

Abstract
Chlorite dismutase carries out the heme-catalyzed decomposition of ClO2– to Cl– and O2, an unusual transformation with biotechnological and bioremediative applications. The enzyme has been successfully overexpressed for the first time in highly functional form in Escherichia coli and its steady state kinetics studied. The purified enzyme is abundant (55 mg/L cell culture), highly active (~4.7 × 103 μmol of ClO2– min–1 mg–1 subunit) and nearly stoichiometric in heme; further, it shares spectroscopic and physicochemical features with chlorite dismutases previously isolated from three organisms. A careful study of the enzyme's steady state kinetics has been carried out. ClO2– consumption and O2 release rates were measured, yielding comparable values of kcat (4.5 × 105 min–1), Km (~215 μM), and kcat/Km (3.5 × 107 M–1 s–1) via either method (4 °C, pH 6.8; all values referenced per heme-containing subunit). ClO2–:O2 stoichiometry exhibited a 1:1 relationship under all conditions measured. Though the value of kcat/Km indicates near diffusion control of the reaction, viscosogens had no effect on kcat/Km or Vmax. The product O2 did not inhibit the reaction at saturating [O2], but Cl– is a mixed inhibitor with relatively high values of KI (225 mM for enzyme and 95.6 mM for the enzyme–substrate complex), indicating a relatively low affinity of the heme iron for halogen ions. Chlorite irreversibly inactivates the enzyme after ~1.7 × 104 turnovers (per heme) and with a half-life of 0.39 min, resulting in bleaching of the heme chromophore. The inactivation KI (Kinact) of 166 μM is similar in magnitude to Km, consistent with a common Michaelis complex on the pathway to both reaction and inactivation. The one-electron peroxidase substrate guaiacol offers incomplete protection of the enzyme from inactivation. Mechanisms in keeping with the available data and the properties of other well-described heme enzymes are proposed.
Activation of O2 is essential to the aerobic way of life. Heme-dependent oxidases and oxygenases that react with O2 or its reduced forms are responsible for a variety of reactions that drive aerobic metabolism, including respiration of O2 (1, 2). Anaerobes live in the absence of O2 but likewise carry out oxidation chemistry and in many cases respiratory energy generation. Such organisms are now known to make use of diverse inorganic oxidants, including NO3– (3, 4), AsO4– (5, 6), SeO42– (7), soluble sulfur oxides (4), elemental sulfur (8), and solid metal oxides (9). Dechloromonas aromatica RCB (sequenced strain) (10) is a beta-proteobacterium that, in addition to O2, uses the thermodynamically strong but kinetically inert oxidant ClO4– (or ClO3–) as a respiratory electron acceptor (11).
The oxochlorates are toxic, primarily anthropogenic pollutants (12). Because of their high water solubility and widespread use as rocket propellants (ClO4–) (12, 13), herbicides (ClO3–), bleaching agents in paper manufacture (ClO2–) and household bleach (ClO–), these anions are now major water contaminants (14). In particular, chlorite has been named a “top ten” water contaminant by the Environmental Protection Agency due to its suspected health risks, especially in childhood anemia (15). Chlorite may also enhance the toxicity of other pollutants, serving as an oxidizing or chlorinating agent. Aside from being able to detoxify these oxochlorates, D. aromatica RCB is the sole organism yet isolated that carries out the complete oxidation of unsubstituted aromatic compounds (including benzene) to CO2 both in the presence and absence of O2 (10, 11, 16). Hence, the organism is of considerable interest for its bioremediative capabilities. The initial oxidant in the benzene oxidation pathway and the relationships between anaerobic oxidation and respiratory chemistries in D. aromatica RCB are unknown.
Complete reduction of ClO4– occurs in three steps and involves at least two enzymes (Scheme 1) (11, 17). Perchlorate reductase is a molybdopterin-dependent enzyme that is proposed to catalyze the successive reductions of ClO4– to ClO3– and ClO3– to ClO2–, with consequent production of a water molecule at each step (18–20). Despite its favorable reduction potential and its well-known use as peroxide-like “shunt”reagent in hemeenzyme and model chemistry (21–23), ClO2– is not reduced. It is, rather, exclusively decomposed to environmentally benign Cl– and O2 in a reaction catalyzed by the heme-containing enzyme chlorite dismutase (Cld)(24–29). Chlorite dismutation is remarkable from a number of perspectives. First, the Cld-catalyzed reaction is the only known enzymatic process for O–O bond formation beyond that catalyzed by photosystem II. Formation of O2 by Cld is curious from a biological standpoint since O2 is a known negative regulator of perchlorate/chlorate respiration (30). Furthermore, the cld gene is necessary for respiratory energy generation from perchlorate or chlorate as ClO2– otherwise accumulates to toxic levels (29); however, many nonrespirers also have cld homologues with unknown functions. Cld may have bioremediative applications in sensing and/or detoxifying chlorite in water. Recent interest in the in situ generation of O2 from salts in medicinal applications suggests a further biotechnological role for Cld (31).
Scheme 1.
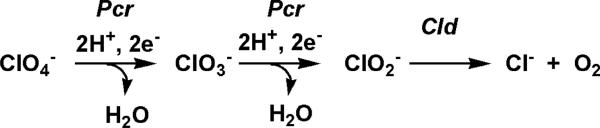
Proposed Pathway for Perchlorate Reduction by D. aromatica RCB
Chlorite dismutases from the β-proteobacteria Ideonella dechloratans (17, 25, 26), strain GR-1 (19, 24, 27), and Dechloromonas agitata (32) have been previously isolated but only minimally characterized. These enzymes are 100–120 kDa homotetramers, with ~1 heme-b per monomer. While they bear intriguing functional similarities to heme-containing catalase, peroxidase, chloroperoxidase, and anion reductase enzymes, no significant primary sequence homologies exist between these enzymes and chlorite dismutase nor between Cld and any protein in the database. Anthropogenic selection pressure may have played a role in the evolution of this novel enzyme, though exactly how it came to be is unknown. The mechanism of chlorite dismutation by Cld and its specificity for this particular substrate and reaction type remain largely unstudied. Rigorous selectivity would be remarkable in light of the known reactions of heme enzymes with related substrates (NO2–, NO, SO3–, H2O2, ClO–, etc.) and other known reactions observed in enzymatic heme/ClO2– systems, including oxidation (33), hydroxylation (34), and chlorination (33, 35).
This report describes the cloning, heterologous expression, purification, chemical/physical characterization, and a steady-state kinetic study of active chlorite dismutase from the model bioremediator Dechloromonas aromatica RCB. This is the first successful effort toward heterologous production of a highly functional chlorite dismutase with close to full heme incorporation. This is also the very first in depth study of the steady state kinetics and mechanism of a Cld from any source. This includes measurements correlating the rates and stoichiometry of ClO2– consumption and O2 release, studies of the influence of product inhibition and viscosity, and an analysis of the enzyme's intriguing chlorite-dependent irreversible inactivation. A mechanism consistent with all of the available data and with the properties of other well-described heme enzymes is proposed.
EXPERIMENTAL PROCEDURES
Engineering Cld for Expression
The cld-containing operon was cloned from a genomic library as previously described (32). SignalP 2.0 HMM predicts a twin-arginine motif secretion peptide preceding the coding sequence for mature Cld. A Quick-Change PCR-mutagenesis kit (Stratagene) was used to insert an NdeI restriction site (underlined) and an in-frame ATG start codon (italics) immediately after the predicted site of cleavage, using the following primer and its reverse complement: 5′-GTTGCGGCGCAGCAGCATATGCAACCCATGCAGTC-3′. The gene with its re-engineered start site (830 bp) was inserted into complementary NdeI/SacI restriction sites in the pET41a expression vector (PT7, KanR, Novagen). The new construct, pBSCld, was commercially sequenced (Fisher) and shown to be free of mutations or errors. The construct was subsequently transformed by electroporation into TunerDE3 Escherichia coli cells (Novagen) for IPTG-regulated1 expression. Cells were maintained as frozen 30% glycerol stocks at –80 °C.
Protein Expression
Cells were inoculated 1:100 from overnight cultures (produced from LB-agar/kanamycin plates streaked from frozen glycerol stocks, kanamycin 50 mg/mL) to 50 mL flasks (LB-kanamycin) and grown with rapid aeration on a rotary shaker until freshly saturated at 30 °C. The 50 mL culture was used to inoculate (1:100) 1 L of terrific broth (TB) supplemented with kanamycin (50 mg/L) in a 2.8 L Fernbach flask. The liter culture was grown at 30 °C to an optical density (at 600 nm) of 0.25. The temperature was lowered to 20 °C, and IPTG and hemin were added to final concentrations of 0.1 mM and 90 μM, respectively. Cells were grown for ~15 h and subsequently collected by centrifugation in 0.5 L Nalgene bottles (JA-10 rotor, 12000g, 30 min, 4 °C). Approximately 24 g of wet cell paste per L of culture was obtained.
Protein Purification
The efficiency of the expression system facilitated the development of a rapid two-column purification procedure. Cell growth, induction of protein expression, and cell collection were carried out as described above on a 1 L scale. Pelleted cells were resuspended in 5 volumes (~150 mL) of 100 mM potassium phosphate buffer, pH 7.4, and placed in a 300 mL glass beaker on ice. Phenylmethyl-sulfonate fluoride (PMSF) was added to 1 mM as a protease inhibitor. Cells were lysed by sonication for 10 min using a microtip-fitted Branson Ultrasonifier at 70% power with 3 s pulses at 2 s intervals. Lysates were centrifuged (JA-20 rotor, 41000g, 30 min, 4 °C) to separate out cellular debris, and the soluble fraction was retained. This fraction was dialyzed in 10,000 molecular weight cutoff [MWCO] dialysis tubing (Fisher) against 20 mM Tris-Cl buffer, pH 8.6, with 3 buffer exchanges and at least 3 h per dialysis cycle.
The deep red dialysate was loaded at 2 mL/min onto a 20 × 5 cm (length by diameter) DEAE-Sepharose ion exchange column (GE Biosciences) equilibrated with 20 mM Tris-Cl buffer, pH 8.6, using an AKTA Prime protein purification system (GE Biosciences). The flow-through fraction (~200 mL) was highly enriched in Cld. This fraction was concentrated 20–50 fold using a stirred-cell N2-gas pressure concentrator (Amicon) with a 10,000 MWCO YM-20 membrane. The concentrated proteins were then loaded onto a 2.6 × 120 cm gel filtration column (S-200 Sephacryl, GE Biosciences) at 0.5 mL/min (100 mM potassium phosphate buffer pH 6.8). Fractions were collected in 1 mL increments and screened for purity using SDS–PAGE. Pure fractions eluting after ~9.5 h were pooled and concentrated in a 10,000 MWCO centrifuge concentrator (Millipore) according to the manufacturer's instructions (JA-10 rotor, 4400g, 25 min, 4 °C).
Bradford and Heme Analyses
Bovine serum albumin was used to generate standard curves with the Bradford dye (Bio-Rad) according to standard methods. Heme type and stoichiometry were determined by the pyridine hemochrome assay, as previously described (36). Horse heart myoglobin (Sigma) was used as a standard.
Metal Analysis
Pure native and expressed proteins were analyzed for metal content via inductively coupled plasma optical emission spectrometry (ICP-OES) using a Perkin-Elmer spectrometer. A standard curve from 0 to 130 ppb metal was generated using ACS-grade optical emission standards (CPI International). Triplicate samples were analyzed and the values averaged.
Mass Spectrometry
The mass of the expressed protein was determined by matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS), carried out at the Notre Dame Mass Spectrometry Facility on a PerSeptive Biosystems Voyager-DE MALDI-TOF instrument. Protein samples to be analyzed were excised from a 12% acrylamide SDS gel. Analytical gel filtration relative to known molecular weight standards was also carried out, using the S-200 Sephacryl column described above (Protein Purification).
O2 Stability
The O2-stability of expressed Cld was assessed. Cell lysate was prepared from 2.23 g of cell paste and clarified by centrifugation. PMSF was added to 1.0 mM. To each of four 50 mL pear shaped flasks was added 1.0 mL of clarified lysate. The headspace gas was briskly purged with N2 for 20 min for two flasks; the remaining two were kept under air. Air/N2-saturated lysates were stored at 4 °C for 24 and 48 h and subsequently aliquotted and snap frozen in liquid N2 for later activity analysis using the discontinuous assay described below. Triplicate samples were analyzed and measured values averaged.
UV/Visible Spectrophotometry
All spectra were measured at 25 °C on a Varian Cary 50 spectrometer with temperature control from a Peltier cooler. Excess sodium dithionite (Sigma) was used to reduce the heme Fe, as previously described (36).
Isoelectric Focusing
The isoelectric point (pI) for chlorite dismutase with its signal peptide cleaved at the predicted location was computed using Compute pI (Swiss-Prot, available on the web through the Swiss Institute of Bioinformatics). The isoelectric point for recombinant Cld was measured by isoelectric focusing polyacrylamide gel electrophoresis (IEF-PAGE) on a pH 3.0–10.0 gradient gel (Bio-Rad) run 60 min at 100 mA, 60 min at 250 mA, and 30 min at 500 mA. Isoelectric focusing standards loaded on the same gel (Bio-Rad) were used to generate a curve of pI versus migration distance, from which the unknown pIs were determined.
Measurements of Activity
Sodium chlorite solutions used in measuring activity (~20 mM sodium chlorite in 100 mM potassium phosphate buffer, pH 7.0) were routinely standardized by iodometric titration (37). Briefly: to 800 μL of a freshly prepared 80–2000 uM chlorite stock was added 100 μL of 12 M HCl (final pH ~0.2) and a >10-fold excess of solid KI (~0.4 g for a ~300 mM final concentration). The resulting I2 formed was titrated by addition of 2–200 μL volumes of a 1–100 mN sodium thiosulfate standard (Titristar) in the presence of 10 μL of a starch indicator (Ricca Chemicals/1.0% w/v) until the blue color (monitored spectroscopically) due to I2 disappeared. For each mole of chlorite initially present, 4 mol of sodium thiosulfate are consumed.
Enzyme activity was measured discontinuously by analyzing quenched reaction mixtures for unreacted chlorite via iodometric titration. Standardized solutions of sodium chlorite (80–2000 μM in 100 mM potassium phosphate buffer, pH 6.8) were incubated with 1.0–5.0 nM enzyme in a 4 °C water bath with constant stirring. A low temperature was used in order to elongate the initial phase of the reaction, yielding a more accurate initial rate. 100 μL aliquots were removed every 15 s and added to tubes containing 7.5 μL of 10 M NaOH, quenching the reaction. Solutions were acidified (pH ~0.2) by the addition of 15 μL of 12 M HCl immediately before the titration. Excess solid KI (to ~300 mM) was added immediately to each tube, and the contents were mixed. Tubes were titrated with 1–4 mN sodium thiosulfate standard, as described above.
Activity was measured continuously using a Clark-type O2 electrode. The electrode was equilibrated to the set temperature for at least 1 h and calibrated to the expected concentration of O2 in air saturated double-distilled Milli-Q water, adjusted for temperature and daily atmospheric pressure. Temperature was maintained at 4 °C by a circulating water bath (Thermo-Fisher Scientific) jacketing the electrode setup. Reactions were carried out in 1.5 mL volumes of 100 mM potassium phosphate buffer, pH 6.8, with 80–2000 μM of sodium chlorite added from a stock made in the same buffer. Reactions were initiated by addition of 1.0–5.0 nM enzyme in 1–5 μL aliquots, via gastight syringe.
Linear rates were fit by least-squares regression to the initial 5–10% of the progress of reaction curves, using Kaleidagraph. Specific activity is defined as (μmol of ClO2– consumed)(mg–1 of enzyme)(min–1) or (μmol of O2 produced)(mg–1 of enzyme)(min–1) at 4 °C and under initial rate conditions (saturating substrate and <10% substrate consumed). To account for small differences in the activities of different enzyme aliquots, initial rates were referenced to the specific activity measured for each aliquot of enzyme used.
Reaction Stoichiometry
The stoichiometry of the reaction was determined at several [ClO2–]initial (Supporting Information, Table S1). The total amount of O2 evolved in a given reaction was determined via Clark electrode. The total amount of ClO2– consumed was determined via iodometric titration. (See above.)
Viscosity Effects
Sodium chlorite solutions (80–2000 μM in 100 mM potassium phosphate buffer, pH 6.8) were prepared with varying weight percents of sucrose, D-(+)-trehalose (microviscosogens) or Ficoll 400 (a macroviscosogen). The relative viscosities (with respect to water) of the solutions were measured using a Carri-Med Controlled Stress Rheometer using a torque of 636 dyne cm, and a shear stress of 15 dyne cm–2 with a gap width of 230 μM at a temp of 4 °C. The relative viscosities (ηrel) of the sucrose and Ficoll 400 solutions used in this study were 30% sucrose, ηrel = 4.85; 5% Ficoll, ηrel = 3.52. Chlorite decomposition in each solution was monitored continuously via a Clark type platinum O2 electrode as described above.
Steady-State Kinetics
Initial reaction velocities were measured as a function of [ClO2–] over a range of 80–1500 μM chlorite, using catalytic amounts (1.0–5.0 nM) of enzyme and the discontinuous and continuous kinetic assays described above (4 °C, 100 mM potassium phosphate buffer pH 7.0). Typical progress of reaction curves (Figure 3) contained at least 8 points when measured via the discontinuous method, or 200 points per minute when measured continuously. Least-squares data fits were carried out using Kaleidagraph 4.0. Plots of vi/[Etotal] versus [ClO2–] were fit to the Michaelis–Menten equation: vi/[Etotal] = kcat[ClO2–]/([ClO2–] + Km).
Figure 3.

(Top) Plots of initial rates of chlorite dismutation per [enzyme] as a function of [ClO2–], measured by monitoring ClO2– depletion (circles) and O2 evolution (diamonds). Points are averages of 3 measurements, and error bars represent ± one standard deviation. The Michaelis–Menten equation was fit to each set of data. (Bottom) Representative progress of reaction curves showing the dismutation of chlorite (circles) and the evolution of O2 (diamonds) measured in parallel reactions over time. For the data shown, [ClO2–]initial = 172 μM. Each data set was fit to an exponential equation, as described in the text.
Product Inhibition
Initial rates of chlorite decomposition were measured continuously via Clark electrode with chlorite as the variable-concentration substrate and NaCl as the fixed-variable inhibitor at 20, 50, 100, 150, and 200 mM. Initial rate plots were likewise made for anaerobic and O2-saturated solutions at 0–1800 μM [ClO2–]. Reactions were made anaerobic by purging the headspace of the stirred reaction chamber with hydrated N2 gas for 3 min. The plunger-style electrode was then lowered into the reaction mixture, displacing the headspace gas. The reading on the O2 meter was subsequently ~0 mV, or 0% O2. O2-saturated reaction mixtures were prepared by similarly purging reaction solutions with O2 gas.
Inactivation, Measurement of Turnover Number, and Guaiacol Rescue
Residual activities after exposure to chlorite were measured after 3 cycles of concentration and resuspension in fresh, chlorite-free buffer, using microcentrifuge concentrators (Amicon), or after 2 h of dialysis against chlorite-free phosphate buffer (Slide-A-Lyzer MINI Dialysis Unit, 10K MWCO, Pierce). Control samples showed negligible activity loss due to either method of buffer exchange. For the plot shown in Figure 6, concentrations of chlorite used were 1073, 870, 650, 435, 109, 43, 11, 630, 360, 180, 72, and 18 μM, and an enzyme concentration of either 1.7 or 2.5 nM, yielding molar ratios of substrate:heme spanning 3.0 × 103 to 4.5 × 104. The reactions were allowed to proceed until O2 evolution ceased. Samples were buffer exchanged and their protein concentrations reassessed by the Bradford assay. The activities of the enzyme samples were then determined. The percent activity retained after incubation with increasing molar ratios of chlorite, relative to the activity of fresh enzyme, was determined for each sample. These values are plotted versus [ClO2–]/[Cld heme], and used to extrapolate the relative molar amount of chlorite needed to completely inactivate the enzyme (38). A similar procedure was followed in the presence of increasing equivalents of guaiacol relative to heme (1.9 × 103 to 1.5 × 104 equiv, see Supporting Information) and in excess guaiacol (600 μM or 2850 equiv per heme).
Figure 6.

Plot of the residual enzymatic activity after exposure to increasing concentrations of chlorite, in the presence (triangles) and absence (circles) of excess/600 μM guaiacol. The turnover number, i.e., the maximal number of chlorite molecules dismutated per heme, is obtained by extrapolating a line fitted through each series of points to the x-axis. Turover numbers are ~1.7 × 104 in the absence of guaiacol and ~1.3 × 10 (5) in its presence.
RESULTS
Protein Expression and Purification
Growth/induction at E. coli's preferred 37 °C resulted in abundant expression of mostly insoluble protein. The soluble protein obtained under these conditions moreover bound only minimal amounts of heme (data not shown). To achieve optimal expression of soluble active protein, pre- and postinduction growth conditions were systematically varied and the resulting protein was screened for solubility. Lower temperatures and reduced concentrations of IPTG (using the TunerDE3 expression host, Novagen) resulted in slower induction of protein expression. The majority of this protein was found in the soluble fraction. Use of a rich growth medium (TB) supplemented with 90 μM hemin resulted in near-stoichiometric heme incorporation.
Published purification procedures for chlorite dismutases use combinations of anion exchange, hydroxyapatite, hydrophobic affinity, and ammonium sulfate precipitation steps (24, 26, 27). A simpler, more rapid method was developed here. The pI Calculator tool (Swiss Proteomics ExPASy server) predicts a fairly basic isoelectric point of 8.9 for Cld. The pI measured by isoelectric focusing of pure enzyme (Supporting Information) is yet higher: >9.6. Since most soluble E. coli proteins are expected to have lower isoelectric points, reverse anion-exchange chromatography was used to capture E. coli protein contaminants. An anion exchange resin (DEAE) equilibrated to a basic pH (8.6) yielded a flow-through fraction that was ~70% Cld as assessed by SDS–PAGE (Figure 1). This fraction was concentrated and further purified to >90–95% homogeneity by high-resolution gel filtration. Following this procedure, a total of ~55 mg of pure protein was obtained per liter of growth culture (Table 1).
Figure 1.

SDS–PAGE of Cld fractions and pure protein: (1) molecular weight size marker (from top: 250, 150, 100, 75, 50, 37, 25, 20, 15, 10 kDa, Bio-Rad); (2) clarified lysate; (3) dialysate; (4) ion exchange flow-through; (5) gel filtrate; (6) second-pass gel filtrate; (7) MW size marker.
Table 1.
Results of Purification for Recombinant Cld
| fraction | [protein] (mg/mL) | volume (mL) | total activity (U) | specific activitya (U/mg) | fold purification |
|---|---|---|---|---|---|
| clarified lysate | 9.0 | 72 | 272,000 | 400 ± 50 | (1) |
| dialysate | 7.3 | 68 | 397,000 | 800 ± 150 | 2.0 |
| anion exchange | 0.83 | 145 | 257,000 | 2,700 ± 300 | 6.8 |
| gel filtration | 7.3 | 7.5 | 206,000 | 4,700 ± 300 | 12 |
Specific activity measured at pH 6.8, 25 °C, according to the iodometric titration procedure (see text), with the exception of the clarified lysate (measured at pH 9.0). Hence, the total units of activity appear to increase after dialysis, but this is likely an artifact of the change in pH.
Protein and Cofactor Analyses
MALDI-TOF-MS predicts a subunit molecular weight of 28.4 ± 0.2 kDa for recombinant Cld, in good agreement with the masses estimated by SDS–PAGE for the same protein (~27 kDa, Figure 1, Table 2), native Cld from D. agitata (30 kDa) (11), strain GR-1 (32 kDa), or I. dechloratans (25 kDa) (25). Analytical gel filtration relative to native molecular weight standards predicts a total molecular weight of 116 kDa relative to molecular weight standards (data not shown). Hence the expressed protein, like the proteins purified from the above native sources, appears to be a homotetramer.
Table 2.
Analyses of Purified Recombinant Chlorite Dismutase
| measurement | method | recombinant Cld |
|---|---|---|
| heme type | pyridine hemochrome assay | heme b |
| heme:tetramer | pyridine hemochrome assaya | 3.7 ± 0.2 |
| Fe:tetramer | ICP-OESa | 3.6 ± 0.2 |
| subunit molecular weight | MALDI-TOF | 28.4 + 0.1 kDa |
| oligomeric state | analytical gel filtration | tetramer: ~116 kDa |
| isoelectric point | isoelectric focusing gel electrophoresis | ~9.6 |
| specific activity | iodometric titration | 3000 ± 500 U/mg |
Numbers are averages of 3 different independent samples, each with 5 averaged measurements at 5 element dependent wavelengths, all done in triplicate. In all cases, errors are reported as the standard deviations of the measurements.
Purified Cld was analyzed for bound heme and Fe (Table 2). Metal analysis by ICP-OES indicates near-stoichiometric Fe:tetramer binding (3.7 ± 0.2 per homotetramer) in the pure protein. This number agrees closely with the heme content (3.8 ± 0.3) predicted by the pyridine hemochrome assay (see below), indicating that all protein-bound iron is heme-associated. The pyridine hemochrome spectrum measured for dithionite-reduced Cld had peaks at 556 and 419 nm, and 556 nm in the reduced minus oxidized difference spectrum. This suggests that the heme incorporated into Cld is protoporphyrin IX (heme b) (36). Using an extinction coefficient of 24 mM–1 cm–1 for the released heme (36) and a Cld molecular weight of 113.6 kDa, a heme stoichiometry of 3.8 ± 0.2 per homotetramer was determined (Table 2). The extinction coefficient for enzyme-bound heme at 392 nm is ε = 99 mM–1 cm–1. Data are referenced per-heme throughout this work.
To determine if the enzyme (produced in its native state only under near-anaerobic conditions) exhibited O2 instability, cell lysates were tested for their activity after incubation at 4 °C under either N2 or air atmospheres for 0, 24, and 48 h. Within experimental error, no loss of activity was observed after either 24 or 48 h of storage.
Activity and Kinetic Analyses
Activity measurements at 25 °C and pH 6.8 were made as described above to assess yields at each step of the purification (Table 1). Purified Cld had a specific activity of 4.7 (±0.3) × 103 μmol ClO2– min–1mg–1 under these conditions.
The ClO2–:O2 stoichiometry was measured for each value of [ClO2–]initial and found to be invariably 1:1, consistent with a dismutation reaction. Stoichiometry was determined by measuring the total O2 evolved (via Clark electrode) and the amount of unreacted ClO2– in the reaction mixture. Only at the lowest chlorite concentrations used in this study was the [ClO2–]initial reduced to zero by the enzyme; otherwise, residual chlorite remained due to irreversible mechanism-based inactivation of the enzyme (Figure 5). Steady-state kinetic analysis of the enzyme showed saturation in the initial rate (4 °C, pH 6.8) with increasing chlorite concentration. This was true whether the reaction was monitored discontinuously via disappearance of chlorite or continuously via the appearance of O2 (Figure 3). Values for kcat and Km obtained by either method were identical within error (Table 3).
Figure 5.

Typical plots showing chlorite-dependent irreversible inactivation. Cld was incubated with chlorite sufficient to inactivate the sample ([heme] = 1.6 nM, [ClO2–] = 2200 μM), and the progress of reaction curve was monitored via oxygen evolution (exponential curve). After buffer exchange, no activity was observed (flat line).
Table 3.
Steady State Kinetic Parameters Measured for Clda
| condition | Km (μM) | kcat (min–1) | kcat/Km (M–1 s–1) |
|---|---|---|---|
| ClO2– consumption, 4 °C, pH 6.8b | 215 ± 25 | 4.53 (±0.2) × 105 | 3.51 × 107 |
| O2 evolution, 4 °C, pH 6.8 | 212 ± 12 | 4.50 (±0.08) × 105 | 3.54 × 107 |
Individual initial rates were measured 3 times and averaged. The Michaelis–Menten equation was fit to the data by least-squares refinement, and the fit error is reported.
Buffer used was 50 mM potassium phosphate, ionic strength = 193 mM. All initial rates used in generating these parameters were referenced per active site heme.
Viscosity Effects
Microscopic (sucrose and trehalose) and macroscopic (Ficoll 400) viscosogens were tested for their effects on kcat/Km. For sucrose, trehalose, and Ficoll 400 the kcat/Km values were 3.57 × 107 M–1 s–1, 3.62 × 107 M–1 s–1, and 3.59 × 107 M–1 s–1. These are very close to the no-viscosogen value (kcat/Km = 3.52 × 107 M–1 s–1). The three viscosogens likewise had little to no impact on either kcat or Km independently. Only minimal changes in kcat/Km in the presence of microviscosogens indicates that diffusional events (substrate binding or product release) are not rate limiting.
Product Inhibition
To test if and to what extent the products of the chlorite decomposition inhibit the reaction, initial rate plots were constructed with fixed-variable concentrations of Cl– or O2. The initial rate plots were insensitive to [O2] at the highest (saturating) and lowest (anaerobic) attainable concentrations of O2, yielding values for kcat, kcat/Km, and Km identical to those measured in air. O2 is therefore not an inhibitor.
Concentrations of Cl– below 20 mM had little apparent effect on the initial rate plot. At higher concentrations, both kcat and Km were observed to decrease (see Supporting Information). This behavior and reciprocal plots of initial rate data (with chloride at fixed-variable concentrations from 20 to 200 mM) suggested a hyperbolic mixed inhibition pattern (39). Mixed inhibition results when both free enzyme E and the enzyme–substrate (ES) complex bind the inhibitor (I), forming EI or ESI complexes with differing affinities defined by the factor α. Both ESI and ES generate product, with rates offset by the coefficient β. (See Supporting Information for a diagram of the kinetic scheme for hyperbolic mixed inhibition.)
From reciprocal plots of the initial rate versus 1/[ClO2–], Vmax (1/y-intercept) and Km(apparent) (1/x-intercept) were determined for each [Cl–]. Slope and intercept replots (versus 1/[I]) were subsequently used to determine the constants α = 0.42, β = 0.48, and KI = 225 mM. The β value below 1.0 indicates that the conversion of the ESI complex is slower than the conversion of ES to E and product P. At saturating ClO2– concentrations, a higher [I] results in a greater proportion of E in the ESI form and hence a lower kcat. The α < 1 value indicates that Cl– facilitates the formation of ESI or EI relative to the ES complex. Notably, a high concentration of Cl– is needed to see any type of inhibition at all, reflected in the high KI values. The highest chlorite concentration in any of the experiments described in this study is 2.5 mM. Product inhibition due to Cl– is therefore not significant for the rate measurements included in this study.
Kinetics of Mechanism-Based Inactivation
Each progress of reaction curve ([O2] or [ClO2–] versus t) depends on two concurrent kinetic phenomena: the reaction of enzyme and chlorite and the time- and chlorite-dependent inactivation of the enzyme. During the initial portion of the reaction, one may approximate that [E] ~ [E]initial and [S] ~ [S]initial, and enzyme inactivation can be ignored. The reaction is in the steady state and the concentrations of intermediate species are constant. Hence, plots of initial velocities reflect the degree of saturation of enzyme with substrate, according to the Michaelis–Menten model.
From the complete progress of reaction curves, it is observed that residual substrate remains after the reaction has gone to completion, even at the lowest values of [S]initial used in Figure 3. Consistent with a pseudo first order process (excess substrate, catalytic amounts of enzyme), all of the kinetic traces could be fit to single-exponential curves ([O2] = [O2]initial[1 – e–kt]) from which kobs values were obtained. A plot of kobs versus chlorite concentration is not linear, as it would be for a simple second order process. Rather, as with the initial rate plots, the plot of kobs versus [ClO2–] approaches a limiting value (Figure 4). An expression for kobs is expected to take the form kobs = kinact(max)[ClO2–]/(Kinact + [ClO2–]), where kinact(max) describes the maximal/saturating rate constant. This is consistent with an equilibrium process preceding the inactivation reaction:
Fitting the kobs versus [ClO2–] plot to this equation yields Kinact = 166 μM and kinact(max) = 1.77 min–1.
Figure 4.

Values for kobs describing the exponential decay of enzymatic activity plotted versus [chlorite]. Data were fit to the expression kobs = kinact(max) [ClO2–]/(Kinact + [ClO2–]), yielding kinact(max) = 1.77 min–1 and Kinact = 166 μM.
Inactivation, Measurement of Turnover Number, and Guaiacol Rescue
Enzyme exposed to chlorite was typically found to have lost activity after exchange into fresh buffer. Depending on the relative amount of chlorite (versus enzyme), inactivation could be complete (Figure 5). The number of turnovers (per heme) that the enzyme can catalyze before inactivation was determined using the method described by Silverman for suicide inactivation (38). Briefly, Cld was incubated with increasing equivalents of ClO2–, and the reaction was allowed to go to completion. The enzyme was then quickly exchanged into fresh buffer and the remaining activity measured via the standard assay. The residual activity is the remaining activity (per mg enzyme) divided by the activity (per mg) of a sample of enzyme before incubation in ClO2–. Residual activity was plotted versus the ClO2–/heme ratio (Figure 6), yielding a straight line with a small amount of curvature at higher ratios. In other cases, such deviations from linearity have been attributed to the ability of accumulating product to protect the enzyme from further inactivation (38). A small amount of product protection is consistent with the observed inhibition due to chloride. A line fit to the linear portion of the data allows for extrapolation to the x-axis, yielding ~1.7 × 104 ClO2– molecules decomposed per active site before inactivation (0% residual activity). The one-electron peroxidase substrate guaiacol was observed to increase the turnover number of the enzyme. A dose–response curve was first constructed in order to gauge the kinds of guaiacol concentrations needed in order to observe an effect, and the extent to which guaiacol could rescue the enzyme (see Supporting Information). A saturating effect was observed by ~500 μM or ~2400 equiv per heme. The turnover number was then remeasured in excess guaiacol (600 μM or 2850 equiv per heme), and found to increase to ~1.3 × 105 (Figure 6).
DISCUSSION
Investigations of anaerobic biochemistry are revealing an intriguing array of novel heme enzymes. Chlorite dismutase, a hallmark of anaerobic perchlorate respiration in bacteria (32), bears no significant homology to any other enzyme in the NCBI database. It catalyzes the formation of the dioxygen bond, a reaction that is known to metalloporphyrin chemistry (23, 40) but novel as the primary function of a heme enzyme, or for that matter any enzyme beyond photosystem II. Intriguingly, chlorite dismutase homologues are found in a broad variety of prokaryotes, the majority of which are not known to respire perchlorate or chlorate. The unique ability of the enzyme's heme to steer chlorite toward this reactivity, its unusual sequence, and its potential for environmental remediation, clean O2-generation, and sensor technology have led us to pursue heterologous expression and characterization of the chlorite dismutase from the sequenced perchlorate respirer and anaerobic benzene oxidizer, Dechloromonas aromatica RCB.
By expressing the protein using the E. coli Tuner host, minimal inducing agent, low temperature (20 °C), a rich growth medium, and hemin supplementation, a large amount of soluble, fully heme-incorporating enzyme was produced (Figure 1, Table 1). The enzyme was expressed without its N-terminal signal peptide for periplasmic export, as prior work indicated that the peptide would likely not be correctly cleaved by E. coli (25). Taking advantage of the protein's unusually high isoelectric point (Table 2), a simple, rapid, and highly efficient two-step chromatographic procedure was developed. Using these methods for expression and purification, large amounts of highly pure protein (~55 mg/L growth culture, Figure 1) are routinely obtained. This compares very favorably to yields of the native protein purified from D. agitata and I. dechloratans, reported as 0.0307 mg of protein per g of wet cell paste and ~1 mg of pure protein per L of cell culture, respectively (25, 41). The native hosts are moreover difficult to grow relative to E. coli and require comparatively lengthy column purification procedures for protein recovery. The purified recombinant enzyme is highly active, with a specific activity of 4.7 × 103 (±0.3 × 103) μmol min–1 mg–1 (pH 7.0, 25 °C). The specific activity measured for the closely related enzyme from D. agitata (62% identity) was of a similar magnitude: 1.9 × 103 μmol min–1 mg–1 (temperature and pH not reported) (41). Chemical analysis (Table 2) indicates near 1:1 Fe:subunit stoichiometry in the purified enzyme (ICP-OES), based on a measured subunit mass of 28.1 kDa (MALDI-MS) and native tetramer mass of 116 kDa. All of the iron could be accounted for as heme-associated, according to the pyridine hemochrome assay (36). While the measured subunit/tetramer masses and the 1:1 heme stoichiometry are in agreement with previous measurements on native chlorite dismutases isolated from I. dechloratans (25), strain GR-1 (27), and Dechloromonas agitata strain CKB (41), the heterologously expressed enzyme from I. dechloratans had only 0.3 heme per subunit (25). Hence, the presently reported expression and purification procedures offer significant advantages over prior approaches, and represent the first successful effort toward expression of a highly functional chlorite dismutase.
Chlorite dismutase has telling similarities and differences with heme-dependent catalases and peroxidases. Like most heme peroxidases, the UV/visible and EPR spectra of Cld indicate an axial histidine ligand to the heme (Figure 2, Table 4) (25, 42), although the Soret band of ferric Cld is notably blue-shifted relative to HRP's. Heme peroxidases, catalases, and Cld all moreover react with oxygen-atom-donor substrates, indicating functional similarities even though Cld shares negligible sequence homology with either of these types of enzyme. Steady state kinetic analysis of Cld further shows saturation in the initial rate with increasing chlorite concentration, allowing for the measurement of values for Km and Vmax/Km (Figure 3, Table 3). By contrast, the heme peroxidases and catalase generally do not exhibit saturation behavior and hence do not adhere to the Michaelis–Menten model.
Figure 2.

UV/visible absorption spectra for oxidized/as-isolated (solid line) and reduced (dashed line) chlorite dismutase. The inset shows the visible region of the spectra on an amplified scale.
Table 4.
UV/Visible Bands Measured for Chlorite Dismutases and Representative Heme Peroxidases
| protein | Soret (nm) | visible bands (nm) | ref |
|---|---|---|---|
| Fe(III) Cld (D. aromatica) | 388 | 510, 644 | this work |
| Fe(III) Cld (I. dechloratans) | 392 | 509, 648 | (25) |
| Fe(III) Cld (GR-1) pH 7 | 394 | NRa | (24) |
| Fe(III) HRPb pH 7 | 402 | 510, 557, 588 | (42) |
| Fe(III) CPOc | 403 | 515, 542, 650 | (49) |
| Fe(II) Cld (D. aromatica) | 434 | 555, 585 | this work |
| Fe(II) Cld (I. dechloratans) | 434 | 555, 586 | (25) |
| Fe(II) Cld (GR-1) pH 7 | 409 | NR | (24) |
| Fe(II) HRP | 437 | 557, 587 | (42) |
| Fe(II) CPO | 409 | 550 | (49) |
NR = not recorded.
HRP = horseradish peroxidase.
CPO = chloroperoxidase (axial heme ligand = cysteine).
Saturation in Cld's initial rate with increasing [chlorite] indicates that an intermediate species accumulates under the given reaction conditions. In the simplest case, this could occur in a two-step process:
| (1) |
where . Mechanism 1 is consistent with the observed 1:1 ClO2–:O2 stoichiometry (see Supporting Information). Further, the lack of a detectable effect of viscosity on kcat/Km indicates that the overall reaction rate is not limited by the diffusional encounter of enzyme and substrate, in keeping with mechanism 1, where the intermediate [E·ClO2–] complex forms prior to a slower, concerted dismutation step (k2).
Alternatively, a mechanism more in keeping with catalases, peroxidases, and their known heme-oxygen intermediates could be proposed (mechanism 2). In this case, chlorite first transfers an oxygen atom to the ferric heme, generating a catalase- or peroxidase-like ferryl-oxo porphyrin cation radical (Compound I) plus hypochlorite as the intermediate. The oxygen atoms of Compound I and hypochlorite then recombine to give the O2 and Cl– products. To account for the Compound I intermediate, the observed saturation in the initial rate plots, and the steps that would be irreversible in a catalase/peroxidase-like system, a more elaborate mechanism must be proposed, e.g.:
| (2) |
Consistent with peroxidases and catalases, the Compound I formation and decay steps are shown as irreversible. Moreover, in these enzymes, Compound I decays more quickly than it forms . This gives rise to the observed lack of saturation in their initial rate plots, as no intermediate accumulates (42). For Cld, the accumulating Michaelis intermediate [E·ClO2–] is therefore modeled as a simple, reversibly forming collision complex, although it could also form via an irreversible step where leads to its accumulation, as in mechanism 1. The lack of a viscosity effect indicates that neither [E·ClO2–] formation nor product release is rate limiting, making conversion of [E·ClO2–] to Compound I + OCl– (k2) the most plausible rate limiting step in mechanism 2. Alternatively, a microscopic step not explicitly accounted for in this mechanism, such as displacement of a water ligand from the heme by the substrate, could be rate limiting. The data presented in this study cohere with either mechanism 1 or 2. Future studies will aim at addressing whether a simple concerted process as in mechanism 1 or stepwise mechanism including oxygen atom transfer (mechanism 2) is at work.
To complete the basic steady-state characterization, we examined the possible inhibitory effects of product Cl– and O2 on the forward reaction. A classic product inhibitor works by competing with substrate for binding to the free enzyme. O2 has no apparent effect at maximal attainable concentrations. Cl–, however, acts as a mixed inhibitor, binding with low affinity to both the free enzyme (KI[E] = 225 mM) and enzyme–substrate complex (KI[ES] = 95.6 mM). The high KI[E] indicates that free enzyme has little affinity for chloride, suggesting that substrate binding is controlled by more than simple electrostatic charge. The relative insensitivity of the reaction to Cl– moreover indicates that this is an appropriate “innocent” counterion to be used in adjusting the ionic strength of buffers. Interestingly, chloride also interacts with greater affinity with ES. If ES is indeed an [E·ClO2–] collision complex, the observed Cl– inhibition suggests that there is additional room in the active site pocket for binding a second ion. A second, non-Fe-coordinating site for ion binding would be necessary if OCl– forms and recombines with Compound I, as in mechanism 2.
Finally, in the course of these steady-state experiments, we observed that some chlorite typically remained in the reaction mixtures once chlorite dismutation ceased. When the reacted enzyme was exchanged into fresh buffer, no activity was observed (Figure 5). The heme absorbance of the inactivated enzyme was moreover abolished, suggesting heme destruction as the means by which the observed irreversible enzyme inactivation occurred. Titrating the enzyme's UV/visible spectrum with chlorite (Figure 7) shows that the heme chromophore is indeed bleached after the addition of fewer than 2.4 × 104 equiv, in good agreement with the measured turnover number of ~1.7 × 104 per heme (Figure 6). Heme peroxidases are known to undergo suicide inactivation under certain conditions. In the presence of excess H2O2 and in the absence of oxidizable substrate, HRP is completely inactivated after only ~600 turnovers (43). Under such conditions, H2O2 acts as a reductant toward the HRP Compound I in a two-electron, catalase-like process (forming water and O2), or in two successive reactions, forming first the ferryl-oxo/neutral-porphyrin Compound II and then a ferric-superoxide species (Compound III). Catalase reactivity is believed to serve a protective function for the enzyme, while Compound III is responsible for enzyme inactivation via heme destruction (44). As little as 2 equiv of the oxygen-donor mCPBA can likewise inactivate HRP in the absence of oxidizable substrate. The first equivalent forms Compound I, which then reacts with a second equivalent of mCPBA (44). An intermediate (P965) forms and decays by competing pathways to give either Compound II (plus mCPBA peroxyl radical) or an inactivated form of the enzyme. Finally, HRP is irreversibly inactivated after incubation with a large excess of Br– and H2O2 at acidic pH (5) (45). HRP's Compound I oxidizes bromide to produce OBr–/HOBr, which then hydroxylates or brominates the vinyl substituents on the heme. Though the heme ring is not disrupted and the Soret absorbance remains, the functionalized HRP is inactive. Interestingly, myeloperoxidases and lactoperoxidases, which generate OCl–/HOCl by a similar mechanism as their primary function, are protected from inactivation by their uniquely functionalized heme side chains (46). These cross-link the heme to the enzyme and give it a distinctive UV/visible spectrum. The Cld heme is neither cross-linked to the enzyme nor similar to myeloperoxidase in its spectrum. Hence we do not expect Cld to have evolved similar self-protection strategies, though it reacts with a similar substrate. Also, because Cld inactivation occurs with quenching of the heme chromophore (Figure 7), we expect that scission of the porphyrin ring and not functionalization of the vinyl groups by OCl– is at work.
Figure 7.

Bleaching of the heme chromophore observed with successive increments of chlorite. The initial spectrum was taken of pure enzyme (10.5 μM heme) that had not been exposed to chlorite. Subsequent spectra were each measured after addition of 1360 equiv of chlorite (3 μL aliquots of 565 mM chlorite solution). The final spectrum where the chromophore appears to be fully bleached corresponds to the addition of 2.4 × 104 equiv of chlorite. Spectra were corrected for dilution effects.
Cld's half-life of 0.39 min in the presence of saturating chlorite is short relative to that of either HRP isoform C (~54 min) or ascorbate peroxidase (~2.5 min) (43, 47) in the presence of excess H2O2. Its exceptional efficiency, however, allows Cld to catalyze close to 20 thousand turnovers (per heme) before self-destructing. In the presence of a large excess of the peroxidase substrate guaiacol (>2,000 equiv or >10× Kinact), the turnover number increases by approximately an order of magnitude, but is never completely rescued (Supporting Information). Peroxidase substrates were likewise shown to rescue both HRP (48) and ascorbate peroxidase from H2O2-dependent inactivation (47). In the latter case, as little as 200 equiv of ascorbate relative to enzyme (lower micromolar concentrations) was sufficient to rescue activity completely. Hence, Cld is much more susceptible to chlorite-dependent inactivation and less amenable to substrate-rescue, relative to these peroxidases. In peroxidases, one-electron substrates such as guaiacol are believed to compete with H2O2 for reaction with Compound II, protecting the peroxidases from inactivation. Together, quenching of the heme chromophore and the protection offered by guaiacol both point toward the involvement of Compound II in Cld inactivation, although it is unclear whether its precursor Compound I is on the pathway toward catalysis. Future work will aim at clarifying the catalytic and inactivation mechanisms, and the nature of possible intermediates in either.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Ching Shang from the laboratory of Prof. J.D. Coates (Department of Plant and Microbial Biology, University of California, Berkeley) for providing the cosmid containing the chlorite dismutase gene. We thank Dr. John Evans for critically reading this manuscript.
Footnotes
Supported by the National Institutes of Health R03 ES014390-01; B.R.S. is supported by an EPA STAR graduate fellowship FP-91690601-0.
Abbreviations: IPTG, isopropyl-beta-d-thiogalactopyranoside; Cld, chlorite dismutase; Pcr, perchlorate reductase; NCBI, National Center for Bioinformatics; MALDI-TOF-MS, matrix assisted laser desorption ionization time-of-flight mass spectrometry; ICP-OES, inductively coupled plasma optical emission spectroscopy; HRP, horseradish peroxidase; mCPBA = meta-chloroperbenzoic acid.
SUPPORTING INFORMATION AVAILABLE
Details of Cld's expression optimization, an image of the isoelectric focusing gel, a table of stoichiometries of O2 evolved/ClO2– decomposed, chloride inhibition data and a complete kinetic analysis, and a figure showing activity rescue versus guaiacol concentration. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Sligar SG, Makris TM, Denisov IG. Thirty years of microbial P450 monooxygenase research: Peroxo-heme intermediatess—The central bus station in heme oxygenase catalysis. Biochem. Biophys. Res. Commun. 2005;338:346–354. doi: 10.1016/j.bbrc.2005.08.094. [DOI] [PubMed] [Google Scholar]
- 2.Capaldi RA. Structure and function of cytochrome-coxidase. Annu. ReV. Biochem. 1990;59:569–596. doi: 10.1146/annurev.bi.59.070190.003033. [DOI] [PubMed] [Google Scholar]
- 3.Moreno-Vivian C, Cabello P, Martinez-Luque M, Blasco R, Castillo F. Prokaryotic nitrate reduction: molecular properties and functional distinction among bacterial nitrate reductases. J. Bacteriol. 1999;181:6573–6584. doi: 10.1128/jb.181.21.6573-6584.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fritz G, Einsle O, Rudolf M, Schiffer A, Kroneck PMH. Key bacterial multi-centered metal enzymes involved in nitrate and sulfate respiration. J. Mol. Microbiol. Biotechnol. 2005;10:223–233. doi: 10.1159/000091567. [DOI] [PubMed] [Google Scholar]
- 5.Oremland RS, Kulp TR, Blum JS, Hoeft SE, Baesman S, Miller LG, Stolz JF. A microbial arsenic cycle in a salt-saturated, extreme environment. Science. 2005;308:1305–1308. doi: 10.1126/science.1110832. [DOI] [PubMed] [Google Scholar]
- 6.Oremland RS, Stolz JF. The ecology of arsenic. Science. 2003;300:939–944. doi: 10.1126/science.1081903. [DOI] [PubMed] [Google Scholar]
- 7.Stolz JF, Oremland RS. Bacterial respiration of arsenic and selenium. FEMS Microbiol. Rev. 1999;23:615–627. doi: 10.1111/j.1574-6976.1999.tb00416.x. [DOI] [PubMed] [Google Scholar]
- 8.Kletzin A, Urich T, Muller F, Bandeiras TM, Gomes CM. Dissimilatory oxidation and reduction of elemental sulfur in thermophilic archaea. J. Bioenerg. Biomembr. 2004;36:77–91. doi: 10.1023/b:jobb.0000019600.36757.8c. [DOI] [PubMed] [Google Scholar]
- 9.Lovley DR, Holmes DE, Nevin KP. Dissimilatory Fe(III) and Mn(IV) reduction. Adv. Microb. Physiol. 2004;49:219–286. doi: 10.1016/S0065-2911(04)49005-5. [DOI] [PubMed] [Google Scholar]
- 10.Coates JD, Chakraborty R, Lack JG, O'Connor SM, Cole KA, Bender KS, Achenbach LA. Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature. 2001;411:1039–1043. doi: 10.1038/35082545. [DOI] [PubMed] [Google Scholar]
- 11.Coates JD, Achenbach LA. Microbial perchlorate reduction: rocket-fuelled metabolism. Nat. Rev. Microbiol. 2004;2:569–580. doi: 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- 12.Urbansky ET, Schock MR. Issues in managing the risks associated with perchlorate in drinking water. J. Environ. Manage. 1999;56:79–95. [Google Scholar]
- 13.Motzer WE. Perchlorate: problems, detection, and solutions. Environ. Forens. 2001;2:301–311. [Google Scholar]
- 14.EPA . Perchlorate environmental contamination: toxicological review and risk characterization (2002 External Review Draft) Washington, D.C.: 2002. [Google Scholar]
- 15. EPA 816-F-03-016, June2003; available for download at http://www.epa.gov/safewater/mcl.html.
- 16.Chakraborty R, Coates JD. Anaerobic degradation of monoaromatic hydrocarbons. Appl. Microbiol. Biotechnol. 2004;64:437–446. doi: 10.1007/s00253-003-1526-x. [DOI] [PubMed] [Google Scholar]
- 17.Danielsson H, Stenklo TK, Karlsson J, Nilsson T. A gene cluster for chlorate metabolism in Ideonella dechloratans. Appl. EnViron. Microbiol. 2003;69:5585–5592. doi: 10.1128/AEM.69.9.5585-5592.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giblin T, Frankenberger WT. Perchlorate and nitrate reductase activity in the perchlorate-respiring bacterium perc1ace. Microbiol. Res. 2001;156:311–315. doi: 10.1078/0944-5013-00111. [DOI] [PubMed] [Google Scholar]
- 19.Kengen SWM, Rikken GB, Hagen WR, van Ginkel CG, Stams AJM. Purification and characterization of (per)chlorate reductase from the chlorate-respiring strain GR-1. J. Bacteriol. 1999;181:6706–6711. doi: 10.1128/jb.181.21.6706-6711.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okeke BC, Frankenberger WT. Molecular analysis of a perchlorate reductase from a perchlorate-respiring bacterium perc1ace. Microbiol. Res. 2003;158:337–344. doi: 10.1078/0944-5013-00213. [DOI] [PubMed] [Google Scholar]
- 21.Collman JP, Tanaka H, Hembre RT, Brauman JI. Metalloporphyrin-catalyzed oxidation of saturated-hydrocarbons with sodium-chlorite. J. Am. Chem. Soc. 1990;112:3689–3690. [Google Scholar]
- 22.Collman JP, Boulatov R, Sunderland CJ, Shiryaeva IM, Berg KE. Electrochemical metalloporphyrin-catalyzed reduction of chlorite. J. Am. Chem. Soc. 2002;124:10670–10671. doi: 10.1021/ja027306l. [DOI] [PubMed] [Google Scholar]
- 23.Slaughter LM, Collman JP, Eberspacher TA, Brauman JI. Radical autoxidation and autogenous O-2 evolution in manganese-porphyrin catalyzed alkane oxidations with chlorite. Inorg. Chem. 2004;43:5198–5204. doi: 10.1021/ic049922j. [DOI] [PubMed] [Google Scholar]
- 24.Hagedoorn PL, de Geus DC, Hagen WR. Spectroscopic characterization and ligand-binding properties of chlorite dismutase from the chlorate respiring bacterial strain GR-1 Eur. J. Biochem. 2002;269:4905–4911. doi: 10.1046/j.1432-1033.2002.03208.x. [DOI] [PubMed] [Google Scholar]
- 25.a Stenklo K, Thorell HD, Bergius H, Aasa R, Nilsson T. Chlorite dismutase from Ideonella dechloratans. J. Biol. Inorg. Chem. 2001;6:601–607. doi: 10.1007/s007750100237. [DOI] [PubMed] [Google Scholar]; b Thorell HD, Karlsson J, Portelius E, Nilsson T. Cloning, characterisation, and expression of a novel gene encoding chlorite dismutase from Ideonella dechloratans. Biochim. Biophys. Acta 1577. 2002:445–451. doi: 10.1016/s0167-4781(02)00446-3. [DOI] [PubMed] [Google Scholar]
- 26.Thorell HD, Beyer NH, Heegaard NHH, Ohman M, Nilsson T. Comparison of native and recombinant chlorite dismutase from Ideonella dechloratans. Eur. J. Biochem. 2004;271:3539–3546. doi: 10.1111/j.0014-2956.2004.04290.x. [DOI] [PubMed] [Google Scholar]
- 27.van Glinkel CG, Rikken GB, Kroon AGM, Kengen SWM. Purification and characterization of chlorite dismutase: A novel oxygen-generating enzyme. Arch. Microbiol. 1996;166:321–326. doi: 10.1007/s002030050390. [DOI] [PubMed] [Google Scholar]
- 28.Xu JL, Logan BE. Measurement of chlorite dismutase activities in perchlorate respiring bacteria. J. Microbiol. Met. 2003;54:239–247. doi: 10.1016/s0167-7012(03)00058-7. [DOI] [PubMed] [Google Scholar]
- 29.O'Connor SM, Coates JD. Universal immunoprobe for (per)chlorate-reducing bacteria. Appl. Environ. Microbiol. 2002;68:3108–3113. doi: 10.1128/AEM.68.6.3108-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaudhuri SK, O'Connor SM, Gustavson RL, Achenbach LA, Coates JD. Environmental factors that control microbial perchlorate reduction. Appl. Environ. Mirobiol. 2002;68:4425–4430. doi: 10.1128/AEM.68.9.4425-4430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison BS, Eberli D, Lee SJ, Atala A, Yoo JJ. Oxygen producing biomaterials for tissue regeneration. Biomaterials. 2007;28:4628–4634. doi: 10.1016/j.biomaterials.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Bender KS, O'Connor SA, Chakraborty R, Coates JD, Achenbach LA. Sequencing and transcriptional analysis of the chlorite dismutase gene of Dechloromonas agitata and its use as a metabolic probe. Appl. Environ. Microbiol. 2002;68:4820–4826. doi: 10.1128/AEM.68.10.4820-4826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahangian S, Hager LP. The reaction of chloroperoxidase with chlorite and chlorine dioxide. J. Biol. Chem. 1981;256:6034–6040. [PubMed] [Google Scholar]
- 34.Gustafsson JA, Rondahl L, Bergman J. Iodosyl-benzene derivatives as oxygen donors in cytochrome-P-450 catalyzed steroid hydroxylations. Biochemistry. 1979;18:865–870. doi: 10.1021/bi00572a020. [DOI] [PubMed] [Google Scholar]
- 35.Jakopitsch C, Spalteholz H, Furtmüller PG, Arnhold J, Obinger C. Mechanism of reaction of horseradish peroxidase with chlorite and chlorine dioxide. J. Inorg. Biochem. 2008;102:293–302. doi: 10.1016/j.jinorgbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Berry EA, Trumpower BL. Simultaneous determination of hemes a, b, and c from pyridine hemocrome spectra. Anal. Biochem. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 37.Jeffery GH, Bassett J, Mendham J, Denney RC. Vogel's Textbook of Quantitative Chemical Analysis. 5th ed. Longman Scientific and Technical; New York: 1989. [Google Scholar]
- 38.Silverman RB. Mechanism-Based Enzyme InactiVation: Chemistry and Enzymology. I. CRC Press, Inc.; Evanston, IL: 1988. [Google Scholar]
- 39.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley and Sons; New York: 1997. [Google Scholar]
- 40.Limburg J, Vrettos JS, Chen HY, de Paula JC, Crabtree RH, Brudvig GW. Characterization of the O-2-evolving reaction catalyzed by [(terpy)(H2O)Mn-III(O)(2)Mn-IV(OH2)(terpy)](NO3) (terpy)2,2′: 6,2′′-terpyridine). J. Am. Chem. Soc. 2001;123:423–430. doi: 10.1021/ja001090a. [DOI] [PubMed] [Google Scholar]
- 41.Coates JD, Michaelidou U, Bruce RA, O'Connor SM, Crespi JN, Achenbach LA. Ubiquity and diversity of dissimilatory (per)chlorate-reducing bacteria. Appl. EnViron. Microbiol. 1999;65:5234–5241. doi: 10.1128/aem.65.12.5234-5241.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunford HB. Heme Peroxidases. Wiley-VCH; New York: 1999. [Google Scholar]
- 43.Arnao MB, Acosta M, Delrio JA, Varon R, Garcia-canovas F. A kinetic-study on the suicide inactivation of peroxidase by hydrogen-peroxide. Biochim. Biophys. Acta. 1990;1041:43–47. doi: 10.1016/0167-4838(90)90120-5. [DOI] [PubMed] [Google Scholar]
- 44.Valderrama B, Ayala M, Vazquez-Duhalt R. Suicide inactivation of peroxidases and the challenge of engineering more robust enzymes. Chem. Biol. 2002;9:555–565. doi: 10.1016/s1074-5521(02)00149-7. [DOI] [PubMed] [Google Scholar]
- 45.Huang LS, Wojciechowski G, Ortiz de Montellano PR. Prosthetic heme modification during halide ion oxidation. Demonstration of chloride oxidation by horseradish peroxidase. J. Am. Chem. Soc. 2005;127:5345–5353. doi: 10.1021/ja050278x. [DOI] [PubMed] [Google Scholar]
- 46.Furtmüller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C. Active site structure and catalytic mechanisms of human peroxidases. Arch. Biochem. Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 47.Hiner ANP, Rodriguez-Lopez JN, Arnao MB, Raven EL, Garcia-Canovas F, Acosta M. Kinetic study of the inactivation of ascorbate peroxidase by hydrogen peroxide. Biochem. J. 2000;348:321–328. [PMC free article] [PubMed] [Google Scholar]
- 48.Arnao MB, Acosta M, del Rio JA, Garcia-Canovas F. Inactivation of peroxidase by hydrogen peroxide and its protection by a reductant agent. Biochim. Biophys. Acta. 1990;1038:85–89. doi: 10.1016/0167-4838(90)90014-7. [DOI] [PubMed] [Google Scholar]
- 49.Morris DR, Hagar LP. Chloroperoxidase. I. Isolation and properties of the crystalline glycoprotein. J. Biol. Chem. 1966;241:1763–1768. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.