

Abstract
In individuals with sickle cell disease (SCD), recognizing the cues to an acute pain episode and responding appropriately are important. The purpose of this mixed-methods pilot study is to identify preliminary factors that influence care seeking for pain in young adults with SCD. Responses were received from 69 young adults with SCD, age 18-35 years. The majority of respondents (88%) wait until the pain intensity is an average of 8.7 (± 1.2) on a scale of 1 to 10 before seeking care. Prominent themes influencing care seeking for pain include: trying to treat pain at home, avoiding the emergency department because of past treatment experiences, the desire to avoid admission to the hospital, and the importance of time in the lives of the young adults with SCD. Young adults with SCD need additional support from family and healthcare providers in order to make timely, appropriate decisions regarding care seeking.
Sickle cell disease (SCD) is a chronic genetic disease that, in the United States affects primarily African Americans (Ely et al., 2002). The hallmark of the disease is pain caused by vaso-occlusion, reperfusion injury, and hypoxemia (Rees, Williams, & Gladwin, 2010). Although advances in SCD treatment have decreased the pain and prolonged survival (Smith et al., 2005), adults with SCD often experience unpredictable painful crises or pain episodes requiring healthcare visits (Waters & Thomas, 1995; Yale, Nagib, & Guthrie, 2000). The pathophysiology of SCD is multifactorial (Rees et al, 2010). However, the polymerization of hemoglobin S (HbS), when it is deoxygenated, is the primary event in the molecular pathogenesis of SCD (Bunn, 1997). Additionally, the adherence of sickle red blood cells as well as other cellular elements to endothelial cells or subendothelial matrix proteins (Stuart & Nagel, 2004) and hypoxia-reoxygenation injury (Solovey et al., 2004) likely contribute to disease pathophysiology.
The clinical manifestations of SCD can lead to unpredictable episodes of acute pain and feelings of inadequacy regarding individuals’ living with SCD care (Strickland, Jackson, Gilead, McGuire, & Quarles, 2001). Individuals with SCD have described feeling less than human when their complaints of pain are ignored or discounted as false by healthcare providers (Jacob & American Pain Society, 2001; Solomon, 2010; Strickland et al., 2001). Young adults with SCD are particularly vulnerable because of inadequate transition from pediatric to adult care and not having the requisite skills to navigate the health care system (Jenerette & Brewer, 2010). Understanding how young adults with SCD seek care is important because young adults with SCD, aged 18-30 years, have the highest rates of emergency department use and highest rates of re-hospitalization for pain (Brousseau, Owens, Mosso, Panepinto, & Steiner, 2010).
Patients with SCD face multiple barriers during care seeking, including being stigmatized as addicted to opioids, disbelieved or mistrusted regarding reports of pain, and not being allowed to participate in their own health care (Haywood et al., 2009; Jenerette & Brewer, 2010; Lattimer et al., 2010; Maxwell, Streetly, & Bevan, 1999). Haywood et al. (2011) reported that clinicians’ attitudes toward patients were considered difficult to change and were a barrier to pain management. In response to a specific case regarding an 18-year-old woman with SCD who had frequent ED visits for pain, Dampier stated “frequent admissions can be frustrating and can lead to mistrust between a patient and his or her provider” wherein “each of them may blame the other for the apparent therapeutic failure” (Dampier, Haywood, & Lantos, 2011, p. 128). There may be additional factors that influence care seeking for young adults with SCD. We have not heard from young adults with SCD as to the reasons why they may delay care seeking for sickle cell pain, and there have been no published reports of factors that impact care seeking for young adults with SCD.
Self-care management is an important part of living with a chronic illness. Recognizing the cues to an evolving SCD-related acute pain episode (usually referred to as a painful crisis) and responding appropriately may be an important part of knowing the body and self-care management, especially for young adults living with this disease. Some young adults with SCD may be overly dependent on emergency departments for care as they transition from pediatric to adult providers (Hemker, Brousseau, Yan, Hoffman, & Panepinto, 2011); they have not yet acquired the self-care pain management skills that older adults have described as “learning their bodies,” and thus, are at particularly high risk for poor pain outcomes (Jenerette & Lauderdale, 2008;Jenerette, Leak, & Sandelowski, 2011). A significant part of learning the body is recognizing the cues to an evolving sickle cell crisis and responding appropriately. A first step may be understanding the phases of a sickle cell crisis and the role it might play in care seeking.
Ballas (1998) and Jacob et al. (2005) describe the four phases of a sickle cell crisis as (1) pre-pain or prodromal, (2) initial or infarctive, (3) established, and (4) resolving, recovery, or post-crisis phase. The prodromal/pre-crisis phase of an evolving pain episode is characterized as a period before the onset of severe pain. The prodromal phase lasts an average of 2–4 days with pain that has been described as low intensity or aches. Researchers have suggested that many adults with SCD predict or have a “premonition” about the prodromal phase of an impending crisis (Murray & May, 1988; Jacobs et al., 2005). Furthermore, those who do not currently recognize cues for the prodromal phase can be taught to do so by keeping a daily diary of symptoms during the steady state of the disease (Stuart, Stone, Akinola, Gallimore, & Pepys, 1994). Once a pain episode evolves beyond the prodromal phase, the pain becomes more intense and difficult to treat. During the infarctive phase, what used to be aches progress gradually to maximum pain. Individuals will have decreased activity. They may also begin to have fear and anxiety about seeking care. The duration of this phase can vary. The next phase is the established phase. This is the phase when most individuals will seek care via the emergency department. The pain is at its peak. It is steady and persistent. There is no relief from home medication. Aches have progressed and joints may now be swollen and stiff. A fever may also be present. The established phase lasts 4–6 days and often requires in-patient hospitalization. The final phase can be referred to as the resolving, recovery, or post-crisis phase. As the name implies, the acute painful crisis is over. Pain is now at a tolerable level. This phase usually lasts 1–2 days and is usually the phase when the individual is discharged from the hospital.
This investigation is guided by the conceptual relationships posited by the revised Theory of Self-Care Management for Sickle Cell Disease (Fig. 1; Jenerette, Brewer, Crandell, & Ataga, 2012; Jenerette & Murdaugh, 2008). This theory focuses on vulnerability factors and self-care management resources that influence health outcomes. In Figure 1, vulnerability factors (lack of sickle cell crisis cue recognition/response, number of complications, number of acute pain episodes per year, and overprotection) negatively influence health outcomes (pain management experience, depressive symptoms, self-esteem, and perceived health-related stigma). Self-care management resources (self-efficacy, coping behaviors, social support, self-care ability, self-care actions, and assertive communications skills) positively mediate the relationship between vulnerability factors and health outcomes. Specifically, this research explores sickle cell crisis cue recognition and response.
Figure 1.
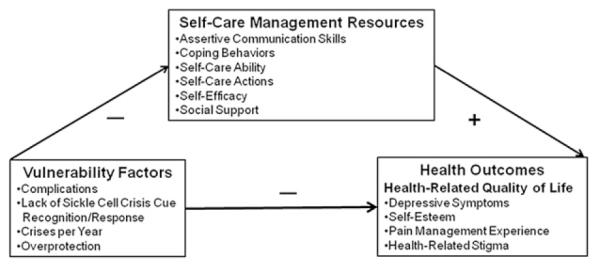
Theory of Self-Care Management for Sickle Cell Disease.
The purpose of this mixed-methods study is to identify factors that influence care seeking for pain in young adults with SCD. The main research questions were as follows:
Research Question 1: On a scale of 1-10 with 10 being the worst pain, what is the average pain score of the young adults with SCD when they seek care?
Research Question 2: What factors influence care seeking for pain in young adults with SCD?
METHODS
Design and Procedure
As a part of a mixed-methods pilot intervention aimed at reducing health-related stigma in young adults with SCD, a convenience sample of young adults with SCD was recruited from the adult outpatient sickle cell clinic affiliated with the Comprehensive Sickle Cell Program at the University of North Carolina at Chapel Hill. Inclusion criteria were a diagnosis of SCD; the ability to read, write, and understand English; and age of 18–35 years. All eligible participants received a letter and were approached for enrollment by the researcher or research assistant. After providing written informed consent, participants completed a baseline questionnaire packet that included a demographic form as well as scales to measure pain, communication, satisfaction with care seeking, and health-related stigma. After completion of the baseline data forms, participants were randomized into either the intervention or control group, with both groups completing two sessions. The intervention group participated in one session focused on sickle cell crises cue recognition and response, and a second session focused on assertive communication. The control group participated in two life-review interviews that were divided into a review of childhood memories to high school and then young adult life. During the first of the two sessions, all participants provided information related to care seeking before their group-specific activity, and this is the information that is presented to address the research questions.
University institutional review board approval was obtained from The University of North Carolina at Chapel Hill before the start of the study. Data collection for session one, the time when preliminary care-seeking data were collected, began in August of 2010 and ended June 2012.
Setting and Sample
After informed consent, baseline data, used to describe the sample, were collected in a private area in the SCD clinic using paper and pencil. For the intervention group session one, data were collected in a private area of the SCD clinic. Control group participants provided responses either in a private area of the clinic or via telephone.
Measures
Demographic Characteristics
The demographic characteristics questionnaire requested information to describe the sample by age, gender, and education and included questions specific to SCD, such as number of crises per year that require hospitalization and the SCD genotype.
Response to Sickle Cell Pain Interview Guide
During session one, participants responded to the Response to Sickle Cell Pain Interview Guide, developed for the study by the first author and two other experts in SCD providing content validity. The interview guide was used to deliberately elicit descriptions of factors that described care seeking among young adults with SCD. The guide focuses on three areas: pain scores, delays in care seeking, and cue recognition/response. Participants were asked about their typical pain level when they seek care for pain during an acute pain episode or crisis and to describe factors that lead to delays in care seeking for pain. Respondents were also asked if they believe that there are phases or stages to a SCD crisis and if they receive cues or signs from their body to indicate an impending crisis.
Data Analysis
Responses were recorded by the interviewer and notes were reviewed with participants for accuracy at the end of the interview. Quantitative data were entered into IBM SPSS Statistics Version 19. Descriptive statistics were calculated to describe the sample and to address research question one or pain score at the time of care seeking. Qualitative data were transcribed verbatim and content analyzed (Hsieh & Shannon, 2005) to determine address research question two or participants’ reasons for delays in care seeking for sickle cell pain. The first two authors independently coded the transcripts initially by the interview questions asked. The results of these independent qualitative analyses were then compared and differences resolved by consensus.
RESULTS
Demographic Characteristics
Data were provided by 69 young adults with SCD (44 females, 25 males) who are enrolled in the mixed-methods pilot intervention study aimed at decreasing health-related stigma. Their average age was 26 years (SD ± 4.9 years, range 19–35 years); average education was a little more than high school or 13.2 years (SD ± 2.2 years, range 8–20 years); and average number of SCD crises per year requiring hospitalization was 3.2 (SD ± 3.6, range 0–20). The majority of participants had the more severe, homozygous form of SCD known as sickle cell anemia (n = 48, 70%).
Research Question 1: Pain at Time of Care seeking
To address research question one, respondents were asked the following question: on a scale of 1 to 10, with 10 being the most severe pain, what is your typical pain level when you seek care for pain during a pain crisis or pain episode? The majority of respondents (88%) waited until pain was an average of 8.7 (SD ± 1.2) on a scale of 1 to 10, with 10 being the worst pain, before seeking care. Pain intensity is commonly reported using a 0–10 numeric rating scale in studies that include measures of pain (Farrar, Polomano, Berlin, & Strom, 2010). Because the participants were asked about their typical pain level when they seek care for pain during an acute pain episode or crisis, a scale of 1–10 was used instead of 0–10.
Using a t test, there was no significant difference in reported pain level at the time of care seeking by gender. Although not statistically significant, there was a modest negative correlation between pain at the time of care seeking and education (r = −0.20, p = .053), suggesting that young adults with more education trended toward seeking care for sickle cell pain at lower pain levels.
Research Question 2: Factors that Influence Delays in Care seeking
To address research question two, respondents were asked to respond to the following statement: tell us what reasons impact putting off or seeking care for your sickle cell pain. Table 1 summarizes these results. The top three reasons are discussed below.
Table 1.
Reasons for Delays in Care in Response to Sickle Cell Pain (Yes Responses)
| Reason | (N = 69) n (%) |
|---|---|
| Try to manage the pain at home | 53 (77%) |
| Avoid the emergency department because of past treatment |
38 (55.7%) |
| Avoid potential hospital admission | 33 (47.8%) |
| No transportation | 14 (19.7%) |
| Poor IV access; anticipating multiple sticks | 14 (19.7 %) |
| Lack of insurance | 9 (13.1%) |
| Just scared | 5 (6.6%) |
| Lack of childcare | 2 (3.3%) |
Try to Manage the Pain at Home
The most common reason (77%) for delays in care seeking for sickle cell pain offered by the young adults was that they tried to manage the pain at home as long as they could. Respondents stated that they were taught to do this by providers or caregivers or learned this on their own. In addition to these explicit responses, respondents were asked if there were any other reasons for delaying care seeking. A 32-year old female stated that she tries to manage pain at home because she wants to “make sure so [she] won’t be discharged only to return.” A 29-year old male stated that he tries to manage his pain at home because he “already knows what will happen” at the emergency department. A 28-year old female noted that she stays at home because of financial reasons as she “don’t want large bill.”
Avoiding the Emergency Department
Young adults reported the second most common reason for delaying care as the desire to avoid the emergency department (ED) (55.7%) because of the way they were treated in the past. Many young adults reported that they just do not like the ED, while others provided more specific reasons. Both a 20-year-old female and a 22-year-old male expressed delaying care because they felt that they were perceived as “drug-seeking” when they sought care in the past. Both females and males also reported that ED staff lacked knowledge to take care of them or behaved in ways that did not encourage them to revisit the ED if warranted.
Avoiding Hospital Admission
Almost as much as young adults with SCD wanted to avoid the ED, they also delayed care in an attempt to avoid a hospital admission (54.1%). A hospital admission did not fit into their lives, as both females and males did not want to miss work, school, or fun.
Time is of the Essence
Time was a theme that was underlying many of the delays in care seeking. A 26-year old female stated she avoids the ED “because I felt I spent so much time in the emergency room all my life.” Another, a 28-year old male, stated that although he believes the staff are “the only ones who know what to do,” traveling for care is time-consuming because of the long distance he lives from the hospital that provides the best care. For those who decide to seek care through the ED, many share the issue of a 25-year–old participant, who stated the “fear that I will be waiting for hours in the waiting room” leads to delays in seeking care. A 29-year old female stated that she waits until the pain increases because she wants the “provider to see [pain], so she gets treatment faster and better.” A 35-year–old participant says providers “don’t think it’s bad unless you are crying, bent over.” A 26-year old female expanded on this by saying that, if she goes to the ED, she wants to make sure that she is “kept at/in the hospital and not discharged only to return later” because she is still hurting. A 33-year old female described that the pain of SCD just did not fit into her schedule. She stated that sometimes she has “other plans that I don’t want to interfere with, like deadlines at work or a school trip.” Finally, a 22-year old male felt that the time that it took to take care of his pain in the ED was a disruption to his family. He does not “want to be a nuisance” and going to the hospital was “no fun.”
Phases and Cues
Young adults were asked if they believe that there are phases or stages involved in a sickle cell crisis. Most (84%) believe that there are phases. A t test did not reveal a significant difference in belief that there are phases of a sickle cell crisis by gender.
Respondents were asked if they receive cues or signs from their body when they are headed toward a sickle cell pain crisis. There were no significant differences by gender, and 84% of respondents reported that they receive cues of an evolving crisis. Cues included a familiar pain, often described as back pain or lower back pain; fatigue or more tired than usual; eyes become yellow or jaundiced; body aches; and decreased appetite. The most common responses to these cues were rest, fluids, and taking pain medication.
DISCUSSION
The Theory of Self-Care Management for Sickle Cell Disease proposes that cue recognition and response are vulnerability factors that can negatively impact health outcomes such as the pain management experience. Because most study participants report that they recognize the cues of an evolving sickle cell crisis, the more problematic issue may be the response. Results suggest that most young adults may be waiting longer than they should to seek care. This is true if one believes that early intervention may lead to a better pain manage experience. For early intervention to take place, the barriers that impact care seeking must be addressed.
Because self-care is an important part of living with chronic disease, it is appropriate that individuals with SCD make every effort to manage pain at home. However, if the crisis evolves, individuals should be encouraged to seek care. The respondents reported that they were told by providers and caregivers to wait by trying to manage their pain at home. It will be important to understand how providers educate young adults with SCD and their family members about when to seek care. In the case of young adults who may be new to adult care, it is very important to involve family members who are an important part of care-seeking decisions. Issues of care seeking should be taught during adolescence and reinforced before the transition from pediatric to adult care.
The study results are supported by previous literature that notes the numerous barriers that individuals with SCD face when they seek care for pain, but the most common barrier noted in the literature, being perceived as drug-seeking, was also a major hindrance for care seeking (Haywood et al., 2009; Jenerette & Brewer, 2011; Lattimer et al., 2010; Maxwell, Streetly, & Bevan, 1999). Few studies are being done to address the issue of clinicians’ attitudes towards individuals with SCD (Haywood et al., 2011). Given the profound effect clinicians’ attitudes can have on the individuals with SCD seeking care, this is an important area for intervention. However, this work focuses on how individuals with SCD can improve their care-seeking experience. Care seeking should be a collaborative process in that both clinicians and patients have a role in developing the helping-trusting relationship that leads to a positive care-seeking experience.
Study results suggest that the stigmatization that individuals with SCD face may lead to delays in care seeking. When individuals delay care because of the way they have been treated in the past, yet eventually must seek care, there is the great potential that they may be unable to communicate their needs effectively. Pain inhibits communication, and because the entire treatment of SCD pain is based on subjective complaints and the provider’s assessment, communication is vital. Moreover, if care is delayed and a sickle cell crisis progresses, the likelihood of hospital admission increases.
The respondents often delay care because they anticipate a hospital admission. Although providers may stereotype individuals as drug-seeking, these young adults report that they do not want to be admitted to the hospital because hospitalization interrupts their lives. Although some hospitalizations cannot be avoided, it may be important to explore the potential of at least decreasing the length of hospital stay with early intervention, which results from earlier care seeking. In the event of hospitalization, once the acute crisis is over, but before discharge, there may also be some teachable moments that can help individuals with SCD to cope with SCD such as mind-body medicine therapies.
Time is of the essence for young adults with SCD. The issue of time was described from two different perspectives. First, these individuals often delayed care seeking because of past treatment, during which they may have experienced perceived long wait times in the ED while care seeking for pain. Wait times may be impacted by clinicians’ attitudes towards individuals with SCD and how their care is prioritized in the ED. Wait times may be amenable to intervention.
Another option for individuals with SCD is outpatient treatment via a sickle cell day hospital that supports early intervention. Day hospitals provide the advantage of care without going to the ED. Unfortunately, many individuals with SCD do not have access to a day hospital. Young adults do not want SCD to interrupt their lives by having to spend time away from their families and activities they enjoy. They also do not want their SCD to impact the lives of others, which one respondent described as being a “nuisance.” Care seeking may be less time consuming if the evolving crisis is halted or at least minimized. This would require seeking care earlier.
Limitations
Although this study has strengths, it also has limitations that must be noted. First, the respondents are from one comprehensive program that serves a large geographic area. Information was not sought about the places that individuals sought care, as some are unable to return to the comprehensive program for their healthcare needs. Thus, the study results are not applicable to any other population, specific institution, or clinician group. Moreover, the data are all self-report, as no pain scores or care-seeking activities were elicited from medical records. Finally, respondents in this study were young adults with SCD aged 18–35 years. Therefore, we do not know if the experience of care seeking is different for other adults with SCD, who may have developed different skills to navigate the healthcare system.
Future Directions
The majority of sickle cell crises are managed outside of the healthcare system. However, when a crisis continues to evolve despite attempts to manage the pain at home, time is of the essence. The preliminary work provides insight into the need for future research and intervention to eliminate the barriers that impact delays in care seeking. Young adults with SCD are particularly vulnerable to poor pain management experiences during care seeking. There may be interventions that can enhance the care-seeking experience by intervening with clinicians as well as individuals with SCD and their families. For individuals with SCD, a chronic, life-long condition, it is important that skills to live with the disease, including appropriate care seeking, are taught early.
Nursing plays an important role in the pain management experience of individuals with SCD. In the ED, nurses triage patients and determine the initial level of care. Because nurses are often the first providers that individuals with SCD come in contact with, the relationship they develop is important in helping to determine satisfaction with the care-seeking experience. It may be important to understand nurses’ attitudes towards individuals with SCD in order to intervene appropriately.
Nurses can also play an important role as educators. Nurses can help patients to better communicate their needs. Nurses in the ED, or perhaps more appropriately in inpatient and outpatient settings, can introduce the communication concept of situation, background, assessment, and recommendation (SBAR) to individuals with SCD. Jenerette and Brewer (2011) suggest that SBAR can be used to improve communication and perhaps satisfaction with the care-seeking experience.
This preliminary work provides additional insight into factors that influence care seeking in young adults with SCD. This study will inform our understanding of the results of the ongoing study to decrease health-related stigma in this population as it informs us about pre-intervention behaviors. Young adults with SCD need to be the focus of additional research in order to design interventions to improve their health outcomes as they live with SCD.
Acknowledgment
This work was supported by a grant from the National Institute of Nursing Research (K23NR011061) to Coretta Jenerette. We would like to thank the young adults living with sickle cell disease who generously shared their experiences.
REFERENCES
- Ballas SK. Sickle cell pain: Progress in pain research and management. Vol. 11. IASP Press; Seattle, WA: 1998. [Google Scholar]
- Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. The Journal of the American Medical Association. 2010;303(13):1288–1294. doi: 10.1001/jama.2010.378. [DOI] [PubMed] [Google Scholar]
- Bunn HF. Pathogenesis and treatment of sickle cell disease. New England Journal of Medicine. 1997;337(11):762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- Dampier C, Haywood C, Jr., Lantos J. A narcotics contract for a patient with sickle cell disease and chronic pain. Pediatrics. 2011;128(1):128–131. doi: 10.1542/peds.2010-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely B, Dampier C, Gilday M, O’Neal P, Brodecki D. Caregiver report of pain in infants and toddlers with sickle cell disease: Reliability and validity of a daily diary. Journal of Pain. 2002;3(1):50–57. doi: 10.1054/jpai.2002.xb30064. [DOI] [PubMed] [Google Scholar]
- Farrar JT, Polomano RC, Berlin JA, Strom BL. A comparison of change in the 0-10 numeric rating scale to a pain relief scale and global medication performance scale in a short-term clinical trial of breakthrough pain intensity. Anesthesiology. 2010;112(6):1464–1472. doi: 10.1097/ALN.0b013e3181de0e6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood C, Jr., Beach MC, Lanzkron S, Strouse JJ, Wilson R, Park H, Witkop C, Bass EB, Segal JB. A systematic review of barriers and interventions to improve appropriate use of therapies for sickle cell disease. Journal of the National Medical Association. 2009;101(10):1022–1033. doi: 10.1016/s0027-9684(15)31069-5. [DOI] [PubMed] [Google Scholar]
- Haywood C, Jr., Lanzkron S, Hughes MT, Brown R, Massa M, Ratanawongsa N, Beach MC. A video-intervention to improve clinician attitudes toward patients with sickle cell disease: The results of a randomized experiment. Journal of General Internal Medicine. 2011;26(5):518–523. doi: 10.1007/s11606-010-1605-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemker BG, Brousseau DC, Yan K, Hoffman RG, Panepinto JA. When children with sickle-cell disease become adults: Lack of outpatient care leads to increased use of the emergency department. American Journal of Hematology. 2011;86(10):863–865. doi: 10.1002/ajh.22106. [DOI] [PubMed] [Google Scholar]
- Hsieh HF, Shannon SE. Three approaches to qualitative content analysis. Qualitative Health Research. 2005;15(9):1277–1288. doi: 10.1177/1049732305276687. [DOI] [PubMed] [Google Scholar]
- Jacob E, American Pain Society Pain management in sickle cell disease. Pain Management Nursing. 2001;2(4):121–131. doi: 10.1053/jpmn.2001.26297. [DOI] [PubMed] [Google Scholar]
- Jacob E, Beyer JE, Miaskowski C, Savedra M, Treadwell M, Styles L. Are there phases to the vaso-occlusive painful episode in sickle cell disease? Journal of Pain & Symptom Management. 2005;29(4):392–400. doi: 10.1016/j.jpainsymman.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Jenerette CM, Brewer C. Health-related stigma in young adults with sickle cell disease. Journal of the National Medical Association. 2010;102(11):1050–1055. doi: 10.1016/s0027-9684(15)30732-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenerette C, Brewer C. Situation, background, assessment, and recommendation (SBAR) may benefit individuals who frequent emergency departments: Adults with sickle cell disease. Journal of Emergency Nursing. 2011;37(6):559–561. doi: 10.1016/j.jen.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Jenerette C, Brewer CA, Crandell J, Ataga KI. Preliminary validity and reliability of the Sickle Cell Disease Health-Related Stigma Scale. Issues in Mental Health Nursing. 2012;33(6):363–369. doi: 10.3109/01612840.2012.656823. [DOI] [PubMed] [Google Scholar]
- Jenerette CM, Lauderdale G. Successful aging with sickle cell disease: Using qualitative methods to inform theory. Journal of Theory Construction & Testing. 2008;12(1):16–24. [PMC free article] [PubMed] [Google Scholar]
- Jenerette C, Leak A, Sandelowski M. Life stories of older adults with sickle cell disease. ABNF Journal: official journal of the Association of Black Nursing Faculty in Higher Education, Inc. 2011;22(3):58–63. [PubMed] [Google Scholar]
- Jenerette CM, Murdaugh C. Testing the theory of self-care management for sickle cell disease. Research in Nursing & Health. 2008;31(4):355–369. doi: 10.1002/nur.20261. [DOI] [PubMed] [Google Scholar]
- Lattimer L, Haywood C, Jr., Lanzkron S, Ratanawongsa N, Bediako SM, Beach MC. Problematic hospital experiences among adults patients with sickle cell disease. Journal of Health Care for the Poor and Underserved. 2010;21(4):1114–1123. doi: 10.1353/hpu.2010.0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell K, Streetly A, Bevan D. Experiences of hospital care and treatment seeking for pain from sickle cell disease: Qualitative study. British Medical Journal. 1999;318(7198):1585–1590. doi: 10.1136/bmj.318.7198.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray N, May A. Painful crises in sickle cell disease—Patients’ perspectives. British Medical Journal. 1988;297(6646):452–454. doi: 10.1136/bmj.297.6646.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Smith WR, Bovbjerg VE, Penberthy LT, McClish DK, Levenson JL, Roberts JD, Gil K, Roseff SD, Aisiku IP. Understanding pain and improving management of sickle cell disease: The PiSCES study. Journal of the National Medical Association. 2005;97(2):183–193. [PMC free article] [PubMed] [Google Scholar]
- Solomon LR. Pain management in adults with sickle cell disease in a medical center emergency department. Journal of the National Medical Association. 2010;102(11):1025–1032. doi: 10.1016/s0027-9684(15)30729-x. [DOI] [PubMed] [Google Scholar]
- Solovey A, Kollander R, Shet A, Milbauer LC, Choong S, Panoskaltsis-Mortari A, Blazar BR, Kelm RJ, Jr., Hebbel RP. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood. 2004;104(3):840–846. doi: 10.1182/blood-2003-10-3719. [DOI] [PubMed] [Google Scholar]
- Strickland OL, Jackson G, Gilead M, McGuire DB, Quarles S. Use of focus groups for pain and quality of life assessment in adults with sickle cell disease. Journal of the National Black Nurses Association. 2001;12(2):36–43. [PubMed] [Google Scholar]
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364(9442):1342–1360. doi: 10.1016/S0140-6736(04)17192-4. [DOI] [PubMed] [Google Scholar]
- Stuart J, Stone PC, Akinola NO, Gallimore JR, Pepys MB. Monitoring the acute phase response to vaso-occlusive crisis in sickle cell disease. Journal of Clinical Pathology. 1994;47(2):166–169. doi: 10.1136/jcp.47.2.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters J, Thomas V. Pain from sickle cell crisis. Nursing Times. 1995;91(16):29–31. [PubMed] [Google Scholar]
- Yale SH, Nagib N, Guthrie T. Approach to the vaso-occlusive crisis in adults with sickle cell disease. American Family Physician. 2000;61(5):1349–1356. [PubMed] [Google Scholar]