

Abstract
Metallo-dithiolene non-innocence is explored in an oxomolybdenum-bis(dithione) complex, [Mo4+O(i-Pr2Pipdt)2Cl][PF6] (where i-Pr2Pipdt is N,N’-piperazine-2,3-dithione), that possesses a piperazine ring as an integral part of the dithiolene ligand. The title complex displays unusual spectroscopic features for a formally reduced Mo(IV) dithiolene complex, namely a low energy metal-to-ligand charge transfer band with appreciable intensity and C-C and C-S stretching frequencies that are markedly different from those of oxomolydenum complexes coordinated to dianionic dithiolene ligands. The electronic structure of the ligand has been described in valence bond terms as a resonance hybrid of dithione and dizwitterionic dithiolene contributing structures.
Keywords: dithiolene, zwitterionic, electronic structure, molybdenum, Raman
Introduction
Metallodithiolenes display an extraordinarily rich metal-ligand redox interplay[1] due to the inherent non-innocent redox behavior of the dithiolene.[2] The fully reduced dianionic ene-1,2-dithiolate can be oxidized by one electron to a radical form,[3] or by two electrons to yield the dithione/dithiete (Scheme 1). Additional possibilities for non-innocent behavior exist when the dithiolene is part of a larger ligand framework. Recently, unusual thiol/thione type dithiolene ligands have been incorporated into oxomolybdenum(IV)-mono(dithiolene) compounds and they were shown to possess low-energy intraligand charge transfer transitions that resulted from the donor-acceptor character present in the extended dithiolene ligand.[4] Dithiolenes are also found in the active sites of pyranopterin molybdenum enzymes[1b, 2, 5]. These enzymes possess a redox active Mo center bound by one or two pyranopterin dithiolene ligands (Fig. 1) and catalyze two-electron redox reactions that are coupled to the formal transfer of an oxygen atom between the active site and substrate.[6] It is believed that Mo-based redox processes are dominate in these enzymes,[6a] with the Mo ion cycling between the Mo(IV) and Mo(VI) redox states. However, the pyranopterin possesses the potential to store up to four redox equivalents by accessing via tetrahydro, dihydro, and oxidized states of the pterin ring. Induced internal redox interconversions in the pyranopterin have been suggested to occur via a combination of pyran ring opening and direct two-electron oxidation of the pterin ring.[2, 7] There is a dearth of experimental studies that have focused on the redox synergy within the coupled pyranopterin-dithiolene moiety or on the role of the pyranopterin-dithiolene in catalysis. In this manuscript, electronic structure calculations are evaluated in the context of electronic absorption and resonance Raman spectroscopies that probe the nature of the piperizine-dithiolene ligand in the oxomolybdenum-bis(dithiolene) complex [Mo4+O(i-Pr2Pipdt)2Cl][PF6] (1, Figure 1) (where i-Pr2Pipdt is N,N’-piperazine-2,3-dithione). Our studies reveal a considerable degree of dizwitterionic ligand character present in the ligands of 1. As a result of this dizwitterionic character, 1 displays unusual spectroscopic features for a formally reduced Mo(IV) dithiolene complex.
Scheme 1.
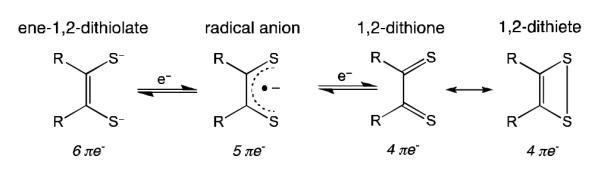
Dithiolene ligand forms.
Figure 1.
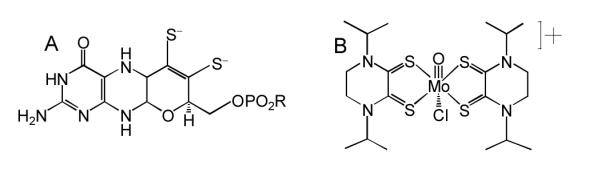
A: Pyranopterin dithiolene coordinated to Mo as the dianion. B: i-Pr2Pipdt ligand coordinated to Mo in 1 as the dithione. Note the presence of a piperazine ring in both structures.
Results and Discussion
Compound 1 possesses a low energy absorption feature at ~13,500 cm−1 (Bands 1 and 2; Figure 2). Low-energy charge transfer transitions are not typically observed in oxomolybdenum (IV) mono- and bis-dithiolene complexes since they possess low-spin (dx2-y2)2 electronic configurations that preclude LMCT transitions to this low-lying Mo(dx2-y2) highest occupied molecular orbital (HOMO). To our knowledge, the only Mo(IV) bis-dithiolene complex to display similar low energy absorption features is Tp*MoO(S2BMOQO), 2, which possesses a 16,400 cm−1 (ε = 5,190 M−1cm−1) intraligand dithiolene(S)→quinoxaline CT transition.[4] The donor-acceptor nature of the S2BMOQO ligand results from an admixture of thiol/thione and dithiol resonance structures, and is responsible for the intensity of the intraligand band in 2.[4] However, the origin of the low energy CT transition in 1 is different than that determined for 2. The lowest unoccupied MOs in 1 (LUMO and LUMO+1) are bis-dithiolene ligand orbitals comprised of antisymmetric (LUMO) and symmetric (LUMO+1) linear combinations of a dithiolene orbital (Fig. 3). The orbital is doubly occupied in reduced dianionic dithiolenes, but is unoccupied in the two electron oxidized form of the ligand. Therefore, electronic transitions to the LUMO and LUMO+1 will directly probe the nature of the dithiolene in 1. Time dependent DFT calculations[8] have allowed for an assignment of Bands 1 and 2 as arising primarily from HOMO→LUMO and HOMO→LUMO+1 one-electron promotions. The MLCT character is clearly evident in the electron density difference map (EDDM) for Band 1, where the acceptor character is delocalized over the N2C2S2 atoms of both ditiholenes (Fig. 2). The 647nm resonance Raman spectrum (Figure S1) of 1 displays bands at 1120, 1226, and 1516 cm−1 that correlate very well with calculated frequencies at 1119, 1224, and 1519 cm−1, which have been scaled with a 2% frequency reduction factor. This has allowed for their respective assignments as ν(C-C + C-S), ν(C-C), and ν(C-N), respectively. The C-C and C-S stretching frequencies for 1 are markedly different from those of oxomolydenum complexes coordinated by dianionic dithiolene ligands, where ν(C-S) ~760 – 860 cm−1 and ν(C-C) ~1,450 – 1,600 cm−1 are typically observed.[9]
Figure 2.
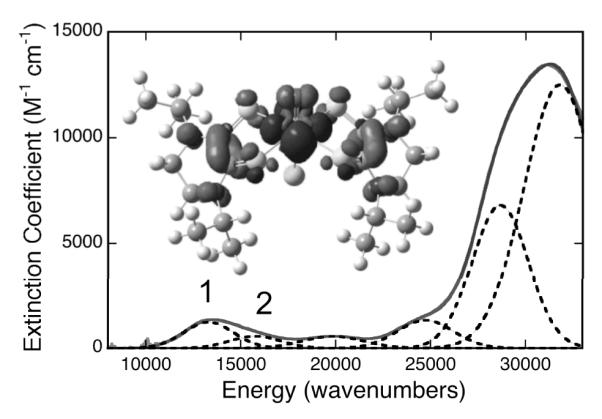
Gaussian resolved electronic absorption spectrum of 1 in acetonitrile. (Inset) Electron density difference map (EDDM) that details the nature of the low-energy MLCT transition (Band 1) in 1 (black: electron density loss in transition, gray: electron density gain in transition).
Figure 3.
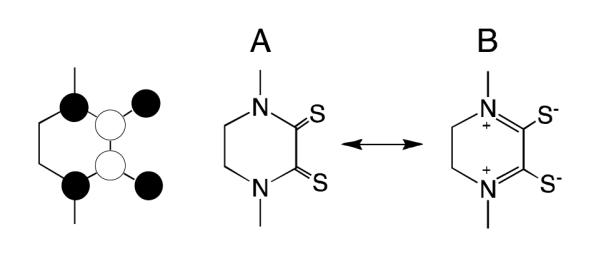
The π delocalized orbital. Right: Two contributing resonance structures for the i-Pr2Pipdt ligand that lead to N2C2C2 π delocalization. A: dithione, B; dizwitterionic dithiol.
The nature of the i-Pr2Pipdt ligands ligands can be further understood by examining the two key resonance structures in Figure 3. Structure A represents the neutral dithione form of i-Pr2Pipdt while structure B represents a dizwitterionic form of the ligand. The nature of the low energy MLCT transitions and the Raman data suggest an admixture of resonance forms A and B in 1 with π delocalization that extends over the entire N2C2S2 ligand framework. The low energy and appreciable intensity of the MLCT band in 1 directly results from the presence of low-lying ligand-based acceptor orbitals that possess extended π conjugation. Additionally, the observation of a high frequency 1519 cm−1 ν(C-N) stretch (aliphatic amine ν(C-N) 1020-1250 cm−1)[10] is fully consistent with a significant contribution from the dizwitterionic resonance form B, which posseses C=N double bond character (ν(C=N) 1471-1689 cm−1)[10]. The contribution of structure B is also clearly evident in the structure of the free ligand, which possesses planar sp2 N atoms (Figure S2). The coefficients of the competing resonance forms of the free ligand can be calculated using a natural bond orbital (NBO) formalism.[11] Our NBO calculations indicate very large donations from the piperazine N lone pairs of resonance form A into C-S π* antibonding orbitals that lead to the formation of resonance structure B. The contributions of these two resonance structures to the ground state valence bond (VB) wavefunction can be approximated through use of NBO derived occupation numbers for the competing resonance forms. This analysis results in a 63:37 ratio of structures A and B contributing to the ground state VB description of the ligand This is consistent with the spectroscopic data that show evidence for extensive N2C2S2 π delocalization in the ligands. Thus, this VB description of the ligands in 1 nicely explains 1) the extended conjugation suggested by the Raman data and observed in the EDDMs of bands 1 and 2, 2) the electron withdrawing nature of the i-Pr2Pipdt ligands that stabilize the Mo(IV) oxidation state, 3) the origin of the unusually low energy MLCT transitions in 1, and 4) the planar sp2 character of the piperazine N atoms.
Conclusions
In summary, our spectroscopic and electronic structure studies point to the i-Pr2Pipdt ligands in 1 as a new type of dithiolene that can be described in VB terms as a resonance hybrid of dithione and dizwitterionic dithiolene contributing structures. This is of fundamental interest with respect to our understanding of the complex coordination chemistry of metallo-dithiolene complexes. In terms of applications, this electronic structure description may provide a guiding principle for designing molecule-based valence-delocalized materials, and for understanding electron transfer regeneration processes in molybdenum enzymes. With respect to the latter, it has been postulated that the piperazine ring component of the pyranopterin cofactor can electronically buffer the Mo center against changes in charge that accompany redox processes.[12] The additional redox non-innocence of the pyranopterin suggests that zwitterionic forms of the cofactor may play a fundamental role in modulating its electron donor ability and facilitating pyranopterin molybdenum enzyme catalysis.[12c]
Experimental Section
(General
Synthesis of the molybdenum complex was carried out in oxygen-free dry argon atmospheres using dry degassed solvents. The ligand, diisopropylpiperazine-2,3-dithione (i-Pr2Pipdt), was synthesized in air.[13] Solvents were purchased either from Aldrich Chemical Co or Acros Organics and were purified by distillation as follows: acetonitrile from CaH2, followed by Li2CO3-KMnO4 and finally from P2O5; CH2Cl2 and CHCl3 from CaH2; diethyl ether and toluene from sodium benzophenone; methanol from sodium ethoxide. MoCl5, N,N- dimethyl ethylene diamine, N,N-diisopropyl ethylene diamine and Lawesson’s reagent were purchased from Aldrich and used without purification. Diethyl oxalate was purchased from Acros Organics and used as received.
Spectroscopic/Spectrometric Measurements
Routine UV-Visible spectra were recorded on a modified temperature-controlled Cary 14 spectrophotometer or a temperature controlled Cary 3 spectrophotometer. 1H, and 13C NMR spectra were collected using either a Bruker 500 MHz or a 400 MHz spectrometer. IR spectra were recorded in reflection mode on a Thermo Electron corporation Nicolet 380 spectrometer with neat samples. Elemental analysis was performed by Midwest Microlab LLC, Indiana, IL. All mass spectra were collected in a Micromass ZMD quadruple spectrometer equipped with an electrospray ionization (ESI) source both negative and positive ion mode, using acetonitrile as the mobile phase. In order to get the molecular ion peak the capillary and the cone voltages were varied between 3.0-4.0 kV and 5-55 V respectively. The desolvation temperature was set at 100 °C and the source bath temperature was set at 80 °C.
Solid state resonance Raman (rR) spectra and associated rR excitation profiles were collected using a system comprised of an PI/Acton SpectraPro SP-2556 500 mm focal length imaging spectrograph with a triple grating turret and a PI/Acton Spec-10:100B back-illuminated 1340 × 100 pixel digital CCD spectroscopy system with a cryogenically cooled camera head. Coherent Innova Ar+ and Kr+ ion lasers were uses as excitation source. Samples were mixed with either NaCl or a NaCl/Na2SO4 mixture with Na2SO4 as an internal calibrant. High resolution electronic absorption spectra were collected using a Hitachi U-3501 UV-Vis-NIR dual-beam spectrophotometer capable of scanning a wavelength region between 185 and 3200 nm. Spectral samples were dissolved in dry, degassed acetonitrile, and the electronic absorption spectra were measured in a 1 cm pathlength, 100 μL, black-masked, quartz cuvette (Starna Cells, Inc.) equipped with a Teflon stopper. All electronic absorption spectra were performed at room temperature and repeated at regular time intervals to ensure the structural stability and integrity of the complex in solution.
Electronic structure and vibrational frequency calculations were performed at the density functional level of theory using the Gaussian 03W[8] and NBO 5.0[11] software packages. All calculations employed the B3LYP hybrid functional and used a LANL2DZ basis set with an effective core potential for Mo. A 6-31G* basis set was used for all light atoms. Input files were prepared using the molecule builder function in the Gaussview software package. Electron density difference maps (EDDMs) were constructed using the GaussSum suite of programs.
Synthetic and characterization details for [(i-Pr2Pipdt)2MoOCl][PF6]
160 mg (0.586 mmol) of MoCl5 was dissolved in 3 mL of dry degassed methanol which led to a vigorous reaction and the reaction mixture turned green. To this mixture 300 mg (1.304 mmol) of i-Pr2Pipdt was added and the color changed to blue green immediately. After stirring the reaction mixture for 15 min, 800 mg (4.74 mmol) of NaPF6 in methanol (10 mL) was added, and stirred for 30 min. The solution was filtered and the precipitate was washed first with methanol to remove excess NaPF6, and then with CHCl3 to remove excess ligand. Drying of the precipitate resulted in analytically pure compound. Yield: 52% (212 mg, 0.306 mmol). Anal. Calcd (Experimental) for C20H36N4S4MoOClPF6: C, 31.89 (31.67); H, 4.82 (4.75); N, 7.44 (7.34); S 17.03 (16.94). IR (neat, cm−1): 1505 (vs, C-N), 1365 (vs, C=S), 1258, 1185, 938 (vs, Mo=O) 831 (vs, PF6), 588 (m), 566 (s). ESI-MS (MeCN): base peak, m/z 609 [M]+. 1H NMR (CD3CN): δ=5.57, 5.42 (sep, 2H, CH), 4.04, 3.48 (s, 4H, CH2). 1.50, 1.25 (d,12H,CH3 ). 13C NMR (CD3CN): δ=180.75 (C=S), 59.10 (CH), 43.16 (CH2), 17.69 (CH3). Conductivity in acetonitrile: 162 ohm−1cm2mol−1.
Supplementary Material
S1. Room temperature solid state resonance Raman spectrum of (i-Pr2Pipdt)2MoOCl][PF6].
S2. X-ray structure of the i-Pr2Pipdt ligand showing planer N atoms (rendered with Mercury 2.4.5).
Acknowledgments
We acknowledge the NIH (GM-057378; MLK and GM-061555; PB), and NSF (CHE-0616190; MLK) for financial assistance.
Footnotes
Supporting Information. Supporting information for this article is available on the WWW under http://www.eurjic.org/ or from the author.
References
- [1] a).Kirk ML, Helton ME, McNaughton RL. In: Prog. Inorg. Chem. Stiefel EI, editor. Vol. 52. John Wiley and Sons, Inc.; Hoboken, New Jersey: 2004. pp. 111–212. [Google Scholar]; b) Stiefel EI. Pure Appl. Chem. 1998;70:889–896. [Google Scholar]; c) Szilagyi RK, Lim BS, Glaser T, Holm RH, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 2003;125:9158–9169. doi: 10.1021/ja029806k. [DOI] [PubMed] [Google Scholar]; d) Sproules S, Benedito FL, Bill E, Weyhermuller T, George SD, Wieghardt K. Inorg. Chem. 2009;48:10926–10941. doi: 10.1021/ic9010532. [DOI] [PubMed] [Google Scholar]; e Deplano P, Pilia L, Espa D, Mercuri ML, Serpe A. Coord. Chem. Rev. 2010;254:1434–1447. [Google Scholar]
- [2].Burgmayer SJN. In: Prog. Inorg. Chem. Stiefel EI, editor. Vol. 52. John Wiley and Sons, Inc.; Hoboken, New Jersey: 2004. pp. 491–538. [Google Scholar]
- [3].Ray K, George SD, Solomon EI, Wieghardt K, Neese F. Chemistry-A European Journal. 2007;13:2783–2797. doi: 10.1002/chem.200601425. [DOI] [PubMed] [Google Scholar]
- [4].Matz KG, Mtei RP, Leung B, Burgmayer SJN, Kirk ML. J. Am. Chem. Soc. 2010;132:7830–7831. doi: 10.1021/ja100220x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stiefel EI. J. Chem. Soc., Dalton Trans. 1997:3915–3923. [Google Scholar]
- [6] a).Hille R. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]; b) Hille R. Molybdenum enzymes containing the pyranopterin cofactor: An overview. Vol. 39. Marcel Dekker, Inc.; New York: 2002. [PubMed] [Google Scholar]
- [7].Helton ME, Gebhart NL, Davies ES, McMaster J, Garner CD, Kirk ML. J. Am. Chem. Soc. 2001;123:10389. doi: 10.1021/ja0100470. [DOI] [PubMed] [Google Scholar]
- [8].Gaussian 03. Revision C.02 Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- [9].Johnson MK. In: Prog. Inorg. Chem. Stiefel EI, editor. Vol. 52. John Wiley and Sons, Inc.; Hoboken, New Jersey: 2004. pp. 213–266. [Google Scholar]
- [10].Silverstein GCBRM, Morrill TC. Spectrometric Identification of Organic Compounds. 4 ed John Wiley and Sons; New York: 1981. [Google Scholar]
- [11].Glendening JED, Badenhoop K, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. Theoretical Chemistry Institute. 5.0 ed University of Wisconsin; Madison: 2001. [Google Scholar]
- [12] a).Inscore F, McNaughton R, Westcott B, Helton M, Jones R, Dhawan I, Enemark J, Kirk M. Inorg. Chem. 1999;38:1401–1410. [Google Scholar]; b) Inscore FE, Knottenbelt SZ, Rubie ND, Joshi HK, Kirk ML, Enemark JH. Inorg. Chem. 2006;45:967. doi: 10.1021/ic0506815. [DOI] [PubMed] [Google Scholar]; c) Westcott BL, Gruhn NE, Enemark JH. J. Am. Chem. Soc. 1998;120:3382–3386. [Google Scholar]
- [13].Perera E, Basu P. Dalton Transactions. 2009:5023–5028. doi: 10.1039/b904113c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1. Room temperature solid state resonance Raman spectrum of (i-Pr2Pipdt)2MoOCl][PF6].
S2. X-ray structure of the i-Pr2Pipdt ligand showing planer N atoms (rendered with Mercury 2.4.5).