AGT TAA TAT ACC ATA TCT AG-3′) and o-fut1-1-r (5′-AAT GAT GTT AGG CAC TTT AGG TAC AGT TTT ATT ACA TTG ATT AAG CTA GAT ATG GTA TAT TAA CTT AGC G-3′) were annealed; to target o-fut1 target site 2, primers o-fut1-2-f (5′-ATC ACA GCC TTT AAT GTC AGG ATT ATC TAC TTA AAT CCT TAA GTT AAT ATA CCA TAT CTA AGT-3′) and o-fut1-2-r (5′-ATG ATG TTA GGC ACT TTA GGT ACC AGG ATT ATC TAC TTA AAT ACT TAG ATA TGG TAT ATT AAC TTA AGG A-3′) were annealed; to target gro target site 1, primers gro-1-f(5′-CTT AAT CAC AGC CTT TAA TGT TAC CAT AAA ACG CTG GCA ACC ATA AGT TAA TAT ACC ATA TCT ATG TT-3′) and gro-1-r (5′ TGA TGT TAG GCA CTT TAG GTA CTA CCA TAA AAC GCT GGC AAA CAT AGA TAT GGT ATA TTA ACT TAT GG-3′) were annealed; to target gro target site 2, primers gro-2-f (5′-TTA ATC ACA GCC TTT AAT GTG GAT CAA CTG CCA TCG AAA CAT TAA GTT AAT ATA CCA TAT CTA ATT T-3′) and gro-2-r (5′-ATG TTA GGC ACT TTA GGT ACG GAT CAA CTG CCA TCG AAA AAT TAG ATA TGG TAT ATT AAC TTA ATG T-3′) were annealed. To build up the miRNAs specific to trk we designed a polycistronic strand of 6 unique miRNAs. To target trk target site 1, primers trk-1-f(5′-CTT AAA CTT AAT CAC AGC CTT TAA TGT GCC AGC CAT GTT TCT GCG TCT ATA AGT TAA TAT ACC ATA TCT ATA TAC-3′) and trk-1-r (5′ TAA TGA TGT TAG GCA CTT TAG GTA CGC CAG CCA TGT TTC TGC GTA TAT AGA TAT GGT ATA TTA ACT TAT AGA-3′) were annealed; To target trk target site 2, primers trk-2-f(5′-CTT AAA CTT AAT CAC AGC CTT TAA TGT AGA AGA GCT ATC ATC TGG GCT ATA AGT TAA TAT ACC ATA TCT ATA TC-3′) and trk-2-r (5′ TAA TGA TGT TAG GCA CTT TAG GTA CAG AAG AGC TAT CAT CTG GGA TAT AGA TAT GGT ATA TTA ACT TAT AGC CC-3′) were annealed; To target trk target site 3, primers trk-3-f(5′-CTT AAA CTT AAT CAC AGC CTT TAA TGT ACT GGA TTG ACT TAG GCT TCA ATA AGT TAA TAT ACC ATA TCT ATT TA-3′) and trk-3-r (5′-TAA TGA TGT TAG GCA CTT TAG GTA CAC TGG ATT GAC TTA GGC TTA AAT AGA TAT GGT ATA TTA ACT TAT TGA-3′) were annealed; To target trk target site 4, primers trk-4-f(5′-CTT AAA CTT AAT CAC AGC CTT TAA TGT ATG CTA TAC TAG TAG CGC CCT TTA AGT TAA TAT ACC ATA TCT AAA TG-3′) and trk-4-r (5′-TAA TGA TGT TAG GCA CTT TAG GTA CAT GCT ATA CTA GTA GCG CCA TTT AGA TAT GGT ATA TTA ACT TAA AGG G-3′) were annealed; To target trk target site 5, primers trk-5-f(5′-CTT AAA CTT AAT CAC AGC CTT TAA TGT TAT ACT AGT AGC GCC ATT TCA TTA AGT TAA TAT ACC ATA TCT AAT TA-3′) and trk-5-r (5′-TAA TGA TGT TAG GCA CTT TAG GTA CTA TAC TAG TAG CGC CAT TTA ATT AGA TAT GGT ATA TTA ACT TAA TGA-3′) were annealed; To target trk target site 6, primers trk-6-f(5′-CTT AAA CTT AAT CAC AGC CTT TAA TGT ATA CAC GTT TAA AAT GTC ACC ATA AGT TAA TAT ACC ATA TCT ATG TT-3′) and trk-6-r (5′-TAA TGA TGT TAG GCA CTT TAG GTA CAT ACA CGT TTA AAA TGT CAA CAT AGA TAT GGT ATA TTA ACT TAT GGT-3′) were annealed. These primary products were all then amplified using primers mir6.1 5′ Not1/Fse1/BglII (5′-TCG GGC GGC CGC ATT TGG CCG GCC AAA GAT CTT TTA AAG TCC ACA ACT CAT CAA GGA AAA TGA AAG TCA AAG TTG GCA GCT TAC TTA AAC TTA ATC ACA GCC TTT AAT GT-3′) and mir6.1 3′ Ecor1/Asc1/BamHI (5′-TGA AGA ATT CAT TGG CGC GCC TTT GGA TCC AAA ACG GCA TGG TTA TTC GTG TGC CAA AAA AAA AAA AAA TTA AAT AAT GAT GTT AGG CAC TTT AGG TAC-3′). These primers add mir6.1 flanking sequences that are thought to promote miRNA processing, as well as several restriction sites. PCR products were purified with Qiagen (Valencia, CA) PCR purification columns, and then digested with several restriction enzymes. For dah-1, o-fut1-1, gro-1 and trk-1 these were EcoRI and BamHI, for dah-2, -fut1-2, gro-2 and trk-2 these were BglII and NotI. Digested products were then ligated into sry-bcd-GMR (7) cut with EcoRI and NotI, generating pBcd-mir6.1-ofut1, pBcd-mir6.1-dah, pBcd-mir6.1-gro and pBcd-mir6.1-trk2. The pBcd-mir6.1-trk2 was then digested with Asc1/Bamh1 and the trk-3 miRNA was digested with BglII/Asc1 and cloned. This digest/cloning was repeated to sequentially clone in trk4, trk5 and trk6 producing a plasmid with 6 miRNAs targeting trk: pBcd-mir6.1-trk6.
Construction of the antidotes and final Medea constructs
Plasmids pBcd-mir6.1-o-fut1, pBcd-mir6.1-dah, pBcd-mir6.1-gro and pBcd-mir6.1-trk6 were cut with Xho-1. The gypsy insulator was amplified from genomic DNA using primers gyp-f (5′-AAG AGA TGT AGA GAT GGC ACA ATT GGT CGA CCT CGA GTC ACG TAA TAA GTG TGC GTT GAA TTT ATT-3′) and gyp-r (5′-ATG AGG CGT CCA GGA TCC CAT GGG GTT CAT CTA ATG TTT AAA CAA TTG ATC GGC TAA ATG GTA TGG CAA-3′). The bnk promoter was amplified similarly using primers bnk-f (5′-TTT TCT TGC CAT ACC ATT TAG CCG ATC AAT TGT TTA AAC ATT AGA TGA ACC CCA TGG GAT CCT GG-3′) and bnk-r (5′-AGA AGT AAG GTT CCT TCA CAA AGA TCC TGG CCG GCC TCG CCG AAT TCG TTG ACG GTT GAA GTA C-3′). The SV40 UTR was amplified using primers SV40-f (5′-CGT ACT TCA ACC GTC AAC GAA TTC GGC GAG GCC GGC CAG GAT CTT TGT GAA GGA ACC TTA CTT C-3′) and SV40-r (5′-ATA ATT TGC GAG TAC GCA AAG CTT GGC TGC AGG TCG ACG GAT CCA GAC ATG ATA AGA TAC ATT GAT G-3′). All three fragments were ligated together using one step recombination technology (54), producing a unique fse-1 cloning site between the bnk promoter and SV40 3′ UTR, resulting in plasmids pBcd-mir6.1-o-fut1-gyp-Bnk-fes1-SV40, pBcd474 mir6.1-gro-gyp-Bnk-fes1-SV40, pBcd-mir6.1-dah-gyp-Bnk-fes1-SV40 and pBcd-mir6.1-trk-gyp-Bnk-fes1-SV40.
The O-fut1 coding region was amplified from a cDNA library using primers o-fut1-anti-f (5′-CAA CAG CAC ATT CGT ACT TCA ACC GTC AAC GAA TTC GGC ATG CAG TGG CTC AAA ATG AAG C-3′) and o-fut1-anti-r (5′-ATG TCA CAC CAC AGA AGT AAG GTT CCT TCA CAA AGA TCC TTT ACA GCT CCT CGT GCA CGT TTG T-3′). One-step recombination technology was used to introduce these into fse-1 digested pBcd-mir6.1-ofut1-gyp-Bnk-fes1-SV40, creating Medeao-fut1. Note that because this o-fut1 transcript lacks a 3′ UTR present in the endogenous o-fut1 transcript, it is not silenced by mir6.1-o-fut-1 or mir6.1-o-fut1-2, which target the o-fut1 3′ UTR. The dah coding region was amplified from a cDNA library using primers dah-anti-f-1 (5′-TCA ACA GCA CAT TCG TAC TTC AAC CGT CAA CGA ATT CGG CAT GCT GAG ATC GTC GGT GCC CGT-3′) and dah -anti-r-1 (5′-GTT GCC CTG TCC AAC TTG TAA TTG GCG TCT TGA TTG AAA TGG CCT AGT TTC TCG CAG GC-3′), and dah -anti-f-2 (5′-GCC TGC GAG AAA CTA GGC CAT TTC AAT CAA GAC GCC AAT TAC AAG TTG GAC AGG GCA AC-3′) and dah -anti-r-2 (5′-ATG TCA CAC CAC AGA AGT AAG GTT CCT TCA CAA AGA TCC TGC TCA CGT GCT GAT GCG CCG CT-3′). One-step recombination was used to introduce these products into fse-1 digested pBcd-mir6.1-dah -gyp-Bnk-fes1-SV40, creating Medeadah. Note that because this dah transcript lacks a 5′ UTR present in the endogenous dah transcript, it is not silenced by mir6.1-dah-1 which targets the dah 5′ UTR. However, mir6.1-dah-2 targets exon 2 of the CDS in dah and therefore in order to make the antidote insensitive to this miRNA we recoded the nucleotide sequence in dah in such that it codes for the same amino acid sequence using different codons. The gro coding region was amplified from cDNA library using primers gro-anti-f (5′-CAT TCG TAC TTC AAC CGT CAA CGA ATT CGG CTT AAT TAA ATG TAT CCC TCA CCG GTG CGC-3′) and gro-anti-r (5′-CAC ACC ACA GAA GTA AGG TTC CTT CAC AAA GAT CCT GCG GCC GCT TAA TAA ATA ACT TCG-3′). One-step recombination was used to introduce these products into fse-1 digested pBcd-mir6.1-gro-gyp-Bnk-fes1-SV40, creating Medeagro. Note that because this gro transcript lacks a 5′ UTR present in the endogenous gro transcript, it is not silenced by mir6.1-gro-1 or mir6.1-gro-2 which are engineered to target gro 5′ UTR. The trk coding region was amplified in 3 fragments to recode target sites from cDNA library using primers for 268bp fragment 1 trk-anti-1-f (5′-GTC AAC GAA TTC GGC TTA ATT AAT GTT CTT AAG AAT TCT GTG TCC AAC TAT GAA ATC CCA AAG-3′) and trk-anti-1-R (5′-GGG CAG CTC GTA CGA CGA TCG CTT ATA GCC TAA GTG GTA TGA TTT TTC CTC GGG CTC GCC C-3′) and 296bp fragment 2 trk-anti-2-f (5′-CGAGCCCGAGGAAAAATCATACCACTTAGGCTATAAGCGATCGTCGTACGAGCTGCCCTTC-3′) and trk-anti-2-R (5′-ATA GCG TGG AAA GTA GTT GAG TCC GAG ATC GAT CCA ATT GAT CTT CGA CGA GCA CTC CCA-3′) and 310bp fragment 3 trk-anti-3-f (5′-TCG TCG AAG ATC AAT TGG ATC GAT CTC GGA CTC AAC TAC TTT CCA CGC TAT ATC CGT TCG-3′) and trk-anti-3-R (5′-CAT TCT AGT TGT GGT TTG TCC AAA CTC ATC AAT GTC TAG TAT AGC ATA ACA CAT TCA CAG-3′). One-step recombination was used to introduce these products into fse-1 digested pBcd-mir6.1-trk -gyp-Bnk-fes1-SV40, creating Medeatrk. Note that because this trk transcript lacks a 5′ and 3′ UTR present in the endogenous trk transcript, it is not silenced by mir6.1-trk-1, which targets the dah 5′ UTR, or mir6.1-trk-4, mir6.1-trk-5, mir6.1-trk-6 which target the 3′UTR. However, mir6.1-trk-2 and mir6.1-trk-3 target exon 1 of the CDS; in order to make the antidote insensitive to these miRNAs we recoded the nucleotide sequence in trk such that it codes for the same amino acid sequence using different codons.
Transgenesis and Population cage experiments
Germline transformants were generated in a w1118 background using standard techniques, by Rainbow Transgenic Flies, Inc (www.rainbowgene.com, Newbury Park, CA). All fly experiments were carried out at 25°C, ambient humidity in 250 ml bottles containing Lewis medium supplemented with live dry yeast. Fly rearing was carried out in a light tight chamber placed in an incubator or in a darkened incubator. For all drive experiments three populations of 50 wildtype w1118 (+/+) males and 50 males heterozygous for either dah (w1118/Y; Medeadah/+) and o-fut1 (w1118/Y; Medeao-fut1/+) were each independently crossed with 100 wildtype w1118 females in separate bottles in triplicate. For all sets of experiments flies were allowed to lay eggs for four days, after which adults were removed. Progeny were allowed to develop, eclose and mate for another 10 days. All adult progeny were collected at this single timepoint and their genotypes determined using eye color as a marker (no Medea, w1118 = white eyed; Medea/+ = yellow/orange eyed; Medea/Medea = darker red eyed). Following counting, progeny were transferred to fresh bottles and allowed to lay eggs for four days, and the cycle repeated.
Embryo and adult viability determination
Adult viability for the crosses presented in Figure 2 was determined as follows. 50 adult males of the indicated genotype were allowed to mate with 50 virgin females in bottles supplemented with dry yeast for three days. 10 bottles were established for each cross. Adults were then removed. Adult progeny from each bottle were collected, genotyped and counted (either directly or by weighing and comparing with a standard) for 10 days following eclosion of the first progeny. For embryo viability counts, 2–4 day old adult virgin females were allowed to mate with males of the relevant genotypes for 2–3 days in egg collection chambers supplemented with wet yeast paste. On the following day, a 3 hr egg collection was carried out, after first having cleared old eggs from the females through a pre-collection period on a separate plate for three hrs. Embryos were isolated into groups of 100 and kept on an agar surface at 25°C for 48–72 hrs. The % survival was then determined by counting the number of unhatched embryos. Four groups of 100 embryos per cross were scored in each experiment, and each experiment was carried out three times. The results presented are averages from these three experiments. Embryo survival was normalized with respect to the % survival observed in parallel experiments carried out with the w1118 strain used for transgenesis.
Total RNA Isolation
Total RNA was extracted from WT (+/+) and heterozygous MedeaM-DAH/+ or MedeaM-O-fut1/+ females stage 14 oocytes and 0–1 hr staged embryos. Samples were flash frozen at specific time points and total RNA was extracted using the Ambion mirVana mRNA isolation kit (Ambion/Applied Biosystems, Austin, TX). All sample collections were staged in the incubator at a relative humidity 60%, 25°C with a 12 hour/12 hour light cycle until the desired time point was reached, then immediately flash frozen. Following the extraction, RNA was treated with Ambion Turbo DNase (Ambion/Applied Biosystems, Austin, TX). The quality of RNA was assessed using the Bioanalyzer 2100 (Aglient Technologies, Santa Clara, CA) and the NanoDrop 1000 UV-VIS spectrophotometer (NanoDrop Technologies/Thermo Scientific, Wilmington, DE). RNA was then prepared for sequencing using the Illumina mRNA-Seq Sample Preparation Kit (Illumina San Diego, CA).
Small RNA Extraction, Cloning, and Sequencing
Twenty micrograms of total RNA from each time point was size fractionated in 15% TBE-Urea polyacrylamide gels. For both the s14-oocytes and 0–1 hr embryo time points, an 18–32 nt band was excised and sequenced. Ethanol precipitated RNA was ligated to an HPLC purified 3′ linker using T4 RNA ligase (Ambion/Applied Biosystems, Austin, TX) and then purified in a 15% TBE-Urea polyacrylamide gel. Ligation products were recovered by high-salt elution following electrophoresis. A HPLC purified 5′RNA linker was ligated to the RNA using T4-RNA ligase, and the RNA was purified in a 15% TBE-Urea polyacrylamide gel. Ligation products were recovered by high-salt elution following electrophoresis. Reverse transcription using SSIII (Invitrogen, Carlsbad, CA) was performed, and cDNA was amplified using Phusion polymerase (Finnzymes Oy, Espoo, Finland). Amplified cDNA libraries were purified using a 2% agarose gel prior to sequencing using the Illumina Genome Analyzer II system. Linker and primer sequences are indicated below: 3′ Ligation linker: Modban (AMP-5′p=5′pCTGTAGGCACCATCAATdideoxyC-3′); 5′ Ligation Solexa Linker (5′-rArCrArCrUrCrUrUrUrCrCrCrUrArCrArCrGrArCrGrC rUrCrUrUrCrCrGrArUrC-3′); 3′ RT primer: BanOne (5′-ATTGATGGTGCCTACAG-3′); 5′ PCR primer Sol_5_SBS3 (5′-AATGATACGGCGACCACCGAACACTCTTTCCCT ACACGACG-3′); 3′ PCR primer Sol_3_Modban (5′-CAAGCAGAAGACGGCATACGATTGATGGTGCCT ACAG-3′).
Poly(A+) Read Alignment and Quantification
The poly(A+) reads were processed and aligned to a reference index we generated for the Drosophila Melanogaster genome (obtained from www.flybase.org), usingTopHat v1.4.1 (55). Reads were aligned using both default parameters. TopHat incorporates the Bowtie v0.12.7 algorithm to perform the alignment (56). The aligned read files were processed by Cufflinks v0.9.3 (57) Cufflinks uses the normalized RNA-Seq fragment counts to measure the relative abundances of transcripts. The unit of measurement is Fragments Per Kilobase of exon per Million fragments mapped (FPKM).
Small RNA Read Alignment and Quantification
The 5′ and 3′ adapter sequences for the small RNA reads were removed using custom pearl scripts requiring a minimal match to the adapter sequence of 6bp, and a minimal size of 18bp and maximum size of 32bp (sequences for the adapters are noted above. The trimmed sequences were aligned to the Drosophila Melanogaster reference genome using bowtie v0.12.7, allowing no mismatches and unlimited alignments/read. We determined the small RNA abundance using custom in house pearl scripts in which we quantified the read count per million of mapped reads (RPM) for each genomic position. Each endogenous miRNA was determined by merging the genomic coordinates from miRbase v19 with our genomic coordinates using custom perl scripts. Levels of all endogenous miRNAs were quantified in terms of RPM and ranked according to expression level. Levels of synthetic miRNAs were quantified in terms of RPM, and in terms of rank and rank percentile with respect to the subset of endogenous miRNAs.
Fitness cost calculation
We modeled Medea dynamics assuming random mating, discrete generations and 100% toxin efficiency. The proportions of the kth generation that are WT, heterozygous and homozygous for Medea are denoted by uk, vk and wk, respectively. Considering all possible mating pairs, and taking into account that WT offspring of heterozygous mothers are unviable, the genotypes of embryos in the next generation are described by the ratio ûk+1: v̂k+1: ŵk+1, where,
| (1) |
| (2) |
| (3) |
Normalizing these ratios and taking into account fitness costs, the genotype frequencies in the next generation are given by:
| (4) |
| (5) |
| (6) |
Here, s and hs represent the fitness costs associated with being homozygous or heterozygous for the element. For additive fitness costs, h = 0.5, and for dominant fitness costs, h = 1. Wk+1 is a normalizing term given by,
| (7) |
The likelihood of the data was calculated by assuming a binomial distribution of WT and red-eyed individuals, and by using the model predictions to generate expected proportions for each fitness cost, i.e. by calculating the log-likelihood,
| (8) |
Here, Ri,k and WTi,k are the number of red-eyed and WT individuals at generation k in experiment i, and the expected genotype frequencies are fitness cost-dependent. The best estimate of the fitness cost is that having the highest log-likelihood. 95% confidence intervals were estimated using a Markov Chain Monte Carlo sampling procedure.
Modeling a Medea-dependent population crash
Population frequency model
Releases of Medea individuals are likely to be all-male, since male mosquitoes don’t bite and male agricultural pests don’t lay eggs. Additionally, the external cues envisaged may induce a gender-specific fitness cost. For this reason, we consider a model allowing for different genotype frequencies among males and females. We denote the proportion of the kth generation that are females having the genotypes mm, Mm and MM by uf,k, vf,k and wf,k, respectively. The corresponding proportions for males are um,k, vm,k and wm,k. By considering all possible mating pairs, the genotype frequencies in the next generation are given by,
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
Here, sf and hfsf represent the fitness costs associated with being homozygous or heterozygous for the Medea element in females, sm and hmsm represent the corresponding fitness costs in males, and σk represents the proportion of embryos that survive to maturity. This is given by,
| (15) |
Using these difference equations, the equilibria, thresholds and time-series dynamics of the Medea element can be calculated. From previous analyses (7) (12), we know that a Medea element released into a population above a certain fitness-dependent threshold will spread to transgene fixation, meaning that all individuals will eventually be either heterozygous or homozygous for the construct. The threshold is particularly difficult to derive, but is 0 for a fitness cost of 0 and increases monotonically with fitness cost magnitude.
Discrete population model
A stochastic model can be used to theoretically explore the idea of using Medea to induce a population crash because, in this way, discrete populations and the concept of 0 population size can be considered. Density-dependence is also an important consideration because, at low population sizes, larval competition is reduced and a single female can produce more offspring that survive to adulthood. We adapt a general density-dependent model (58) of the form,
| (16) |
Here, Nk represents the absolute population size at generation k, R0 represents the average number of offspring produced per insect that survive to adulthood in a density-independent population (or the average number of female offspring that each female gives birth to), and α represents the strength of density-dependence. This last term is related to the carrying capacity, K, of the habitat by,
| (17) |
For agricultural pests under field conditions, R0 typically ranges between 3 and 11; but could be much higher in some populations (50) (59). We assume a default value of R0 = 7, and a carrying capacity of 10,000.
We adapt this model to account for the fact that the number of offspring in the next generation is determined by the number of adult females – an important consideration as we consider male-only releases of Medea homozygotes, and the external cue we envisage causes female-specific lethality. We also adapt the model to account for the fact that the maternal toxin is early-acting, leading to offspring lethality prior to larval competition, and hence unviable offspring do not contribute to density-dependence. On the other hand, we assume that fitness costs associated with the construct are late-acting, manifesting themselves at the adult stage and hence not reducing the strength of density-dependence. Similar assumptions are made in models of other genetic population suppression systems (60).
Taking these considerations into account, the total expected population size at generation k+1 is given by,
| (18) |
Here, Nf,k represents the female population size at generation k, and 2R0Nf,k represents the number of male and female offspring that females give birth to, neglecting offspring lethality and density-dependence. The population size that is subject to density-dependence is given by 2R0Nf,kσl,k, where σl,k represents the proportion of offspring that survive to the larval stage. Finally, σk represents the proportion of offspring that survive to mate, and is given in equation 15. It is worth noting that, for an entirely wild-type population, we recover the general density-dependent model in equation 16 since 2Nf,k = Nk and σk = σl,k = 1.
With this density-dependent framework in place, we integrate the population genetics of the Medea element into the model. At generation k, we denote the number of females having genotypes mm, Mm and MM at generation k by Uf,k, Vf,k and Wf,k, respectively. The corresponding numbers for males are Um,k, Vm,k and Wm,k. This framework allows us to express the female population size at generation k by,
| (19) |
and the total population size at generation k by,
| (20) |
The genotype frequencies at generation k are therefore,
| (21) |
Equations 1–7 then describe the expected genotype frequencies in the next generation, and the proportion of offspring that survive to the larval stage is essentially equation 15 without fitness costs, i.e.
| (22) |
Having calculated the expected total population size at generation k+1, using equation 18, the expected numbers of individuals having each genotype at generation k+1 is given by,
| (23) |
| (24) |
Stochastic effects are modeled by sampling the number of individuals having each genotype from a Poisson distribution with a mean equal to the expected number of individuals having this genotype. This has the effect of discretizing the population, which is appropriate for modeling a population crash. Illustrating this calculation for Mm females, an absolute population size of Vf,k+1 can be calculated from an expected population size of Ȳf,k+1 by sampling from the Poisson distribution,
| (25) |
This procedure can be repeated for all six genotypes, so that the total population size at generation k+1 becomes,
| (26) |
Considering a release of Xm,0 males homozygous for the Medea element at generation 0, the initial condition is given by,
| (27) |
| (28) |
Subsequent releases of Xm,k male homozygotes at generation k, can be modeled by making the following substitution,
| (29) |
We consider a single, all-male release representing 50% of the population at the time of release, i.e. Xm,0 = K.
Supplementary Material
MicroRNA expression profiles for s14 ovary and 0–1 hr embryo.
The genomic location for each detected annotated miRNAs (guide and passenger if both were detected) is provided along with the read-count and the normalized expression value in read count per million mapped reads (RPM).
Top 20 miRNAs in s14 ovary
The top 20 miRNAs with respect to the WT s14 ovary sample are listed. The Chromosome start and stop site, strand, read count and RPM are all shown for each ovary sample.
Top 20 miRNAs in 0–1 hr embryo.
The top 20 miRNAs with respect to the WT 0–1 hr-embryo sample are listed. The Chromosome start and stop site, strand, read count and RPM are all shown for each embryo sample.
Acknowledgments
We would like to thank Lorain Schaeffer and Vijaya Kumar for help with library preparations and sequencing. This work was supported by grants to BAH from the NIH (DP10D003878), to JMM from the Medical Research Council, UK, and in part by a grant from the Foundation for the NIH through the Grand Challenges in Global Health initiative (Anthony A. James, PI, UC Irvine).
References
- 1.Nicholson GM. Fighting the global pest problem: preface to the special Toxicon issue on insecticidal toxins and their potential for insect pest control. Toxicon. 2007;49:413–422. doi: 10.1016/j.toxicon.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 2.Schmid-Hempel P. Evolutionary ecology of insect immune defenses. Annu Rev Entomol. 2005;50:529–551. doi: 10.1146/annurev.ento.50.071803.130420. [DOI] [PubMed] [Google Scholar]
- 3.Tripet F, Aboagye-Antwi F, Hurd H. Ecological immunology of mosquito-malaria interactions. Trends Parasitol. 2008;24:219–227. doi: 10.1016/j.pt.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lambrechts L, Koella JC, Boete C. Can transgenic mosquitoes afford the fitness cost? Trends Parasitol. 2008;24:4–7. doi: 10.1016/j.pt.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Braig HRaYG. The spread of genetic constructs in natural insect populations. In: Letourneau DKaBBE., editor. Genetically Engineered Organisms: Assessing Environmental and Human Health Effects. CRC Press; 2001. pp. 251–314. [Google Scholar]
- 6.Sinkins SP, Gould F. Gene drive systems for insect disease vectors. Nat Rev Genet. 2006;7:427–435. doi: 10.1038/nrg1870. [DOI] [PubMed] [Google Scholar]
- 7.Chen CH, Huang H, Ward CM, Su JT, Schaeffer LV, Guo M, Hay BA. A synthetic maternal-effect selfish genetic element drives population replacement in Drosophila. Science. 2007;316:597–600. doi: 10.1126/science.1138595. [DOI] [PubMed] [Google Scholar]
- 8.Beeman RW, Friesen KS, Denell RE. Maternal-effect selfish genes in flour beetles. Science. 1992;256:89–92. doi: 10.1126/science.1566060. [DOI] [PubMed] [Google Scholar]
- 9.Hastings IM. Selfish DNA as a method of pest control. Philos Trans R Soc Lond B Biol Sci. 1994;344:313–324. doi: 10.1098/rstb.1994.0069. [DOI] [PubMed] [Google Scholar]
- 10.Wade MJ, Beeman RW. The population dynamics of maternal-effect selfish genes. Genetics. 1994;138:1309–1314. doi: 10.1093/genetics/138.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith NG. The dynamics of maternal-effect selfish genetic elements. J Theor Biol. 1998;191:173–180. doi: 10.1006/jtbi.1997.0579. [DOI] [PubMed] [Google Scholar]
- 12.Ward CM, Su JT, Huang Y, Lloyd AL, Gould F, Hay BA. Medea selfish genetic elements as tools for altering traits of wild populations: a theoretical analysis. Evolution. 2011;65:1149–1162. doi: 10.1111/j.1558-5646.2010.01186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maxton-Kuchenmeister J, Handel K, Schmidt-Ott U, Roth S, JackleJackle H. Toll homologue expression in the beetle tribolium suggests a different mode of dorsoventral patterning than in drosophila embryos. Mech Dev. 1999;83:107–114. doi: 10.1016/s0925-4773(99)00041-6. [DOI] [PubMed] [Google Scholar]
- 14.Zhang CX, Lee MP, Chen AD, Brown SD, Hsieh T. Isolation and characterization of a Drosophila gene essential for early embryonic development and formation of cortical cleavage furrows. J Cell Biol. 1996;134:923–934. doi: 10.1083/jcb.134.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin H, Tan S, Hermanowski J, Bohm S, Pacheco S, McCauley JM, Greener MJ, Hinits Y, Hughes SM, Sharpe PT, Roberts RG. The dystrotelin, dystrophin and dystrobrevin superfamily: new paralogues and old isoforms. BMC Genomics. 2007;8:19. doi: 10.1186/1471-2164-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasamura T, Sasaki N, Miyashita F, Nakao S, Ishikawa HO, Ito M, Kitagawa M, Harigaya K, Spana E, Bilder D, Perrimon N, Matsuno K. neurotic, a novel maternal neurogenic gene, encodes an O-fucosyltransferase that is essential for Notch-Delta interactions. Development. 2003;130:4785–4795. doi: 10.1242/dev.00679. [DOI] [PubMed] [Google Scholar]
- 17.Okajima T, Irvine KD. Regulation of notch signaling by o-linked fucose. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- 18.Okajima T, Xu A, Lei L, Irvine KD. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science. 2005;307:1599–1603. doi: 10.1126/science.1108995. [DOI] [PubMed] [Google Scholar]
- 19.Okajima T, Reddy B, Matsuda T, Irvine KD. Contributions of chaperone and glycosyltransferase activities of O-fucosyltransferase 1 to Notch signaling. BMC biology. 2008;6:1. doi: 10.1186/1741-7007-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci U S A. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schejter ED, Wieschaus E. bottleneck acts as a regulator of the microfilament network governing cellularization of the Drosophila embryo. Cell. 1993;75:373–385. doi: 10.1016/0092-8674(93)80078-s. [DOI] [PubMed] [Google Scholar]
- 22.Shelton CA, Wasserman SA. pelle encodes a protein kinase required to establish dorsoventral polarity in the Drosophila embryo. Cell. 1993;72:515–525. doi: 10.1016/0092-8674(93)90071-w. [DOI] [PubMed] [Google Scholar]
- 23.Letsou A, Alexander S, Orth K, Wasserman SA. Genetic and molecular characterization of tube, a Drosophila gene maternally required for embryonic dorsoventral polarity. Proc Natl Acad Sci U S A. 1991;88:810–814. doi: 10.1073/pnas.88.3.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michellod MA, Forquignon F, Santamaria P, Randsholt NB. Differential requirements for the neurogenic gene almondex during Drosophila melanogaster development. Genesis. 2003;37:113–122. doi: 10.1002/gene.10233. [DOI] [PubMed] [Google Scholar]
- 25.Johnson K, Knust E, Skaer H. bloated tubules (blot) encodes a Drosophila member of the neurotransmitter transporter family required for organisation of the apical cytocortex. Dev Biol. 1999;212:440–454. doi: 10.1006/dbio.1999.9351. [DOI] [PubMed] [Google Scholar]
- 26.Parks S, Wieschaus E. The Drosophila gastrulation gene concertina encodes a G alpha-like protein. Cell. 1991;64:447–458. doi: 10.1016/0092-8674(91)90652-f. [DOI] [PubMed] [Google Scholar]
- 27.Field CM, Coughlin M, Doberstein S, Marty T, Sullivan W. Characterization of anillin mutants reveals essential roles in septin localization and plasma membrane integrity. Development. 2005;132:2849–2860. doi: 10.1242/dev.01843. [DOI] [PubMed] [Google Scholar]
- 28.Sprenger F, Stevens LM, Nusslein-Volhard C. The Drosophila gene torso encodes a putative receptor tyrosine kinase. Nature. 1989;338:478–483. doi: 10.1038/338478a0. [DOI] [PubMed] [Google Scholar]
- 29.Paroush Z, Finley RL, Jr, Kidd T, Wainwright SM, Ingham PW, Brent R, Ish-Horowicz D. Groucho is required for Drosophila neurogenesis, segmentation, and sex determination and interacts directly with hairy-related bHLH proteins. Cell. 1994;79:805–815. doi: 10.1016/0092-8674(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 30.Casanova J, Furriols M, McCormick CA, Struhl G. Similarities between trunk and spatzle, putative extracellular ligands specifying body pattern in Drosophila. Genes Dev. 1995;9:2539–2544. doi: 10.1101/gad.9.20.2539. [DOI] [PubMed] [Google Scholar]
- 31.Casali A, Casanova J. The spatial control of Torso RTK activation: a C-terminal fragment of the Trunk protein acts as a signal for Torso receptor in the Drosophila embryo. Development. 2001;128:1709–1715. doi: 10.1242/dev.128.9.1709. [DOI] [PubMed] [Google Scholar]
- 32.Ross S. A first course in probability. 8. Prentice Hall; 2009. [Google Scholar]
- 33.Moussian B, Roth S. Dorsoventral axis formation in the Drosophila embryo--shaping and transducing a morphogen gradient. Curr Biol. 2005;15:R887–899. doi: 10.1016/j.cub.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 34.Mazumdar A, Mazumdar M. How one becomes many: blastoderm cellularization in Drosophila melanogaster. Bioessays. 2002;24:1012–1022. doi: 10.1002/bies.10184. [DOI] [PubMed] [Google Scholar]
- 35.Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012 doi: 10.1038/nrg3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 37.Benoit P, Papin C, Kwak JE, Wickens M, Simonelig M. PAP- and GLD-2-type poly(A) polymerases are required sequentially in cytoplasmic polyadenylation and oogenesis in Drosophila. Development. 2008;135:1969–1979. doi: 10.1242/dev.021444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, Zhao Y, Zamore PD, Hannon GJ, Marra MA, Weng Z, Perrimon N, Lai EC. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res. 2011;21:203–215. doi: 10.1101/gr.116657.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ameres SL, Horwich MD, Hung JH, Xu J, Ghildiyal M, Weng Z, Zamore PD. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ameres SL, Hung JH, Xu J, Weng Z, Zamore PD. Target RNA-directed tailing and trimming purifies the sorting of endo-siRNAs between the two Drosophila Argonaute proteins. RNA. 2011;17:54–63. doi: 10.1261/rna.2498411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 42.Okamura K, Liu N, Lai EC. Distinct mechanisms for microRNA strand selection by Drosophila Argonautes. Mol Cell. 2009;36:431–444. doi: 10.1016/j.molcel.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forstemann K, Horwich MD, Wee L, Tomari Y, Zamore PD. Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell. 2007;130:287–297. doi: 10.1016/j.cell.2007.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ballarino M, Pagano F, Girardi E, Morlando M, Cacchiarelli D, Marchioni M, Proudfoot NJ, Bozzoni I. Coupled RNA processing and transcription of intergenic primary microRNAs. Mol Cell Biol. 2009;29:5632–5638. doi: 10.1128/MCB.00664-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009;461:546–549. doi: 10.1038/nature08349. [DOI] [PubMed] [Google Scholar]
- 47.Clark MS, Worland MR. How insects survive the cold: molecular mechanisms-a review. Journal of comparative physiology B, Biochemical, systemic, and environmental physiology. 2008;178:917–933. doi: 10.1007/s00360-008-0286-4. [DOI] [PubMed] [Google Scholar]
- 48.Hahn DA, Denlinger DL. Energetics of insect diapause. Annu Rev Entomol. 2011;56:103–121. doi: 10.1146/annurev-ento-112408-085436. [DOI] [PubMed] [Google Scholar]
- 49.Clements AN. The biology of mosquitoes. Vol 1, Development, nutrition and reproduction. Chapman & Hall; 1992. [Google Scholar]
- 50.Dyck VA, Hendrichs J, Robinson AS, editors. Sterile Insect Technique: Principles and Practice in Area-Wide Integrated Pest Management. Springer; Dordrecht, The Netherlands: 2005. [Google Scholar]
- 51.Fu G, Condon KC, Epton MJ, Gong P, Jin L, Condon GC, Morrison NI, Dafa’alla TH, Alphey L. Female-specific insect lethality engineered using alternative splicing. Nat Biotechnol. 2007;25:353–357. doi: 10.1038/nbt1283. [DOI] [PubMed] [Google Scholar]
- 52.Fu G, Lees RS, Nimmo D, Aw D, Jin L, Gray P, Berendonk TU, White-Cooper H, Scaife S, Kim Phuc H, Marinotti O, Jasinskiene N, James AA, Alphey L. Female-specific flightless phenotype for mosquito control. Proc Natl Acad Sci U S A. 2010;107:4550–4554. doi: 10.1073/pnas.1000251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schetelig MF, Handler AM. A transgenic embryonic sexing system for Anastrepha suspensa (Diptera: Tephritidae) Insect Biochem Mol Biol. 2012 doi: 10.1016/j.ibmb.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 54.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 55.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA- Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10 doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bellows TS. The descriptive properties of some models for density dependence. Journal of Animal Ecology. 1981;50:139–156. [Google Scholar]
- 59.Dye C. Models for the population dynamics of the yellow fever mosquito, Aedes aegypti. J Anim Ecol. 1984;53:247–268. [Google Scholar]
- 60.Alphey N, Bonsall MB, Alphey L. Modeling resistance to genetic control of insects. J Theor Biol. 2011;270:42–55. doi: 10.1016/j.jtbi.2010.11.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MicroRNA expression profiles for s14 ovary and 0–1 hr embryo.
The genomic location for each detected annotated miRNAs (guide and passenger if both were detected) is provided along with the read-count and the normalized expression value in read count per million mapped reads (RPM).
Top 20 miRNAs in s14 ovary
The top 20 miRNAs with respect to the WT s14 ovary sample are listed. The Chromosome start and stop site, strand, read count and RPM are all shown for each ovary sample.
Top 20 miRNAs in 0–1 hr embryo.
The top 20 miRNAs with respect to the WT 0–1 hr-embryo sample are listed. The Chromosome start and stop site, strand, read count and RPM are all shown for each embryo sample.