

Abstract
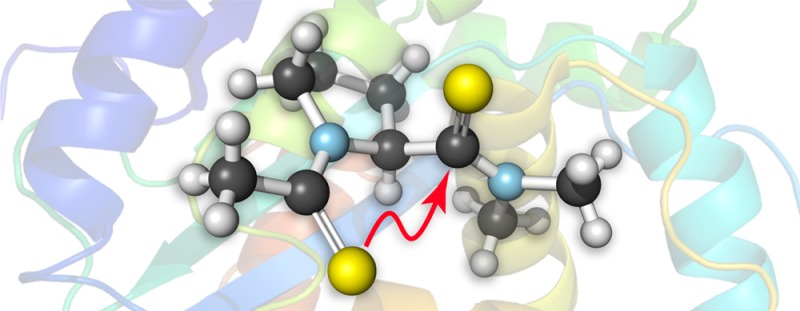
Carbonyl–carbonyl interactions between adjacent backbone amides have been implicated in the conformational stability of proteins. By combining experimental and computational approaches, we show that relevant amidic carbonyl groups associate through an n→π* donor–acceptor interaction with an energy of at least 0.27 kcal/mol. The n→π* interaction between two thioamides is 3-fold stronger than between two oxoamides due to increased overlap and reduced energy difference between the donor and acceptor orbitals. This result suggests that backbone thioamide incorporation could stabilize protein structures. Finally, we demonstrate that intimate carbonyl interactions are described more completely as donor–acceptor orbital interactions rather than dipole–dipole interactions.
Protein architecture is mediated by a suite of noncovalent interactions within and between polypeptide chains, including the hydrophobic effect, hydrogen bonding, Coulombic interactions, and van der Waals interactions.1 We have put forth an n→π* interaction as an additional means by which peptide bonds themselves interact.2 In this n→π* interaction, a carbonyl oxygen donates lone pair (n) electron density into another carbonyl group (Figure 1). Such donation occurs when the donor and acceptor form a short contact along the Bürgi–Dunitz trajectory for nucleophilic addition.3 These interactions have been implicated in many systems, including small molecules,4 peptides,5 proteins,6 peptoids,7 and nucleic acids.8
Figure 1.

(A) Notion of a carbonyl–carbonyl n→π* interaction in amide 3. (B) Three-dimensional orbital rendering and (C) contour plot showing overlap of n and π* orbitals in the trans exo conformation of 3. Images were rendered with NBOView 1.1.
We and others have used a torsion balance in a proline model system to characterize energetic relationships of importance to peptide and protein structure (Figure 2).2,9,5c,5g,5j,5m,10 Both the cis and trans isomers of the N-acetyl proline peptide bond are populated at room temperature and can be distinguished by NMR spectroscopy due to their slow interconversion. As the n→π* interaction is only possible in the trans isomer, the ratio of isomers (Ktrans/cis) reports on the energy of the interaction. All previous studies employed esters as the n→π* acceptor (1); because esters are more electrophilic than the amides found in proteins, those studies overestimated the strength of n→π* interactions at 0.7 kcal/mol.2c,9 Hence, we sought to determine the energy of a prototypical n→π* interaction between two amides. Primary and secondary amides can both donate hydrogen bonds to the acetyl group, obscuring the n→π* interaction in our analysis;11 thus, we elected to examine the tertiary dimethyl amide (3).
Figure 2.

Compounds used to evaluate n→π* interactions in torsion balance analyses.
In D2O, the value of Ktrans/cis for amide 3, like that for ester 1, is greater than unity (Figure 3A). We then employed density functional theory (DFT) calculations at the B3LYP/6-311+G(2d,p) level of theory with natural bond orbital (NBO) analysis to estimate the energy of the n→π* interaction.12 We optimized the geometry of 3 in both the Cγ-endo and Cγ-exo puckers of its pyrrolidine ring (Figure 4) and found the corresponding n→π* energies to be 0.14 and 0.53 kcal/mol, respectively. At room temperature, proline exists ∼66% in the endo pucker and ∼34% in the exo pucker.2b Based on this ratio, we estimate the energy of the n→π* interaction in 3 to be En→π* = 0.27 kcal/mol (Figure 3B). This interaction is weaker than that in 1, which is consistent with the lower electrophilicity of the amide acceptor relative to the ester. Importantly, because the tertiary amide is less electrophilic and more stericially encumbered than the secondary amides common in proteins, the values we report here are likely to underestimate the energy of an n→π* interaction between most peptide bonds. Thus, we expect that n→π* interactions in proteins contribute ≥0.27 kcal/mol of stabilization per interaction.
Figure 3.

(A) Values of Ktrans/cis for compounds 1–6 in D2O at 25 °C. (B) Energy of n→π* interactions in 1–6 from second-order perturbation theory. (C) Overlap integrals between the n and π* orbitals of 3–6. (D) Reciprocal of the energy gap between the n and π* orbitals of 3–6. Data for esters 1 and 2 are from ref (9).
Figure 4.
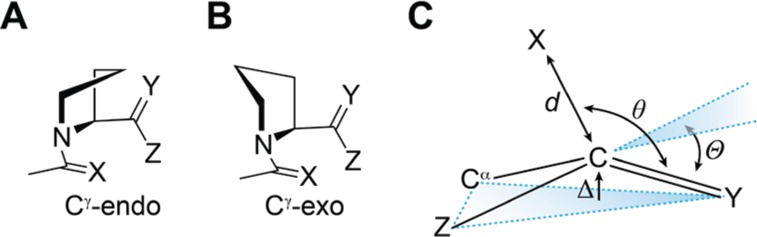
(A) Cγ-endo and (B) Cγ-exo pyrrolidine ring pucker of compounds 1–6. (C) Parameters that denote pyramidalization of carbonyl groups due to n→π* donation.
Previously, we demonstrated that the substitution of an amide donor (i.e., 1) with a thioamide (2) increases n→π* donation to an ester carbonyl (Figure 3A).9,13 This increase arises from sulfur being a better electron-pair donor than its oxygen congener. This finding suggested to us that backbone thioamide substitution could enhance the n→π* interaction between carbonyl groups and stabilize the folded structures of proteins. Still, the quality of a thioamide as an n→π* acceptor has been predicted only computationally.14 Hence, we synthesized 4–6 to evaluate thioamides as both donors and acceptors of the n→π* interaction for the first time.
Replacing the donor of 3 with the larger thioamide to yield 4 increased the value of Ktrans/cis, despite the added steric clash, confirming that sulfur is a stronger n→π* donor than oxygen (Figure 3A). Interestingly, whereas replacing the acceptor of 3 with a thioamide to yield 5 reduced Ktrans/cis, replacing the acceptor of 4 with a thioamide to yield 6 led to an increase in Ktrans/cis. Our NBO analyses show that replacing the acceptor with a thioamide reduces orbital overlap with the donor (Figure 3C), providing a rational basis for the value of Ktrans/cis for 5 being lower than that for 3.
NBO analysis of thioamides 3 and 5 revealed another pertinent quantum mechanical attribute. The π* orbital of a thioamide is lower in energy than that of an amide, reducing the energy gap between donor and acceptor orbitals (Figure 3D). From second-order perturbation theory, the energy released by the mixing of two orbitals is proportional to the reciprocal of the energy gap between those orbitals. Thus, though a thioamide acceptor overlaps less with an n→π* donor (Figure 3C), a smaller energy gap between the donor and acceptor can produce more effective orbital mixing and a stronger interaction overall. The consequences are apparent in thioamide 6, in which the n→π* interaction is particularly strong at 0.88 kcal/mol (Figure 3B), demonstrating that pairs of thioamides engage in significantly stronger n→π* interactions than do pairs of analogous amides.
As the n→π* interaction populates the π* orbital of the acceptor, it induces pyramidalization of the acceptor toward the donor (Figure 4C). This distortion is detectable by X-ray diffraction analysis and can provide a signature of n→π* interactions in small molecules and peptides.4,5k,9,13,15 Hence, we conducted X-ray diffraction analysis of crystalline 4–6 to search for this signature of an n→π* interaction. Thioamide 4 crystallized as its cis isomer and therefore does not show an n→π* interaction, leaving the acceptor nearly planar (Table 1). Thioamides 5 and 6 both crystallized as their trans isomer, with 6 crystallizing in both pyrrolidine ring puckers. In both 5 and 6, the acceptor carbon is pyramidalized toward the donor significantly more than in 4, denoting a stronger n→π* interaction. In addition, pyramidalization of the acceptor in both conformations of 6 is greater than in 5, which is consistent with the stronger n→π* interaction in 6. Moreover, the greater pyramidalization of the acceptor in 6-exo than in 6-endo confirms that the exo ring pucker of proline promotes stronger n→π* interactions.16 Indeed, the pyramidalization in 6-exo is among the largest observed to date in this proline model system.9,17 These observations are also consistent with the pyramidalization in crystal structures of thioamide-containing peptides (see: Figure S1).
Table 1. Conformational Parameters of Thioamidesa.
| compound | conformation | d (Å) | θ (deg) | Δ (Å) | Θ (deg) |
|---|---|---|---|---|---|
| 4 | cis, endo | 4.6158(14) | 66.96(7) | 0.0035(14) | 0.43(17) |
| 5 | trans, endo | 3.2529(12) | 92.19(4) | 0.0237(8) | 2.61(9) |
| 6 | trans, endo | 3.4248(16) | 96.11(6) | 0.0243(17) | 2.70(19) |
| 6 | trans, exo | 3.2433(15) | 101.92(7) | 0.0392(16) | 4.36(18) |
From X-ray diffraction analysis of the crystalline compound. Parameters are defined in Figure 4.
These data further establish the quantum mechanical nature of intimate carbonyl–carbonyl interactions. Some have argued that these interactions are primarily dipolar in nature.18 The dipole moment of an amide is greater than that of an ester,19 so if a dipolar interaction is dominant, replacing the ester of 1 with the amide in 3 should cause an increase in Ktrans/cis, which is contrary to observation (Figure 3A). Moreover, as a thioamide has a still larger dipole moment than an amide,20 a dipolar origin would predict a larger value of Ktrans/cis in thioamide 5 than in amide 3. That was not observed in our experiments. Finally, in a dipolar interaction, neither of the participating groups has a defined role; rather, they interact symmetrically. Thus, if intimate carbonyl interactions are dipolar in nature, then substituting either amide with a thioamide should have a comparable effect on Ktrans/cis. The conformational preferences of 4 and 5 suggest otherwise: substituting the n→π* donor amide with a thioamide increases Ktrans/cis, whereas replacing the acceptor decreases Ktrans/cis. These data affirm that a dipolar mechanism is insufficient to describe intimate interactions between carbonyl groups. Instead, the data are more consistent with an electronic donor–acceptor effect like the n→π* interaction.
Individual n→π* interactions between amides are relatively weak. In abundance, however, they could make a significant contribution to the conformational stability of a protein. We note another implication as well. Shifts in the equilibrium between α-helices and β-sheets have been implicated in amyloid fibrillogenesis.21 Hydrogen bonding, which is operative in both α-helices and β-sheets,22 is unlikely to affect this equilibrium decisively. In contrast, n→π* interactions are common in α-helices but not β-sheets,6b and thus could play a critical role in the maintenance of protein homeostasis. In addition, our finding that the n→π* interaction between two thioamides is 3-fold stronger than that between two amides (Figure 3B) encourages efforts to exploit thioamides to enhance conformational stability in peptides and proteins.23,24 Finally, as these interactions are not included in conventional force fields, we argue that accounting for the n→π* interaction could improve the accuracy of computational investigations of proteins.
□. Acknowledgments
We are grateful to Brian Dolinar (University of Wisconsin—Madison) for technical assistance and to Amit Chouhdary (Harvard University) and Grant Krow (Temple University) for contributive discussions. R.W.N. was supported by NIH Biotechnology Training Grant T32 GM008349. This work was Supported by grants R01 AR044276 (NIH) and CHE-1124944 (NSF). High-performance computing was supported by grant CHE-0840494 (NSF).
Supporting Information Available
Synthetic and analytical procedures, and computational data on compounds 3–6. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Anfinsen C. B. Science 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]; b Dill K. A. Biochemistry 1990, 29, 7133–7155. [DOI] [PubMed] [Google Scholar]
- a Bretscher L. E.; Jenkins C. L.; Taylor K. M.; DeRider M. L.; Raines R. T. J. Am. Chem. Soc. 2001, 123, 777–778. [DOI] [PubMed] [Google Scholar]; b DeRider M. L.; Wilkens S. J.; Waddell M. J.; Bretscher L. E.; Weinhold F.; Raines R. T.; Markley J. L. J. Am. Chem. Soc. 2002, 124, 2497–2505. [DOI] [PubMed] [Google Scholar]; c Hinderaker M. P.; Raines R. T. Protein Sci. 2003, 12, 1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hodges J. A.; Raines R. T. Org. Lett. 2006, 8, 4695–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgi H. B.; Dunitz J. D.; Shefter E. Acta Crystallogr. 1974, B30, 1517–1527. [Google Scholar]
- a Choudhary A.; Kamer K. J.; Raines R. T. J. Org. Chem. 2011, 76, 7933–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kamer K. J.; Choudhary A.; Raines R. T. J. Org. Chem. 2013, 78, 2099–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jenkins C. L.; Lin G.; Duo J.; Rapolu D.; Guzei I. A.; Raines R. T.; Krow G. R. J. Org. Chem. 2004, 69, 8565–8573. [DOI] [PubMed] [Google Scholar]; b Horng J. C.; Raines R. T. Protein Sci. 2006, 15, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sonntag L.-S.; Schweizer S.; Ochsenfeld C.; Wennemers H. J. Am. Chem. Soc. 2006, 128, 14697–14703. [DOI] [PubMed] [Google Scholar]; d Kumin M.; Sonntag L.-S.; Wennemers H. J. Am. Chem. Soc. 2007, 129, 466–467. [DOI] [PubMed] [Google Scholar]; e Shoulders M. D.; Guzei I. A.; Raines R. T. Biopolymers 2008, 89, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Dai N.; Wang X. J.; Etzkorn F. A. J. Am. Chem. Soc. 2008, 130, 5396–5397. [DOI] [PubMed] [Google Scholar]; g Chiang Y. C.; Lin Y. J.; Horng J. C. Protein Sci. 2009, 18, 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Dai N.; Etzkorn F. A. J. Am. Chem. Soc. 2009, 131, 13728–13732. [DOI] [PubMed] [Google Scholar]; i Kuemin M.; Schweizer S.; Ochsenfeld C.; Wennemers H. J. Am. Chem. Soc. 2009, 131, 15474–15482. [DOI] [PubMed] [Google Scholar]; j Kuemin M.; Nagel Y. A.; Schweizer S.; Monnard F. W.; Ochsenfeld C.; Wennemers H. Angew. Chem., Int. Ed. 2010, 49, 6324–6327. [DOI] [PubMed] [Google Scholar]; k Choudhary A.; Raines R. T. Protein Sci. 2011, 20, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Pollock S. B.; Kent S. B. Chem. Commun. 2011, 47, 2342–2344. [DOI] [PubMed] [Google Scholar]; m Erdmann R. S.; Wennemers H. J. Am. Chem. Soc. 2012, 134, 17117–17124. [DOI] [PubMed] [Google Scholar]
- a Gao J.; Kelly J. W. Protein Sci. 2008, 17, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bartlett G. J.; Choudhary A.; Raines R. T.; Woolfson D. N. Nat. Chem. Biol. 2010, 6, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fufezan C. Proteins 2010, 78, 2831–2838. [DOI] [PubMed] [Google Scholar]
- a Gorske B. C.; Bastian B. L.; Geske G. D.; Blackwell H. E. J. Am. Chem. Soc. 2007, 129, 8928–8929. [DOI] [PubMed] [Google Scholar]; b Gorske B. C.; Stringer J. R.; Bastian B. L.; Fowler S. A.; Blackwell H. E. J. Am. Chem. Soc. 2009, 131, 16555–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Kamer K. J.; Powner M. W.; Sutherland J. D.; Raines R. T. ACS Chem. Biol. 2010, 5, 655–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Gandla D.; Krow G. R.; Raines R. T. J. Am. Chem. Soc. 2009, 131, 7244–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a DeTar D. F.; Luthra N. P. J. Am. Chem. Soc. 1977, 99, 1232–1244. [DOI] [PubMed] [Google Scholar]; b Renner C.; Alefelder S.; Bae J. H.; Budisa N.; Huber R.; Moroder L. Angew. Chem., Int. Ed. 2001, 40, 923–925. [PubMed] [Google Scholar]; c Williams K. R.; Adhyaru B.; German I.; Alvarez E. J. Chem. Educ. 2002, 79, 372–373. [Google Scholar]; d Taylor C. M.; Hardre R.; Edwards P. J. B.; Park J. H. Org. Lett. 2004, 5, 4413–4416. [DOI] [PubMed] [Google Scholar]; e Taylor C. M.; Handre R.; Edwards P. J. B. J. Org. Chem. 2005, 70, 1306–1315. [DOI] [PubMed] [Google Scholar]; f Meng H. Y.; Thomas K. M.; Lee A. E.; Zondlo N. J. Biopolymers 2006, 84, 192–204. [DOI] [PubMed] [Google Scholar]; g Cadamuro S. A.; Reichold R.; Kusebauch U.; Musiol H. J.; Renner C.; Tavan P.; Moroder L. Angew. Chem., Int. Ed. 2008, 47, 2143–2146. [DOI] [PubMed] [Google Scholar]; h Zhang K.; Teklebrhan R. B.; Schreckenbach G.; Wetmore S.; Schweizer F. J. Org. Chem. 2009, 74, 3735–3743. [DOI] [PubMed] [Google Scholar]; i Ivanova G.; Yakimova B.; Angelova S.; Stoineva I.; Enchev V. J. Mol. Struct. 2010, 975, 330–334. [Google Scholar]; j Cipolla L.; Airoldi C.; Bini D.; Gregori M.; Marcelo F.; Jiménez-Barbero J.; Nicotra F. Eur. J. Org. Chem. 2011, 2011, 128–136. [Google Scholar]; k Erdmann R. S.; Wennemers H. Org. Biomol. Chem. 2012, 10, 1982–1986. [DOI] [PubMed] [Google Scholar]; l Torbeev V. Y.; Fumi E.; Ebert M.-O.; Schweizer W. B.; Hilvert D. Helv. Chim. Acta 2012, 95, 2411–2420. [Google Scholar]; m Lee K. K.; Park K. H.; Joo C.; Kwon H. J.; Jeon J.; Jung H. I.; Park S.; Han H.; Cho M. J. Phys. Chem. B 2012, 116, 5097–5110. [DOI] [PubMed] [Google Scholar]; n Pandey A. K.; Naduthambi D.; Thomas K. M.; Zondlo N. J. J. Am. Chem. Soc. 2013, 135, 4333–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Higashijima T.; Tasumi M.; Miyazawa T. Biopolymers 1977, 16, 1259–1270. [DOI] [PubMed] [Google Scholar]; b Liang G.-B.; Rito C. J.; Gellman S. H. Biopolymers 1992, 32, 293–301. [DOI] [PubMed] [Google Scholar]; c Improta R.; Benzi C.; Barone V. J. Am. Chem. Soc. 2001, 123, 12568–12577. [DOI] [PubMed] [Google Scholar]; d Benzi C.; Improta R.; Scalmani G.; Barone V. J. Comput. Chem. 2002, 23, 341–350. [DOI] [PubMed] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- Jakobsche C. E.; Choudhary A.; Miller S. J.; Raines R. T. J. Am. Chem. Soc. 2010, 132, 6651–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Raines R. T. ChemBioChem 2011, 12, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Pua K. H.; Raines R. T. Amino Acids 2011, 41, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders M. D.; Raines R. T. Annu. Rev. Biochem. 2009, 78, 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzei I. A.; Choudhary A.; Raines R. T. Acta Crystallogr. 2013, E69, o805–o806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Paulini R.; Muller K.; Diederich F. Angew. Chem., Int. Ed. 2005, 44, 1788–1805. [DOI] [PubMed] [Google Scholar]; b Fischer F. R.; Wood P. A.; Allen F. H.; Diederich F. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17290–17294. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Worley B.; Richard G.; Harbinson G. S.; Powers R. PLoS One 2012, 7, e42075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lide D. R. In CRC Handbook of Chemistry & Physics; 93rd ed.; CRC Press: Boca Raton, FL, 2013. [Google Scholar]
- a Wiberg K. B.; Rablen P. R. J. Am. Chem. Soc. 1995, 117, 2201–2209. [Google Scholar]; b Wiberg K. B.; Rush D. J. J. Am. Chem. Soc. 2001, 123, 2038–2046. [DOI] [PubMed] [Google Scholar]
- a Chiti F.; Stefani M.; Taddei N.; Ramoni G.; Dobson C. M. Nature 2003, 424, 805–808. [DOI] [PubMed] [Google Scholar]; b Chiti F.; Dobson C. M. Annu. Rev. Biochem. 2006, 75, 333–366. [DOI] [PubMed] [Google Scholar]; c Eisenberg D.; Jucker M. Cell 2012, 148, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pauling L.; Corey R. B.; Branson H. R. Proc. Natl. Acad. Sci. U.S.A. 1951, 37, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pauling L.; Corey R. B. Proc. Natl. Acad. Sci. U.S.A. 1951, 37, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fleming P. J.; Rose G. D. Protein Sci. 2005, 14, 1911–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For example, the absolute values of the φ and ψ dihedral angles for 6-endo (−77°,161°) and 6-exo (−70°,149°) are similar to those for proline residues in the Xaa (−75°,164°) and Yaa (−60°,152) position, respectively, of a collagen triple helix (Berisio R.; Vitagliano L.; Mazzarella L.; Zagari A.. Protein Sci. 2002, 11, 262–270). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thioamides can be installed into proteins by native chemical ligation (Batjargal S.; Want Y. J.; Goldberg J. M.; Wissner R. F.; Petersson E. J.. J. Am. Chem. Soc. 2012, 134, 9172–9182). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.