

Abstract
Replication-dependent chromosomal breakage suggests that replication forks occasionally run into nicks in template DNA and collapse, generating double-strand ends. To model replication fork collapse in vivo, I constructed phage λ chromosomes carrying the nicking site of M13 bacteriophage and infected with these substrates Escherichia coli cells, producing M13 nicking enzyme. I detected double-strand breaks at the nicking sites in λ DNA purified from these cells. The double-strand breakage depends on (i) the presence of the nicking site; (ii) the production of the nicking enzyme; and (iii) replication of the nick-containing chromosome. Replication fork collapse at nicks in template DNA explains diverse phenomena, including eukaryotic cell killing by DNA topoisomerase inhibitors and inviability of recombination-deficient vertebrate cell lines.
Keywords: replication fork collapse, single-strand DNA breaks, double-strand DNA breaks
DNA-damaging treatments are more lethal if they are administered during chromosomal replication (1–4), suggesting that encounters of replication forks with DNA lesions aggravate these lesions. Most types of one-strand DNA damage are removed by excision repair, which proceeds via a common intermediate, single-strand DNA interruption (reviewed in ref. 5). What happens when, during an ongoing excision repair, a replication fork runs into a single-strand interruption in template DNA? There may be a natural safeguard mechanism preventing such an eventuality, because replication fork progress appears to depend on the presence of negative supercoiling in the DNA segment to be replicated. Indeed, partial loss of DNA supercoiling because of coumermycin inhibition of DNA gyrase activity in Escherichia coli parallels severe inhibition of DNA synthesis (6). Because the nick in the downstream template relieves the negative supercoiling in front of the replication fork, the loss of supercoiling should inhibit fork progress.
Quite a different idea is that a replication fork reaches the nick and collapses, producing a double-strand end (Fig. 1 A–C) (2, 7–10). If two replication forks converge on a single nick, a double-strand break will form (Fig. 1 B–D). This idea is based on two observations: (i) replication-induced chromosomal fragmentation (double-strand breakage) in mutants or in conditions that elevate the number of single-strand interruptions in DNA (11–14) and (ii) preferential degradation of the newly synthesized DNA in these conditions (2, 15). The most economical interpretation of these observations is that persistent nicks in replicating DNA in vivo are converted into double-strand ends by replication forks (reviewed in ref. 16). The relevance of this process to the formation of lethal DNA damage is underscored by the uncontrollable fragmentation of chromosomes in Rad51-deficient vertebrate cell lines (17, 18), which apparently underlies the inviability of rad51 mutant mice (17).
Figure 1.

The idea of replication fork collapse at a single-strand interruption in template DNA. (A) A DNA segment with a long-lived nick. (B) A replication fork is approaching the nick. (C) The replication fork has reached the nick and collapsed; another replication fork is approaching the remaining single-strand interruption from the opposite direction. (D) The second replication fork has reached the interruption and collapsed; as a result, one of the daughter chromosomes has a double-strand break. (E) An alternative scenario of double-strand breakage at replication forks: replication fork “explosion” at the nick.
The idea of replication fork collapse at single-strand interruptions is not new, but its experimental testing was delayed by difficulties with induction of persistent and site-specific single-strand scissions in replicating chromosomes in vivo. A class of DNA-nicking enzymes that collaborate with the DNA replication machinery comprises proteins initiating rolling-circle DNA replication in conjugative plasmids and filamentous bacteriophages: gpII of phages M13, f1 or fd, or TraI of the F factor (19). Intriguingly, there is genetic evidence that the transfer origin (the nicking site) of F factor is a site of double-strand breakage in vivo (7, 20). I decided to use a cloned M13 nickase (gpII) to deliver controlled nicks in substrate chromosomes replicating in E. coli cells.
Materials and Methods
Media, Growth Conditions, and General Methods.
Cells were grown in LB broth or on LB plates (21). When cells were carrying plasmids, the media were supplemented with 100 μg/ml of ampicillin. For the wild-type gpII induction off pCL475, the temperature shift-up from 28 to 42°C was used; for the mutant gpII induction off pK125 or pK133, 0.5 mM isopropyl β-d-thiogalactoside (IPTG) was used.
Small-scale and preparative plasmid DNA isolation was done as described (22). λ phages were isolated from plate lysates by two rounds of polyethylene glycol-3400 precipitation (21). TM buffer is 10 mM Tris⋅HCl, pH 8.0/10 mM MgSO4; TE buffer is 10 mM Tris⋅HCl, pH 8.0/1 mM EDTA.
Bacterial Strains.
Bacterial strains are E. coli K-12; they are derivatives of AB1157 (23), except AK98 and AK99, which are derivatives of R594 (23). recBCD mutants were confirmed by their ability to plate T4 2 mutant phage (=ExoV−) (24); rep mutants were confirmed by their inability to plate M13 phage (25).
recB270 recC271 is SK129 (26); recB268 is N2101 (27); BT125 and WA800 are described (28), as well as JJC213 and JJC735 (29). Δrep∷kan recD1011 is AK92 (BT125 × P1 JJC213, selection for kanamycin resistance, inability to support M13 replication). Δrep∷cam recD1011 is AK93 (BT125 × P1 JJC735, selection for chloramphenicol resistance, inability to support M13 replication). Δrep∷cam recB270 recC271 is AK95 (SK129 × P1 JJC735, selection for chloramphenicol resistance, inability to support M13 replication, inviability at 42°C). AK98 is Su− Δrep∷cam (R594 × P1 JJC735, selection for chloramphenicol resistance, inability to support M13 replication). Su− Δrep∷cam recC1010 is AK99 (AK98 × P1 WA800, selection for tetracycline resistance, T4 2 permissivity).
Plasmids.
pK107 is a pBR322 derivative carrying the λ cI and PaeR7I restriction-modification system (XhoI specificity) (30). pCL475 is an M13 wild-type gpII-producing plasmid (31). pK125 carries G73A mutant gpII under the control of a strong tacI promoter and lacIq gene; gpII synthesis is, therefore, IPTG-inducible. pK125 was constructed by combining three fragments: the 1,580-bp-long EcoRI-EcoRI from pDG117IIA.H11 carrying PtacI-gpII G73A (32), the 1,260-bp-long EcoRI-HindIII from pAH3 carrying lacIq (32), and the 4,328-bp-long HindIII-EcoRI backbone of pBR322. pK133 is a shrink of pK125, from which the 882-bp-long NgoMI fragment of nonessential DNA has been deleted.
Bacteriophages.
λ phages MMS2660 and MMS2663 are described (33). Because they carry the Psus80 mutation, they can replicate in AB1157 (SuII) background, but cannot replicate in R594 (Su−) background. The sequence of the nicking site-containing oligo corresponds to “complex II” of ref. 34:
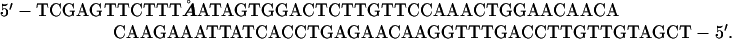 |
The bold Å in the top strand marks the 5′-end of the nick. Note that, when inserted into a XhoI-site as written, this sequence will restore the XhoI site on its left. Insertion of this oligo as written into the natural XhoI site of MMS2660 gives λAK2 (nick in the top strand), whereas its insertion in the opposite orientation gives λAK3 (nick in the bottom strand). Likewise, insertion of this oligo, as written, into the SstII∷XhoI site of MMS2663 gives λAK4 (nick in the top strand), whereas its insertion in the opposite orientation gives λAK5 (nick in the bottom strand).
Isolation of Phage λ DNA.
For isolation of big quantities of λ DNA, concentrated phage stocks are prepared from 8–10 plates by the standard technique (see above), but with pure agarose instead of agar. To 450 μl of an agarose λ stock, 50 μl of 10% SDS are added, and the aqueous phase is extracted once with 500 μl of phenol, once with 500 μl of phenol/chloroform (1:1), and once with 500 μl of chloroform. Then 20 μl of 4 M KCl is added, and the phage DNA is precipitated with 1 ml of ethanol and dissolved in 200 μl of TE. After addition of 300 μl of 6 M LiCl, the tube is vortexed vigorously and, after chilling on ice for 15 min and spinning for 2 min in an Eppendorf centrifuge, the supernatant is transferred into a fresh tube. Nucleic acids are precipitated with 1 ml of ethanol, dissolved in 500 μl of TE, and, after addition of 20 μl of 4 M KCl, reprecipitated with 1 ml of ethanol and dissolved in 500 μl of TE.
The Standard Cross.
A 2-ml LB culture, supplemented with 100 μg/ml of ampicillin, is inoculated with a fresh colony and incubated with shaking at 28°C overnight. The next morning, 200–400 μl of the saturated culture is used to inoculate 20 ml of LB, supplemented with 100 μg/ml of ampicillin; the diluted culture is grown with shaking at 28°C to mid-logarithmic phase. Cells are collected by centrifugation, resuspended in 1 ml of TM, and counted under the microscope in a Petroff–Houser chamber. In a 10-ml glass tube on ice, 4 × 108 cells in 200 μl of TM are mixed with the appropriate quantity of phage in 50 μl of TM to produce the indicated multiplicity of infection (moi); the adsorption mixture is vortexed gently and chilled on ice for 15 min. The glass tube is placed at 37°C for 3 min to effect phage injection, and the whole adsorption mixture is transferred into 2 ml of prewarmed LB culture, supplemented or not with 0.5 mM IPTG. Incubation is at the indicated temperatures. At the indicated time points, a 1.5 ml aliquot is taken for DNA isolation.
Preparation of Total DNA from E. coli.
The cells are pelleted in a microcentrifuge, the supernatant is removed with a pipet, and the cells are resuspended in 50 μl of 30% sucrose in TE buffer. The cells are lysed by adding 350 μl of 2% SDS in TE, mixing by inversion and incubating at 70°C for 5 min. To degrade the protein, 20 μl of 10 mg/ml Proteinase K is added, and the lysates are incubated for 15 min at 70°C. After chilling on ice, the lysates are extracted once with 200 μl of phenol, once with 200 μl of phenol/chloroform (1:1), and once with 300 μl of chloroform. The aqueous phase is transferred into a fresh tube, and the nucleic acids are precipitated, after thorough mixing with 20 μl of 4 M KCl and 1 ml of ethanol, by centrifugation in a microcentrifuge. The supernatant is discarded, the pellet is dissolved in 500 μl of TE, and nucleic acids are reprecipitated as above. The pellet is eventually dissolved in 200 μl of TE; 1–50 μl of this solution is used for subsequent analysis.
DNA Analysis.
DNA is cut with specified restriction endonucleases, separated in 0.7% agarose gels, either in 40 mM Tris⋅acetate, pH 8.5/2 mM EDTA (neutral gels) or in 30 mM NaOH, 1 mM EDTA (denaturing gels), vacuum transferred to Zeta-Bind nylon membrane (Cuno), and probed with λ-specific probes, as described (33). Probe 1 covers both sides of the XhoI site in phages MMS2660, λAK2, and λAK3, and is λ NruI-NheI fragment (32409–34679). Probe 2 covers both sides of the SstII∷XhoI site in phages MMS2663, λAK4, and λAK5, and is λ HincII fragment (39836–40942).
Results
The Rational and Experimental Systems.
To test ideas about replication fork encounters with persistent single-strand breaks in template DNA in vivo, one needs a substrate chromosome (i) that can be efficiently introduced into cells; (ii) that can be nicked at will at a specific location once inside the cell; and (iii) whose replication can be regulated in vivo. Because phage λ already proved itself in this “in vivo biochemistry” approach (30, 33), I have constructed λ phages carrying the minimal nicking site of the filamentous bacteriophage M13. Although the “functional origin” of M13 covers some 140 bp, increased production of the cognate nicking enzyme reduces the sequence requirements of the reaction to 40 bp (35). I inserted this 40-bp double-stranded fragment (see Materials and Methods) in either orientation at the unique XhoI site in the λ chromosome (Fig. 2A Upper), generating three phages (Fig. 2B): the original control phage without the nicking site, and the two experimental phages, carrying the nicking site in either the top or the bottom strand. Because the phages with the nicking site retain the XhoI site, I used in vivo cutting with XhoI (injecting the phages into XhoI-producing cells) to generate molecular weight markers for the double-strand break at the nicking site (Figs. 3B and 4B, lanes a–c).
Figure 2.

Experimental substrates. (A) The two λ phages used to construct the nicking site-containing phages. The XhoI-sites at which the nicking sites were inserted and the restriction sites used in Southern analysis are shown, together with their coordinates on wild-type λ chromosome and the sizes of the generated fragments. (Upper) MMS2660, the progenitor of λAK2 (nick in the top strand) and λAK3 (nick in the bottom strand). These three phages are used in Figs. 3 and 4. (Lower) MMS2663, the progenitor of λAK4 (nick in the top strand) and λAK5 (nick in the bottom strand). These three phages are used in Fig. 5. (B) A set of three phage substrates (either MMS2660, λAK2, and λAK3 or MMS2663, λAK4, and λAK5) run in parallel in one experiment. All three phages within a set have a unique XhoI site at approximately the same location. The control phage (marked “—”) has nothing else; the middle phage carries the nicking site in the top strand and is therefore marked “t;” the bottom phage carries the nicking site in the bottom strand and is therefore marked “b.”
Figure 3.

The interplay between λ and M13 DNA replication. (A) Sigma replication in Rep+ cells is predicted to generate the “half-break” pattern at the nicking sites. 3′-ends are indicated by arrows. (A) The substrate chromosomes carrying nicking sites at the same location, either in the top or in the bottom strand. (B) The nicking sites are nicked by gpII (open circles attached to the 5′-sides of the nicks). (C) The Rep helicase unwinds the 5′-ends of the nicks, attracting replisomes to the replication fork structures. (D) Sigma replication from the nicks. (E) Restriction cutting in vitro (at sites indicated by vertical lines in D) reveals the “half-break” pattern. (B) Interference between λ DNA replication and nicking by the wild-type nicking enzyme. Phages MMS2660, λAK2, or λAK3, indicated in the entry “substrate” as “—”, “t,” or “b,” respectively, were infected at moi = 6 into the strains described below; the cells were incubated as indicated, and the phage DNA was extracted and analyzed by restriction digestion with EcoRI and blot hybridization with probe 1. Molecular weight markers for double-strand break at the natural XhoI site were generated in vivo (lanes a–c). The strains and conditions are as follows: lanes a–c, recB268 pK107, 10 min at 28°C; lanes d–f, recB270 recC271 pCL475, 30 min at 42°C; lanes g–i, Δrep∷cam recB270 recC271 pCL475, 30 min at 42°C; lanes j–l, recB270 recC271 pCL475, incubated for 30 min at 42°C before phage injection and 60 min at 28°C after phage injection; lanes m–o, Δrep∷cam recB270 recC271 pCL475, incubated for 30 min at 42°C before phage injection and 60 min at 28°C after phage injection.
Figure 4.

Replication-induced double-strand breaks at persistent nicks at the natural XhoI-site. (A) Replication of a circular λ chromosome containing a nick is predicted to generate one circular chromosome and one linear chromosome with a double-strand break at the nick. Nicking enzyme is shown as an open circle attached to the 5′-end of the nick; 3′-ends are indicated by half-arrows, and RNA primers are marked by wavy lines. (A) Initiation of theta replication in the circular λ chromosome. (B) The nicking site is nicked while the theta replication continues. (C) The first replication fork runs into the nick and collapses, switching the chromosome to sigma replication. (D) The second replication fork reaches the single-strand interruption, resulting in a double-strand break in one of the replicated chromosomes. (B) Phages MMS2660, λAK2, or λAK3, indicated in the entry “substrate” as “—”, “t,” or “b,” respectively, were infected at moi = 10 into the strains described below; the cells were incubated at 37°C for 40 min (unless indicated otherwise), and the phage DNA was extracted and analyzed by restriction digestion with EcoRI and blot hybridization with probe 1 under both neutral (Upper gel) and denaturing (Lower gel) conditions. Molecular weight markers for double-strand break at the XhoI site were generated in vivo (lanes a–c). The strains and conditions are as follows: lanes a–c, recB268 pK107, incubated for 10 min at 37°C; lanes d–f, Δrep∷kan recD1011 pK125 + IPTG; lanes g–i, Δrep∷kan recD1011 pK125, no induction; lanes j–l, Su− Δrep∷cam recC1010 pK125 + IPTG. To compensate for the smaller amount of λ DNA because of the inhibition of λ replication, 25 times more total DNA has been loaded in the case of Su− samples (lanes j–l).
In the experiment reported in Fig. 3B, the wild-type gene for the nicking enzyme of the phage M13 is under the control of a temperature-sensitive λ repressor, so both the production of the nicking enzyme and λ replication are effected by temperature shift-up (31). When I wanted to produce the nicking enzyme without λ replication, cells were preinduced at 42°C for 30 min before phage injection but kept at 28°C after phage injection.
The M13 nicking enzyme is the only phage protein required to start rolling-circle DNA replication from the nicking site (36). The enzyme does this by attracting the host Rep helicase, which then unwinds the 5′-side of the nick and assembles a regular replication fork on the unwound intermediate (Fig. 3A) (37). In rep mutants, the nicking still occurs, but no rolling-circle DNA replication follows (25). Thus, in Rep+ strains, the nicking sites in λ were expected to stimulate rolling-circle DNA replication, producing a pattern of “half-breaks,” with a breakage product on one side of the nick only, and the complementary breakage product generated by the substrate with the nick in the other DNA strand (Fig. 3A). Indeed, the “half-break” pattern was observed in Rep+ cells and was especially clear when λ's own DNA replication was disallowed (Fig. 3B, compare lanes d–f with j–l). As expected, this half-break pattern, indicative of rolling-circle DNA replication, was completely suppressed in rep mutants (Fig. 3B, lanes g–i and m–o).
Therefore, allowing λ replication in rep mutants should reveal double-strand breakage because of replication fork encounters with nicks (Fig. 4A). However, no double-strand breakage was detected in this case (Fig. 3B, lanes g–i), suggesting either that replication forks never reach the nicks or that λ DNA replication interferes with the nicking. The nicking by the wild-type nicking enzyme requires DNA supercoiling (19); because replicating λ DNA loses supercoiling, the nicking reaction should be inhibited by λ's replication. And yet, for the purpose of this experiment, it was critical to have an efficient nicking of replicating chromosomes. To increase the efficiency of nicking of replicating λ substrates, I used G73A mutant nickase, which shows a reduced superhelicity requirement (32). To further increase sensitivity of the system, I conducted all subsequent experiments in rep mutant cells, thus preventing rolling-circle DNA replication from the nicking sites.
Replication-Dependent Double-Strand Breaks.
I found that, in the presence of the supercoiling-insensitive nickase, replication of λ DNA carrying the cognate nicking site causes double-strand breakage at the site (Fig. 4B Upper, lanes e and f). Similar patterns of double-strand breaks form with either orientation of the nicking site. Importantly, no double-strand breaks are observed in the parental phage lacking the nicking site (Fig. 4B Upper, lane d).
When the expression of the nicking enzyme is not induced, double-strand breaks do not form in replicating λ phages (Fig. 4B Upper, lanes g–i), indicating dependence of the double-strand breakage on the nickase. In vitro, the nicking enzyme of filamentous phages catalyzes a low level of double-strand breaks at the nicking site when Mg2+ is replaced with Mn2+ (19). Therefore, the double-strand breaks (Fig. 4B Upper, lanes e and f) might have been because of an infrequent double-strand cutting by the nickase in vivo. However, preventing λ DNA replication (in a Su− host) eliminates almost all double-strand breakage (Fig. 4B Upper, lanes k and l), although the nicks, as monitored by alkaline agarose gel electrophoresis, still form efficiently in these cells (Fig. 4B Lower, compare lanes e and f with k and l). The alkaline gel also shows no nicking when the nicking enzyme is not produced (Fig. 4B Lower, lanes h and i). I conclude that, at this particular location in the λ chromosome, nicking sites are prone to double-strand breakage in the presence of the cognate nicking enzyme if the chromosome is undergoing replication (Fig. 4A).
Locus Independence of the Breaks.
To make sure that the observed phenomenon is not restricted to a particular location on the λ chromosome, I inserted the nicking site at the unique SstI∷XhoI site in a different λ phage (Fig. 2A Lower). Because λ has bidirectional DNA replication, it was important to place the nicking site on the other side of the λ replication origin [λ ori is around position 39100 on the λ chromosome (38)] to see whether the findings at the first location would hold. In this experiment, in vivo molecular weight markers are generated by using sigma replication of the same set of substrates in Rep+ cells; therefore, the markers are “half-breaks” (compare Fig. 5, lanes a–c, with Fig. 3B, lanes j–l) rather than regular double-strand breaks.
Figure 5.

Replication-induced double-strand breaks at persistent nicks at the XhoI-site on the other side of the λ replication origin. Phages MMS2663, λAK4, or λAK5, indicated in the entry “substrate” as “—”, “t,” or “b,” respectively, were infected at moi = 6 into the strains described below; the cells were incubated at 28°C for 80 min, and the phage DNA was extracted and analyzed by restriction digestion with PstI + NcoI and blot hybridization with probe 2 under both neutral (Upper gel) and denaturing (Bottom gel) conditions. Molecular weight markers for “half-breaks” at the XhoI site were generated in vivo (lanes a–c). The strains and conditions are as follows: lanes a–c, recB268 pK133 + IPTG; lanes d–f, Δrep∷cam recD1011 pK125 + IPTG; lanes g–i, Δrep∷cam recD1011 pK125, no induction; lanes j–l, Su− Δrep∷cam recC1010 pK125 + IPTG. To compensate for the smaller amount of λ DNA because of the inhibition of λ replication, 25 times more total DNA has been loaded in the case of Su− samples (lanes j–l).
A similar pattern of breakage was obtained at the new location, that is, double-strand breaks formed efficiently in replicating λ carrying the nicking sites in the presence of the nicking enzyme (Fig. 5 Upper, lanes e and f). No breaks were formed if the enzyme was not produced (Fig. 5 Upper, lanes h and i), or if the nicking sites were absent (in the original control phage) (Fig. 5 Upper, lanes d, g, and j), or if λ's DNA replication was inhibited (Fig. 5 Upper, lanes k and l). The alkaline counterpart indicated an efficient cleavage of the nicking sites whenever the nicking enzyme was produced (Fig. 5 Lower, lanes b and c, e and f, and k and l). Therefore, the overall phenomenon looks similar at two separate locations on opposite sides of the λ replication origin. I conclude that, when a replication fork runs into a persistent single-strand break in template DNA, a double-strand break at the nick results.
Discussion
The main purpose of these experiments was to test ideas about replication fork interaction with persistent nicks in template DNA. One idea predicted that a nick relaxes DNA supercoiling and therefore would inhibit the replication fork progress in its vicinity. Another idea predicted that the replication fork would run into the nick and collapse, generating a double-strand end (Fig. 1 A → B → C). In the case of two replication forks converging on the same nick, a double-strand break would be generated (Fig. 1 C → D). My observations are consistent with the replication fork collapse scenario: persistent nicks, introduced by a nicking enzyme in the replicating λ chromosome, cause the formation of double-strand breaks (Fig. 4A). It should be noted that these experiments were not designed to rule out the replication fork inhibition scenario; indeed the majority of replication forks could be inhibited by the nick-associated relaxation and never reach the nick.
The nicking enzymes of bacteriophages are related to type I DNA topoisomerases and, in this respect, the reported experiments have their predecessors. Camptothecin is a progenitor of a family of potent anticancer drugs (39, 40); camptothecin kills eukaryotic cells during S-phase (3) by preventing the closing part of the DNA topoisomerase I cycle (41). Persistent nicks, caused by camptothecin-inhibited DNA topoisomerase I, were proposed to cause double-strand breaks in replicating simian virus 40 (SV40) molecules (9, 14, 42), but because the sites of the original single-strand interruptions were difficult to map, the question remained as to the link between topoisomerase-induced nicks and detected double-strand ends. Electron microscopy studies of replicating SV40 DNA, extracted from camptothecin-treated cells, produced spectacular pictures of circular chromosomes with what looked like disintegrated replication forks (43), but the results were again only suggestive. In my experiments, replication-dependent double-strand breaks form at preexisting single-strand scissions and nowhere else.
What could be the molecular mechanisms of this DNA replication-induced double-strand breakage at persistent nicks? One widely discussed possibility (2, 7–10, 39, 44–46) is that when a replication fork reached a single-strand interruption, it collapses, and if a converging replication fork soon comes to the same nick, a double-strand break results (Fig. 1 A–D), exactly what is observed in the experiments reported here. An alternative explanation for the nick-induced double-strand breakage is that a replication fork encounters the nick and “explodes,” generating two replicated branches and one unreplicated branch (40) (Fig. 1E). The reported in vivo results, as well as earlier in vitro results of others (47), do not distinguish between the “collapse” versus “explosion” ideas. Although the possibility of replication fork explosion is less specified, it will be a legitimate contender until distinct predictions of the two ideas are tested experimentally.
One limitation of the used experimental system is that nicks produced by specialized replicative enzymes are not generic single-strand interruptions of the type generated during excision repair. Rather, the 5′-end of the nick is likely to be bound by the M13 nicking enzyme [may be covalently (48)], which creates a certain “microenvironment” around the nick and likely even around the subsequent double-strand end. Therefore, nicks introduced by other means [for example, by “nicking” mutants of restriction endonucleases (49, 50)] could better approximate spontaneous single-strand interruptions encountered by replication forks in vivo.
Apart from inevitable limitations of the experimental system, this work illustrates that replication not only duplicates DNA strands but also “duplicates” any lesion in these strands. Fragmentation and loss of chromosomes in rapidly dividing rad51 mutant vertebrate cells (17, 18), apparently because of normal levels of single-strand DNA damage, dramatically illustrate this point. As a result, one-strand DNA damage, which before replication would have been fixed by excision repair, is aggravated by DNA replication and now requires a more sophisticated repair system. There is good evidence that two-strand DNA damage repair is based on homologous recombination (16). Experiments to detect homologous exchange in response to the replication-dependent double-strand breaks in the λ chromosome should be the next step in characterization of two-strand damage formation and repair.
Acknowledgments
I am indebted to Frank Stahl for encouragement and generous financial support during the initial stages of the project. I thank Nick Dixon, Kentsuke Horiuchi, Bob Lloyd, Bénédicte Michel, and Wilfred Wackernagel for providing strains and plasmids. Frank Stahl, Bénédicte Michel, and Elena Kouzminova critically read the manuscript. This work was supported by National Science Foundation Grants MCB-9402695 to Frank Stahl and MCB-0196020 to A.K.
Abbreviations
- IPTG
isopropyl β-d-thiogalactoside
- moi
multiplicity of infection
Footnotes
This paper results from the National Academy of Sciences colloquium, “Links Between Recombination and Replication: Vital Roles of Recombination,” held November 10–12, 2000, in Irvine, CA.
References
- 1.Ginsberg D M, Jagger J. J Gen Microbiol. 1965;40:171–184. doi: 10.1099/00221287-40-2-171. [DOI] [PubMed] [Google Scholar]
- 2.Hanawalt P C. Photochem Photobiol. 1966;5:1–12. [PubMed] [Google Scholar]
- 3.Li L H, Fraser T J, Olin E J, Bhuyan B K. Cancer Res. 1972;32:2643–2650. [PubMed] [Google Scholar]
- 4.Tyrrell R M, Moss S H, Davies D J G. Mutat Res. 1972;16:1–12. doi: 10.1016/0027-5107(72)90058-9. [DOI] [PubMed] [Google Scholar]
- 5.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 6.Drlica K, Snyder M. J Mol Biol. 1978;120:145–154. doi: 10.1016/0022-2836(78)90061-x. [DOI] [PubMed] [Google Scholar]
- 7.Kuzminov A. Mol Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 8.Skalka A. In: Mechanisms in Recombination. Grell R F, editor. New York: Plenum; 1974. pp. 421–432. [Google Scholar]
- 9.Snapka R M. Mol Cell Biol. 1986;6:4221–4227. doi: 10.1128/mcb.6.12.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strauss B S. In: DNA Repair Mechanisms. Altmann H, editor. Stuttgart: Schattauer; 1972. pp. 151–171. [Google Scholar]
- 11.Bonura T, Smith K C. J Bacteriol. 1975;121:511–517. doi: 10.1128/jb.121.2.511-517.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merrill B J, Holm C. Genetics. 1999;153:595–605. doi: 10.1093/genetics/153.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thoms B, Wackernagel W. J Bacteriol. 1998;180:5639–5645. doi: 10.1128/jb.180.21.5639-5645.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsao Y-P, Russo A, Nyamuswa G, Silber R, Liu L F. Cancer Res. 1993;53:5908–5914. [PubMed] [Google Scholar]
- 15.Horii Z I, Suzuki K. Photochem Photobiol. 1968;8:93–105. doi: 10.1111/j.1751-1097.1970.tb05976.x. [DOI] [PubMed] [Google Scholar]
- 16.Kuzminov A. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim D-S, Hasty P. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonoda E, Sasaki M S, Buerstedde J-M, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer T F, Geider K. J Biol Chem. 1979;254:12642–12646. [PubMed] [Google Scholar]
- 20.Foster P L, Trimarchi J M, Maurer R A. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arber W, Enquist L, Hohn B, Murray N, Murray K. In: Lambda II. Hendrix R W, Roberts J W, Stahl F W, Weisberg R A, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. pp. 433–466. [Google Scholar]
- 22.Birnboim H C. Methods Enzymol. 1983;100:243–255. doi: 10.1016/0076-6879(83)00059-2. [DOI] [PubMed] [Google Scholar]
- 23.Bachmann B J. In: Cellular and Molecular Biology. Neidhardt F C, editor. Washington, DC: Am. Soc. Microbiol.; 1987. pp. 1190–1219. [Google Scholar]
- 24.Silverstein J L, Goldberg E B. Virology. 1976;72:212–223. doi: 10.1016/0042-6822(76)90324-x. [DOI] [PubMed] [Google Scholar]
- 25.Fidanián H M, Ray D S. J Mol Biol. 1972;72:51–63. doi: 10.1016/0022-2836(72)90067-8. [DOI] [PubMed] [Google Scholar]
- 26.Kushner S R. J Bacteriol. 1974;120:1213–1218. doi: 10.1128/jb.120.3.1213-1218.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd R G, Buckman C, Benson F E. J Gen Microbiol. 1987;133:2531–2538. doi: 10.1099/00221287-133-9-2531. [DOI] [PubMed] [Google Scholar]
- 28.Rinken R, Thoms B, Wackernagel W. J Bacteriol. 1992;174:5424–5429. doi: 10.1128/jb.174.16.5424-5429.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bidnenko V, Seigneur M, Penel-Colin M, Bouton M-F, Ehrlich S D, Michel B. Mol Microbiol. 1999;33:846–857. doi: 10.1046/j.1365-2958.1999.01532.x. [DOI] [PubMed] [Google Scholar]
- 30.Kuzminov A, Stahl F W. Genes Dev. 1999;13:345–356. doi: 10.1101/gad.13.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love C A, Lilley P E, Dixon N E. Gene. 1996;176:49–53. doi: 10.1016/0378-1119(96)00208-9. [DOI] [PubMed] [Google Scholar]
- 32.Higashitani A, Greenstein D, Horiuchi K. Nucleic Acids Res. 1992;20:2685–2691. doi: 10.1093/nar/20.11.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stahl M M, Thomason L, Poteete A R, Tarkowski T, Kuzminov A, Stahl F W. Genetics. 1997;147:961–977. doi: 10.1093/genetics/147.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenstein D, Horiuchi K. J Mol Biol. 1987;197:157–174. doi: 10.1016/0022-2836(87)90115-x. [DOI] [PubMed] [Google Scholar]
- 35.Dotto G P, Zinder N D. Proc Natl Acad Sci USA. 1984;81:1336–1340. doi: 10.1073/pnas.81.5.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pratt D, Erdahl W S. J Mol Biol. 1968;37:181–200. doi: 10.1016/0022-2836(68)90082-x. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi S, Hours C, Iwaya M, Lane H E D, Denhardt D T. In: The Single-Stranded DNA Phages. Denhardt D T, Dressler D H, Ray D S, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1978. pp. 393–400. [Google Scholar]
- 38.Daniels D L, Schroeder J L, Szybalski W, Sanger F, Blattner F R. In: LAMBDA II. Hendrix R W, Roberts J W, Stahl F W, Weisberg R A, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. pp. 469–517. [Google Scholar]
- 39.D'Arpa P, Liu L F. Biochim Biophys Acta. 1989;989:163–177. doi: 10.1016/0304-419x(89)90041-3. [DOI] [PubMed] [Google Scholar]
- 40.Froelich-Ammon S J, Osheroff N. J Biol Chem. 1995;270:21429–21432. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 41.Redinbo M R, Stewart L, Kuhn P, Champoux J J, Hol W G J. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 42.Hsiang Y-H, Lihou M G, Liu L F. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 43.Avemann K, Knippers R, Koller T, Sogo J M. Mol Cell Biol. 1988;8:3026–3034. doi: 10.1128/mcb.8.8.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cox M M. Genes Cells. 1998;3:65–78. doi: 10.1046/j.1365-2443.1998.00175.x. [DOI] [PubMed] [Google Scholar]
- 45.Gourlie B B, Pigiet V P. J Virol. 1983;45:585–593. doi: 10.1128/jvi.45.2.585-593.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saffhill R, Ockey C H. Chromosoma. 1985;92:218–224. doi: 10.1007/BF00348697. [DOI] [PubMed] [Google Scholar]
- 47.Howard M T, Neece S H, Matson S W, Kreuzer K N. Proc Natl Acad Sci USA. 1994;91:12031–12035. doi: 10.1073/pnas.91.25.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asano S, Higashitani A, Horiuchi K. Nucleic Acids Res. 1999;27:1882–1889. doi: 10.1093/nar/27.8.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heitman J, Ivanenko T, Kiss A. Mol Microbiol. 1999;33:1141–1151. doi: 10.1046/j.1365-2958.1999.01556.x. [DOI] [PubMed] [Google Scholar]
- 50.Heitman J, Model P. Proteins Struct Funct Genet. 1990;7:185–197. doi: 10.1002/prot.340070207. [DOI] [PubMed] [Google Scholar]