

Abstract
The lamina serves to maintain the nuclear structure and stiffness while acting as a scaffold for heterochromatin and many transcriptional proteins. Its role in endothelial mechanotransduction, specifically how nuclear mechanics impact gene regulation under shear stress, is not fully understood. In this study, we successfully silenced lamin A/C in bovine aortic endothelial cells to determine its role in both glucocorticoid receptor (GR) nuclear translocation and glucocorticoid response element (GRE) transcriptional activation in response to dexamethasone and shear stress. Nuclear translocation of GR, an anti-inflammatory nuclear receptor, in response to dexamethasone or shear stress (5, 10, and 25 dyn/cm2) was observed via time-lapse cell imaging and quantified using a Bayesian image analysis algorithm. Transcriptional activity of the GRE promoter was assessed using a dual-luciferase reporter plasmid. We found no dependence on nuclear lamina for GR translocation from the cytoplasm into the nucleus. However, the absence of lamin A/C led to significantly increased expression of luciferase under dexamethasone and shear stress induction as well as changes in histone protein function. PCR results for NF-κB inhibitor alpha (NF-κBIA) and dual specificity phosphatase 1 (DUSP1) genes further supported our luciferase data with increased expression in the absence of lamin. Our results suggest that absence of lamin A/C does not hinder passage of GR into the nucleus, but nuclear lamina is important to properly regulate GRE transcription. Nuclear lamina, rather than histone deacetylase (HDAC), is a more significant mediator of shear stress-induced transcriptional activity, while dexamethasone-initiated transcription is more HDAC dependent. Our findings provide more insights into the molecular pathways involved in nuclear mechanotransduction.
Keywords: glucocorticoid receptor, nuclear lamina, shear stress, dexamethasone, endothelial cells
cardiovascular diseases such as atherosclerosis are the leading cause of death in the United States. The initiation and development of atherosclerosis are typically associated with endothelial dysfunction (20, 51). Endothelial cells at the lumen of blood vessels are continuously exposed to both hemodynamic shear stress and cyclic stretch due to blood flow (16). The distinct local hemodynamic profiles influence endothelial behaviors in the vasculature. Atherosclerotic lesions tend to develop at regions of disturbed flow and low shear stress at bifurcation sites and curvatures, while regions with uniform high laminar shear stress are protected (10, 48, 66). Endothelial responses to shear stress can be categorized as immediate [rapid release of nitric oxide (NO); (49)] as well as short-term [upregulation of tissue plasminogen activator gene (27)]. Shear stress has been shown to activate a variety of signaling molecules such as heat shock proteins, transcription factors, mitogen-activated protein (MAP), and phosphatidylinosital 3 kinases (40, 45, 63). Ongoing research is mapping out the pathways involved in endothelial mechanotransduction.
Endothelial cells, as do most cell types, possess nuclear receptors such as the glucocorticoid receptor (GR) that respond to steroid hormones under physiological conditions (28). GR is part of the superfamily of nuclear receptors that function as DNA-binding, transcription regulators. Following passive transport through the cell membrane, glucocorticoids such as dexamethasone bind to GR and initiate conformational change, dimerization, and release of heat shock proteins. GR has also been shown to become activated in a ligand-independent manner via phosphorylation of specific serine residues at the N terminus by ERK, MAPK, glycogen synthase kinase-3, and protein kinases A and C (1, 2, 46). Subsequently, the GR nuclear localization signal is exposed, which allows the complex to translocate into the nucleus and bind to glucocorticoid response elements (GRE) to regulate target genes (7). Specifically, it can induce transcription of anti-inflammatory genes via a process known as transactivation or suppress inflammatory gene expression by inhibitory protein-protein interactions, referred to as transrepression (8).
Underneath the nuclear envelope is the nuclear lamina, a meshwork of type V intermediate filaments made of lamin proteins that provides structural support to the nucleus (13). Furthermore, the lamina serves as a scaffold for nuclear proteins, influences gene expression, supports chromatin organization and DNA replication, and links the cytoskeleton through nesprin and SUN proteins in the linker of nucleoskeleton and cytoskeleton (LINC) complex (18, 42, 64). Mutations of the lamin protein cause a wide range of human diseases called laminopathies (9, 11, 15, 21, 29, 30). Research on the disease mechanisms of laminopathies suggests a crucial role for lamins as a load-bearing structure necessary for structural integrity and normal nuclear mechanics (19, 50). Cells with defective lamina demonstrate deformed nuclei, disorganized heterochromatin, and a defective DNA damage response pathway (19, 62). It is hypothesized that signaling cascades are hindered when the lamina is disrupted and the nucleus becomes fragile and deformed. Specifically, a defective lamina, cytoskeleton, and integrin relay may affect the proper transduction of extracellular stress, thus leading to altered transcriptional regulation or deregulation. One type of laminopathy, Hutchinson-Gilford Progeria Syndrome, a disease caused by abnormal lamin A due to mutations, leads to premature atherosclerosis in children (14, 37, 43). Endothelial cells of patients with laminopathies show hindered regeneration after injury, resulting in suboptimal intimal integrity (4). While long-term sustained exposure to shear stress leads to both endothelial cell realignment with flow and changes in structural organization, mechanical properties, and nuclear activities (26), the role of lamins in endothelial mechanotransduction is not yet understood.
We have already identified the glucocorticoid receptor as an anti-inflammatory nuclear receptor regulated by shear stress (47). Using a unique expectation-maximization algorithm based on Bayesian statistics, we quantified GR subcellular movement based on changes of fluorescently labeled GR [green fluorescence protein (GFP)-GR]. We showed that GR translocates to the nucleus within 20 min following activation by dexamethasone and within 2 h under shear stress, independent of an intact cytoskeleton (57). The finding that arterial shear stress may act similarly to glucocorticoids to induce GR nuclear localization through a steroid-independent mechanism is significant because it suggests that mechanical forces alone can trigger intracellular movement of GR into the nucleus. The nuclear matrix has been shown to serve as a scaffold and stabilizer for GR and other transcription factors upon the formation of protein complexes (52). It is not known, however, how GR nuclear functions would be affected by the absence of lamina. Understanding the role of endothelial lamina in shear-induced GR nuclear localization and transcription regulation would help elucidate how force transduction and nuclear mechanics impact gene regulation.
In this study, we have utilized short hairpin (sh)RNA to selectively silence the lamin A/C gene. Following chemical or mechanical stimulation, we quantified variations in nuclear and cytoplasmic brightness over time using our image analysis method described previously. Microscopy results are supported by endogenous protein analysis of GR in the cytoplasm and nuclear fractions. Furthermore, we utilized a luciferase reporter assay to examine glucocorticoid-responsive gene expression in control and lamin-deficient cells. Our results have led to further understanding of endothelial mechanotransduction at the nuclear level, and using the GR regulatory pathway, we continue to investigate the impact of fluid shear stress on transcriptional regulation in the nucleus.
MATERIALS AND METHODS
Reagents.
Dexamethasone (Sigma, St. Louis, MO) was dissolved in ethanol and stored at −20°C at a stock concentration of 25 mM. Puromycin (Sigma) was dissolved in water, sterile filtered, and stored at a stock concentration of 10 mg/ml at −20°C. Hexadimethrine bromide (Sigma) was dissolved in water to a stock solution of 250 mg/ml and stored at 4°C. Trichostatin A (TSA), 5 mM in DMSO from Sigma, was diluted to 500 μmol/l in ethanol and used at a working concentration of 500 nM. In the GFP-GR plasmid vector, the mouse GR cDNA N terminus was fused in frame to the C terminus of GFP on the pEGFP-C3 plasmid (Clontech) (47). The GRE luciferase reporter (Qiagen CCS-006L) was stored at −20°C. Plasmid purification was done using GeneElute Maxiprep kit (Sigma), and purified plasmids were stored at −20°C.
Lamin A/C gene silencing with shRNA.
Silencing of the lamin A/C gene was accomplished using shRNA in stable transfection with lentiviral particles. The lamin shRNA Bacterial Glycerol Stock (Sigma SCHLNG NM-170707) was chosen after confirming conservation of sequence to bovine lamin A/C gene and stored at −80°C. The Non-Target shRNA Control Plasmid (Sigma SHC002) was chosen as a control and stored at −20°C. Lentiviral particles were generated using the MISSION Lentiviral Packaging Mix (Sigma SHP001) and extracted based on the manufacturer's protocol. Briefly, HEK293T (ATCC) cells were grown to 70% confluency and transfected with the Lentiviral Packaging Mix, plus either lamin A/C shRNA or nontargeting control vector, and FuGene 6 reagent (Roche, Indianapolis, IN). The first and second harvests of viral particles were performed 2 and 3 days posttransfection, respectively, and particles were stored at −80°C. The concentration of viral particles was determined using the HIV p24 Antigen ELISA assay (ZeptoMetrix, Buffalo, NY) per the manufacturer's protocol. Bovine aortic endothelial cells (BAECs) were then grown to 70% confluency and treated with lentiviral particles at a multiplicity of infection of either 1 or 5 in the regular growth media with 8 μg/ml hexadimethrine bromide to improve lentiviral uptake. Puromycin was used to select for shRNA-positive BAECs at 2 μg/ml working concentration, which were then passed to larger culture vessels upon reaching confluency.
Cell culture and transfection.
Following selection of shRNA-positive cells, BAECs, passages 15 or less, were cultured in DMEM supplemented with 10% heat-inactivated FBS (JR Scientific, Woodland, CA), 1% l-glutamine, 2% penicillin streptomycin, and 2 μg/ml puromycin (all Sigma) and incubated at 37°C in a humidified 5% CO2 environment. Wild-type and lamin-deficient (Lmn A−/−) mouse embryonic fibroblasts (MEFs), were provided by Jan Lammerding at Cornell University. These cells were cultured under similar conditions in media with no puromycin added. Cells were seeded on 38 × 75 mm glass slides coated with fibronectin (1 mg/ml) at a density of 8–10 ×105 cells per slide. BAECs and MEFs on slides were transfected for 4 h with pGFP-GR and Metafectene Pro reagent (Biontex, San Diego, CA) at a plasmid to reagent ratio of 1:3 (5 μg plasmid to 15 μl reagent), and allowed to recover overnight. For the GRE reporter assay, shRNA cells were transfected with GRE plasmid (Qiagen, Valencia, CA) and Metafectene Pro at 1:4 ratio (0.6 μg plasmid to 2.4 μl reagent). After transfection, cells were maintained in phenol-red free DMEM (Sigma) culture media supplemented with 10% charcoal/dextran-treated FBS (Thermo HyClone, Rockford, IL) for reduced background steroid and growth factor concentrations. Before shear imaging experiments, cells were incubated with blue nuclear chromatin Hoechst 33342 stain (1:1,000 dilution; Molecular Probes, Grand Island, NY) for 15 min for nuclear fluorescence imaging.
Shear stress exposure and live cell imaging.
For shear experiments, cells on slides were placed in a parallel plate flow chamber attached to a sterile, laminar flow system as previously described (32, 58) in an environmental chamber kept at 37°C with 5% CO2. The magnitude of shear stress (τ) on the cell monolayer is calculated based on the Navier-Stokes equation for a Newtonian fluid in a parallel plate geometry. The equation for wall shear stress simplifies to 6μQ/bh2, where μ is the viscosity of the media (0.01 dyn-s/cm2), Q is the volumetric flow rate (∼0.3, 0.6 or 1.2 ml/s), b is the width of the flow chamber (2.5 cm), and h is the separation distance between the chamber and the glass slide (0.025 cm). With the use of this system, cells were exposed to 5, 10, or 25 dyn/cm2 laminar wall shear stress. Flow experiments were carried out in regular growth media (10% charcoal/dextran-treated FBS). For live cell imaging, the flow chamber is placed flat on the microscope stage of a Leica DMI 6000-B fluorescence microscope enclosed inside a 37°C chamber. Phase contrast and fluorescence images were captured live through a CCD camera (Leica). Microscope and image acquisition were controlled by Leica AF6000 software. Images were captured at ×40 magnification.
Quantitative image analysis.
Image analysis was performed as described previously using an algorithm based on Bayesian statistics and Expectation-Maximization/Maximization of Posterior Marginals (57). Briefly, fluorescence GFP-GR and Hoechst images were first converted to 8-bit black and white, and the Hoechst images were thresholded to obtain the nuclear outline. At each time point, the GFP-GR and nuclear images were processed using our novel algorithm to produce three segmentations: background, cytoplasm, and nucleus. The nuclear brightness value was divided by the cytoplasm value to provide a normalized nuclear brightness that accounts for brightness changes as a result of GFP-GR brightness focus drift. The value obtained for each time point was then normalized to the initial brightness at t = 0.
Protein analysis.
To confirm lamin A/C silencing, whole cell lysates were obtained using RIPA buffer with 0.5 mM PMSF, 150 mM protease inhibitor, and 1 mM DTT. After shear experiments, BAECs were lysed and separated into cytoplasm and nuclear fractions using a nuclear extraction kit (Active Motif, Carlsbad, CA) following the manufacturer's instruction. Protein concentrations were determined using the Bradford protein assay (Bio-Rad, Hercules, CA). Protein samples were then separated by SDS-PAGE on 4–12% Bis-Tris NuPage gels (Invitrogen, Grand Island, NY), transferred to a nitrocellulose membrane by electro-blotting, and blocked for 1 h at room temperature with 5% nonfat milk in PBS with 0.1% Tween (Sigma) (PBS-T). Incubation with the primary mouse monoclonal anti-GR IgG2b (Millipore 05–827, Billerica, MA) at 1:200 ratio or mouse monoclonal anti-lamin A/C (Cell Signaling 4777, Danvers, MA) at 1:700 ratio was done in blocking solution for 2–4 h while shaking at room temperature or overnight at 4°C. The blot was then washed three times (10 min each) with PBS-T before incubating with horseradish peroxidase-conjugated goat anti-mouse IgG (Bio-Rad) at 1:4,000 dilution for 1.5 h. Goat polyclonal lamin A/C (Santa Cruz sc-6215, 1:300 ratio, Santa Cruz, CA) and mouse monoclonal transcription factor IID (Santa Cruz sc-374035) primary antibodies served as nuclear controls, while goat GAPDH (Santa Cruz sc-48166, 1:200 ratio in 5% BSA/PBS-T) served as the cytoplasmic control. Membranes were illuminated using SuperSignal West Pico Chemiluminescent Substrate Reagents (Thermo). Imaging was done using Bio-Rad Molecular Imager ChemiDoc XRS+ System and acquired using Quantity One Image Analysis Software.
Dual-luciferase reporter assay.
Luciferase activity in BAECs was determined using the Dual-Luciferase Reporter Assay System kit (Promega, Madison, WI) following the manufacturer's protocol. Briefly, 18–20 h following shear stress or dexamethasone treatment, cells were lysed using passive lysis buffer for 15 min and collected into a 1.5-ml tube to be centrifuged. The supernatant was collected, and 100 μl of Luciferase Assay Reagent II were then added to 20 μl of the cell lysate in a white 96-well plate, and firefly luciferase activity was measured. Immediately following, 100 μl of Stop & Glo reagent were added to each sample to measure Renilla luciferase activity. Firefly values were normalized to Renilla values.
Histone acetylase/deacetylase assays.
Activity of histone proteins was determined using both histone acetylase (HAT) and deacetylase (HDAC) assay kits (Epigentek, Farmingdale, NY) per the manufacturer's instruction. Briefly, between 7–10 μg of nuclear extracts were used for the histone deacetylase assay, and 4 μg of nuclear extracts were used for the HAT assay. Histone activity was determined based on absorbance measured at 450 nm and the amount of nuclear extract used. Data are presented as average of fold change over control for each group.
Quantitative real-time PCR.
Immediately following dexamethasone or shear stress treatment, RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA was turned into cDNA using a reverse transcription kit (Applied Biosystems, Grand Island, NY). Real-time PCR and analysis were performed using the 7500 Real Time PCR system (Applied Biosystems). Primer kits for NF-κB inhibitor alpha (NF-κBIA; Bio-Rad, Hercules, CA) and dual specificity phosphatase 1 (DUSP1; SABiosciences, Valencia, CA) were used in amplification conditions of 95°C (15 s) denature, 60°C (60 s) anneal/extend, 40 cycles. Data were analyzed using the ΔΔCt method.
Statistics.
All experiments were carried out for a minimum of three times, and each time sample was prepared and analyzed independently. Results of image, luciferase, and PCR analysis are expressed as means ± SE. Standard error of each group was calculated to verify the statistical significance of the results. Paired Student t-test was used to compare one condition between two sample sets. P < 0.05 was considered sufficient for statistical significance.
RESULTS
Lamin A/C gene silencing using shRNA.
We successfully silenced lamin A/C in BAECs using stable transfection with shRNA lentiviral particles. Western blot of whole cell lysate revealed an absence of lamin A/C proteins in cells treated with lentiviral particles containing the lamin shRNA plasmid (Fig. 1A). Cells treated with nontargeting plasmid still demonstrated consistent lamin A/C expression, indicating specificity of the shRNA gene silencing. Fluorescence labeling of cell nuclei with lamin shRNA using nuclear Hoechst stain also revealed distorted nuclear shapes compared with cells with control shRNA (Fig. 1B) that showed the characteristic circular nuclear shape. Deformed nuclear shape is a marker of defective nuclear lamina (19). Thus images of Hoechst-stained nuclei support our Western blot results and further demonstrate the absence of nuclear lamina in lamin shRNA cells.
Fig. 1.

Confirmation of lamin A/C silencing. A: Western blot of whole cell protein extractions from control and lamin short hairpin (sh)RNA bovine aortic endothelial cells (BAECs) confirm the presence and absence of lamin A/C, respectively. Actin was used as the loading control. B: Hoechst staining of live cell nuclei showed that cells with lamin shRNA have irregularly shaped nuclei, and cells with non-targeting control shRNA had the characteristic circular nuclei. Images were taken at ×40 magnification. Scale bar = 15 μm.
Dexamethasone treatment induces GFP-GR translocation in the absence of nuclear lamina.
Cells with either control nontargeting shRNA or lamin A/C shRNA were then transfected with GFP-GR plasmid and treated with 25 μM dexamethasone. Again, Hoechst staining revealed that nuclei of lamin-deficient cells appeared deformed while nuclei of control shRNA cells maintained the characteristic oval shape. In both cases, before dexamethasone treatment, GFP-GR was distributed evenly in the cytoplasm and nucleus. Immediately following addition of dexamethasone, we observed the onset of GFP-GR nuclear translocation; within 20 min, GFP-GR was mostly localized inside of the nucleus (Fig. 2A). Both green (GFP-GR) and blue (Hoechst) fluorescence time-lapse images were used in our unique Bayesian image analysis algorithm (57) to quantify the dynamic nuclear localization of GFP-GR based on fluorescence brightness (Fig. 2B). Both cell types, with or without lamin A/C, demonstrated similar rates of nuclear movement of GFP-GR. After 20 min, we noticed a 50.4 ± 3.8% increase in nuclear brightness in the control shRNA cells and a 53.3 ± 4.4% increase in the lamin shRNA cells. These data show that the absence of nuclear lamina did not hinder GFP-GR nuclear localization under dexamethasone activation.
Fig. 2.

Lamin A/C silencing does not hinder green fluorescence protein (GFP)- glucocorticoid receptor (GR) translocation following dexamethasone (Dex) treatment. A: following treatment with 25 μM Dex, GFP-GR nuclear translocation was evident within 20 min in BAECs with lamin shRNA (left) and control shRNA (right). GFP-GR became primarily localized in the nucleus outlined by Hoechst staining. Images were taken at ×40 magnification. Scale bar = 15 μm. B: results of our quantitative image analysis of GFP-GR nuclear movement show a 53.4 ± 4.4 and 50.4 ± 3.8% increase in nuclear brightness for lamin (n = 5) and control (n = 13) shRNA BAECs, respectively. C: Western blot of endogenous GR from control and lamin shRNA BAECs shows localization of GR in both the cytoplasmic (C) and nuclear (N) fractions of untreated cells, whereas 20 min of Dex induction resulted in primarily nuclear localized GR. Lamin A/C and transcription factor IID (TFIID) served as the nuclear protein controls, and GAPDH was used as control for cytoplasmic protein. D: quantitative analysis of cytoplasmic and nuclear GR fractions based on repeated Western blots shows similar distribution of GR in the absence of treatment (n > 3). There was a significant increase of GR in the nuclear over cytoplasmic fraction following Dex treatment in both cell types: control (top) and lamin shRNA (bottom) (*P < 0.05, compared with untreated cells).
Western blot confirms GR nuclear localization is independent of nuclear lamina.
Western blots of endogenous GR also correlated to our live cell images (Fig. 2C). Again, we confirmed lamin A/C gene silencing in lamin shRNA-treated cells: lamin A/C proteins were only present in nuclear fractions of control cells but not lamin shRNA cells. In lamin-deficient cells, transcription factor IID (TFIID) was used as the control for nuclear protein, and GAPDH was used as control for cytoplasmic fractions in both cell types. In untreated control and lamin shRNA cells, GR was present in both the cytoplasm and nucleus. After dexamethasone treatment, GR significantly increased in the nuclear fraction indicating nuclear movement. Quantitative analysis of repeated Western blots revealed that the shift of GR from cytoplasmic to nuclear fraction was statistically significant (Fig. 2D). Cytoplasmic GR decreased from 38.6 ± 1.5% in control shRNA cells to 5.4 ± 1.7% after dexamethasone treatment (P < 0.05), while nuclear GR increased from 61.4 ± 1.5 to 94.6 ± 1.7% (P < 0.05; Fig. 2D, top). For lamin shRNA cells following dexamethasone treatment, cytoplasmic GR decreased from 49.9 ± 3.8 to 9.5 ± 3.3%, while nuclear GR increased from 50.1 ± 3.8 to 90.5 ± 3.3% (Fig. 2D, bottom).
Lamin silencing did not hinder GR nuclear translocation under shear stress.
We have already reported that shear stress activates GR to nuclear localize. Here we determine the effect of nuclear lamina on ligand-independent GR translocation under shear stress at 10 and 25 dyn/cm2 for up to 2 h. Lamin A/C gene silencing was confirmed by deformed nuclear shape (Figs. 3A and 4A). Our image analysis algorithm tracked and quantified GFP-GR nuclear movement over time. At 10 dyn/cm2, cells lacking lamin protein demonstrated slightly higher rate of GR nuclear localization early. GFP-GR brightness in the nucleus increased at a linear rate in cells with or without lamina, reaching a maximum after 2 h: 22.2 ± 2.1% for control shRNA and 22.9 ± 3.4% for lamin shRNA (Fig. 3B). At 25 dyn/cm2, both types of cells demonstrated more consistent GFP-GR nuclear movement. By 90 min, however, GFP-GR brightness in lamin shRNA cells approached its maximum value before leveling out at 22.2 ± 1.8% at 120 min. Control shRNA cells reached a maximum of 24.1 ± 3.0% increase in nuclear brightness at 120 min (Fig. 4B). Thus, even in the absence of lamin A/C, GFP-GR was still able to translocate into the nucleus under shear stress within 2 h, independent of agonist binding. Furthermore, when we compared nuclear brightness over time for different shear stress levels, we observed that for control shRNA cells nuclear GFP-GR increase was consistently greater under 25 dyn/cm2 than 10 dyn/cm2. This finding agrees with what was observed for untreated BAECs (57). The difference in nuclear brightness was statistically significant at 40 and 90 min (P < 0.05), but this distinction in shear level was not observed in lamin shRNA cells.
Fig. 3.

Lamin deficiency did not prevent GFP-GR nuclear movement under shear stress at 10 dyn/cm2. A: live cell imaging of GFP-GR in BAECs transfected with lamin shRNA showed increasingly nuclear localized GR after 120 min of shear stress at 10 dyn/cm2 (left). Similar GFP-GR movement was also observed in cells with control shRNA (right). Nuclei were labeled with Hoechst stain. Images were taken at ×40 magnification. Scale bar = 15 μm. B: quantitative image analysis of GFP-GR subcellular movement shows a 22.9 ± 3.4% and 22.2 ± 2.1% increase in nuclear brightness after 2 h of shearing in BAECs with lamin (n = 7) and control (n = 14) shRNA respectively. *P < 0.05 at t = 20 min for control vs. lamin shRNA-treated BAEC. C: Western blot of endogenous GR shows similar increase of GR in the nuclear (N) protein fractions compared with cytoplasmic (C) in both cell types after 120 min of shear. Lamin A/C and TFIID served as the nuclear protein controls, and GAPDH was used as control for cytoplasmic protein. D: quantitative analysis of cytoplasmic and nuclear GR fractions based on repeated Western blots shows a significant increase of GR in the nuclear fraction compared with cytoplasmic proteins (n = 3).
Fig. 4.

GFP-GR nuclear movement is independent of lamin A/C under shear stress of 25 dyn/cm2. A: live cell imaging of GFP-GR in BAECs transfected with lamin shRNA showed increasingly nuclear localized GR after 120 min of shear stress at 25 dyn/cm2 (left). Similar GFP-GR movement was also observed in cells with control shRNA (right). Nuclei were labeled with Hoechst stain. Images were taken at ×40 magnification. Scale bar = 15 μm. B: quantitative image analysis of GFP-GR subcellular movement shows a 22.2 ± 1.8% and 24.1 ± 3.0% increase in nuclear brightness for lamin (n = 7) and control (n = 9) shRNA-treated BAECs respectively, after 120 min. C: Western blot of endogenous GR shows similar increase of GR in the nuclear (N) protein fractions compared with cytoplasmic (C) in both cell types following 120 min of shearing at 25 dyn/cm2. Lamin A/C and TFIID served as the nuclear protein controls, and GAPDH was used as control for cytoplasmic protein. D: quantitative analysis of cytoplasmic and nuclear GR fractions based on repeated Western blots shows a significant increase of GR in the nuclear compared with cytoplasmic fraction following shear in both cell types (n = 3).
Western blots of endogenous GR protein agreed with our time-lapse image analysis results. We saw a corresponding increase of GR in the nuclear fraction after shearing at 10 and 25 dyn/cm2 after 2 h (Figs. 3C and 4C). Quantitative analysis of Western bands also revealed a redistribution of GR from cytoplasmic to nuclear fraction (Figs. 3D and 4D). Control and lamin shRNA cells showed a GR cytoplasm/nuclear distribution of 20.0 ± 7.8/80.0 ± 7.8 and 21.2 ± 1.7/78.8 ± 1.7%, respectively, after exposure to 10 dyn/cm2 shear stress. For 25 dyn/cm2 we observed cytoplasm/nuclear distributions of 11.2 ± 0.6/88.7 ± 0.6 and 16.3 ± 2.3/83.7 ± 2.3% for control and lamin shRNA cells, respectively. These results indicate that shear-induced GFP-GR nuclear localization occurred independent of lamin A/C proteins; and for control cells, GFP-GR nucleus brightness was greater over time at 25 dyn/cm2 compared with 10 dyn/cm2. The overall trends and the final GFP-GR nuclear brightness at 120 min correspond well with what we reported previously for control cells.
Lower shear stress reduced GR nuclear translocation in control but not lamin shRNA cells.
To determine if GR translocation also occurs in the presence of a lower shear stress level, BAECs were sheared at 5 dyn/cm2. Fluorescence images again demonstrated deformed nuclei in lamin shRNA cells, as well as GFP-GR nuclear localization for either cell types with shear (Fig. 5A). Quantitative image analysis revealed that greater nuclear localization was observed in lamin shRNA cells than control cells. Lamin shRNA cells reached a maximum of 20% increase in nuclear brightness vs. 13% for control cells after 100 min (Fig. 5B). The difference was significant at t = 40, 100, and 120 min (*P < 0.05). Compared with higher shear magnitudes, control shRNA cells with an intact lamina demonstrated a lower nuclear brightness change within the same 2-h time frame (13.3 ± 1.6%), while the nuclear brightness change observed in lamin shRNA cells was similar to that treated with 10 or 25 dyn/cm2 shear stress (19.4 ± 2.2%). Western blots of endogenous GR agreed with our image analysis results (Fig. 5C): sheared cells showed increased GR in the nuclear fractions. Quantification of these Western blots showed a 34.1 ± 7.2 cytoplasmic and 65.9 ± 7.2% nuclear distribution for control cells. On the other hand, Western blots of lamin shRNA cells reflected the image analysis results: 20.2 ± 5.1 cytoplasmic and 79.8 ± 5.1% nuclear distribution (Fig. 5D).
Fig. 5.

GR nuclear translocation is reduced in control compared with lamin shRNA cells under shear stress of 5 dyn/cm2. A: live cell imaging of GFP-GR in BAECs transfected with lamin shRNA showed nuclear localized GR after 120 min of shear stress at 5 dyn/cm2 (left). Similar GFP-GR movement was also observed in cells with control shRNA (right). Nuclei were labeled with Hoechst stain. Images were taken at ×40 magnification. Scale bar = 15 μm. B: quantitative image analysis of GFP-GR subcellular movement shows a 19.4 ± 2.2 and 13.3 ± 1.6% increase in nuclear brightness for lamin (n = 11) and control (n = 11) shRNA-treated BAECs respectively, after 120 min. *P < 0.05 at t = 40, 100, and 120 min for control vs. lamin shRNA-treated BAEC. C: Western blot of endogenous GR shows an increase of GR in the nuclear (N) protein fractions compared with cytoplasmic (C) in both cell types following 120 min of shearing at 5 dyn/cm2. Lamin A/C and TFIID served as the nuclear protein controls, and GAPDH was used as control for cytoplasmic protein. D: quantitative analysis of cytoplasmic and nuclear GR fractions based on repeated Western blots shows an increase of GR in the nuclear compared with cytoplasmic fraction following shear in both cell types (n = 3). However, the GR shift to nuclear fraction under shear is less obvious than the high shear levels.
GR fails to nuclear localize under shear stress in MEFs.
To evaluate GR nuclear localization in different nonendothelial cells, we used MEFs from wild-type and lamin-deficient mice. MEF cells were exposed to dexamethasone (25 μM) for 20 min or shear stress (25 dyn/cm2) for up to 2 h as was done for BAECs, and analyzed for GR content in the nuclear and cytoplasmic fractions using Western blot analysis (Fig. 6). Western blots of wild-type MEFs initially show more cytoplasmic localization (63.4 ± 1.9%) and less nuclear localization (36.6 ± 1.9%), compared with BAECs, under static conditions. Following 20 min of dexamethasone treatment, GR becomes highly nuclear localized, up to 95.4 ± 0.9% in the nuclear fraction vs. 4.6 ± 0.9% cytoplasmic. Lmn A−/− MEFs also showed higher cytoplasmic than nuclear GR, similar to wild-type MEFs (66.4 ± 8.4% cytoplasmic vs. 33.6 ± 8.4% nuclear). Treatment with 25 μM dexamethasone also induced significant nuclear localization within 20 min: 93.0 ± 1.0% cytoplasmic vs. 7.0 ± 1.0% nuclear. Interestingly, shear stress had the opposite effect than dexamethasone in both cell types, such that GR remained or became more localized in the cytoplasmic fraction instead. GR failed to show significant nuclear translocation under shear. When sheared for 2 h at 25 dyn/cm2, both wild-type and Lmn A−/− MEFs did not show the same degree of GR nuclear localization that was observed in BAECs. The cytoplasmic GR distribution was similar for both cell types: 88.5 ± 1.5 vs. 11.5 ± 1.5% for wild-type, and 89.3 ± 3.6 vs. 10.7 ± 3.6% for Lmn A−/− MEFs. Interestingly, GR cytoplasmic fractions after shear were even higher than control cells.
Fig. 6.

Mouse embryonic fibroblasts (MEFs) show different glucocorticoid receptor behavior under shear stress compared with endothelial cells. Western blots of GR in either wild-type (WT; A) or lamin A-deficient (−/−; B) MEFs, treated with 25 μM Dex for 20 min or 25 dyn/cm2 shear stress for 2 h (Shear) and separated into cytoplasmic (C) and nuclear (N) fractions. Lamin A/C and TFIID served as the nucleus loading controls, and GAPDH served as the cytoplasmic loading control. C: quantitative analysis of cytoplasmic and nuclear GR fractions based on repeated Western blots shows the relative GR fractions in control and Dex- and shear-treated cells for both wild-type and lamin-deficient cells (n = 3).
Luciferase expression based on GRE promoter is upregulated in the absence of lamin A/C.
Using a dual-luciferase reporter plasmid, we further examined if subsequent transcription from GRE promoters was also increased. We hypothesized that activated GR in the nucleus binds to GRE promoters and initiates transcription of the reporter gene. For cells with control, nontargeting shRNA and intact nuclear lamina, treatment with dexamethasone (25 μM) resulted in a 7.6 ± 1.3-fold increase in luciferase expression over untreated cells, while cells sheared at 25 dyn/cm2 showed a 3.4 ± 0.6-fold increase (Fig. 7A). We saw significantly higher luciferase expression under dexamethasone induction compared with shear stress, which is consistent with the levels of nuclear localization we observed in our image and Western analyses. Since dexamethasone induced greater GR nuclear localization, these data suggest that dexamethasone also yielded stronger activation of the GRE promoter compared with shear stress. On the other hand, in lamin A/C-silenced BAECs, we observed a significantly higher increase in luciferase for cells treated with both dexamethasone and shear stress compared with control shRNA (P < 0.05): 18.9 ± 2.4- and 16.5 ± 1.6-fold increases, respectively (Fig. 7B). There was no significant difference in luciferase expression between dexamethasone and shear stress induction. These data suggest the expression of the GRE promoter is enhanced in the absence of nuclear lamina, regardless of induction.
Fig. 7.
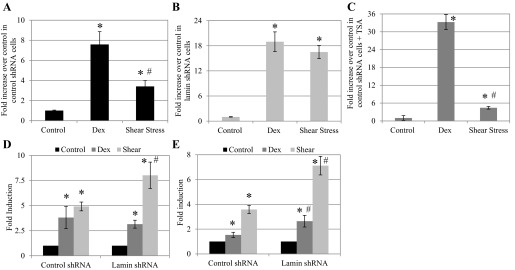
Dex- and shear-induced GRE promoter activation is affected by nuclear lamina and HDAC. A: in BAECs with control shRNA, Dex (25 μM), and shear stress (25 dyn/cm2) upregulated luciferase expression based on GRE reporter plasmid by 7.6 ± 1.3- and 3.4 ± 0.6-fold compared with untreated cells, respectively. For all cases, n = 3. *P < 0.05, compared with control. In addition, Dex-induced luciferase expression was significantly higher than that of shear stress. #P < 0.05, compared with Dex. B: In BAECs with lamin shRNA, treatment with Dex (n = 5) and shear stress (n = 6) significantly increased luciferase expression: 18.9 ± 2.4- and 16.5 ± 1.6-fold increases, respectively, compared with untreated cells (n = 5). *P < 0.05 compared with control. C: after pretreatment with 500 nM TSA, BAECs with control or lamin shRNA were treated with Dex or shear stress as before. We observed a significant 33.3 ± 2.5-fold increase in Dex-treated cells (n = 5) over untreated control TSA cells (n = 5), similar to lamin shRNA cells. TSA-treated and sheared cells showed a 4.4 ± 0.4-fold increase of luciferase over untreated control cells (n = 3). *P < 0.05, compared with control. Dex-induced luciferase expression was significantly higher than that of shear stress. #P < 0.05, compared with Dex. D: fold induction of NF-κBIA expression in control and lamin shRNA cells. Control and lamin shRNA BAECs were treated with Dex for 20 min or 25 dyn/cm2 for 2 h before RNA extraction. Compared with untreated cells, Dex resulted in 3.81 ± 1.11- and 3.15 ± 0.40-fold increase of NF-κBIA expression, while shear stress induced 4.91 ± 0.76- and 8.02 ± 1.31-fold increase, in control and lamin shRNA cells, respectively (n = 3). E: fold induction of DUSP1 expression in control and lamin shRNA cells. Compared with untreated cells, Dex treatment in control and lamin shRNA cells showed 1.53 ± 0.22- and 2.64 ± 0.47-fold increases, respectively. For shear stress, control and lamin shRNA cells showed fold expression increases of 3.58 ± 0.33 and 7.12 ± 0.75, respectively (n = 3 and 4 for control shRNA and lamin shRNA, respectively). *P < 0.05, compared with control. #P < 0.05, compared with same treatment of control shRNA cells.
Inhibiting HDAC activity increased luciferase expression in control cells under dexamethasone, not shear stress, induction.
We investigated further the role of nuclear lamina in GRE-based transcription. The nuclear lamina is home to nuclear membrane proteins that function in chromatin organization and transcriptional regulation (36). We hypothesize that in the absence of lamins, chromatin disorganization and histone acetylation are increased, resulting in increased transcriptional activity. Lamina-associated proteins interact with histone modifiers such as HDAC. TSA selectively inhibits class I mammalian HDAC. Control shRNA BAECs were pretreated with 500 nM TSA for 8 h before addition of dexamethasone (25 μM) or shear stress (25 dyn/cm2). After 18–20 h, luciferase activity was determined, and data are presented as fold change over the no treatment group (Fig. 7C). We saw that with the addition of TSA, control cells with intact lamin A/C demonstrated a significant 33.3 ± 2.5-fold increased in luciferase expression (P < 0.05), a similar trend to that observed in lamin-deficient cells under dexamethasone induction. Under shear induction, however, TSA pretreatment yielded a slight yet significant increase of 4.4 ± 0.4 (P < 0.05) compared with untreated cells. Thus treating cells with TSA and dexamethasone replicated the increased GRE promoter expression of luciferase that was observed in cells lacking lamin A/C. On the other hand, TSA treatment on sheared cells did not mimic the change in luciferase expression observed in lamin-deficient cells.
GR-based transcriptional activation is confirmed in NF-κBIA and DUSP1 using quantitative RT-PCR.
To further support our GRE promoter results with functional significance, we measured transcription changes of genes that are relevant to cardiovascular disease and have been shown to be responsive to GR activation. These genes primarily fall into the category of anatomical structural development, stress response, apoptosis regulation, and transcription factor activity (60). Using quantitative RT-PCR, we examined transcription changes of the NF-κBIA gene (Fig. 7D), also known as IκB-α, which is reported to contain two GRE consensus sequences. In control shRNA BAECs, fold induction increased by 3.81 ± 1.11- and 4.91 ± 0.76-fold with dexamethasone and 25 dyn/cm2 treatment, respectively. In lamin shRNA BAECs, fold induction increased by 3.15 ± 0.40 and 8.02 ± 1.31 for dexamethasone and shear treatment, respectively (*P < 0.05, compared with untreated control). Dexamethasone yielded similar increase in NF-κBIA transcription in control and lamin shRNA cells. However, shear stress caused a significant increase in NF-κBIA in lamin-deficient cells compared with control cells (#P < 0.05). Thus our quantitative RT-PCR results support our luciferase promoter study and showed that both dexamethasone and shear stress increased transcription of GR-responsive genes. Shear stress, in fact, produced greater gene transcription in lamin shRNA cells than control cells. We also examined the transcription of DUSP1, also known as mitogen-activated protein kinase phosphatase 1 (MKP-1), with three GRE consensus sequences (60) (Fig. 7E). In control shRNA BAECs, fold induction increased by 1.53 ± 0.22 and 3.58 ± 0.33 with dexamethasone and shear stress, respectively. In lamin shRNA BAECs, fold induction increased by 2.64 ± 0.47 and 7.12 ± 0.75, respectively (*P < 0.05, compared with control; #P < 0.05, compared with same treatment in control shRNA). In contrast to NF-κBIA, we did see significantly increased transcription in DUSP1 under dexamethasone treatment in lamin shRNA cells, which correlates with our luciferase data. Sheared lamin shRNA cells also showed a significant increase in DUSP1 upregulation compared with those sheared with intact lamin.
Dexamethasone and shear stress increased HDAC and HAT activities in control cells but not in absence of nuclear lamina.
To further examine the role of HDAC in transcriptional regulation and mechanotransduction at the nucleus, we also analyzed HAT activities in control and lamin-deficient cells. Using nuclear extracts from both cell types, we measured differences in histone acetylase and deacetylase activities using respective assays. In cells with intact lamina, dexamethasone treatment induced a 2.19 ± 0.35-fold increase in HDAC activity, while shear stress at 25 dyn/cm2 induced a greater 2.95 ± 0.52-fold increase over untreated control (both P < 0.05; Fig. 8A). This significant increase in HDAC activity under dexamethasone and shear stress treatment was not present in cells with compromised lamina following treatment with lamin shRNA. Compared with untreated lamin shRNA BAECs, dexamethasone induced a 1.68 ± 0.25-fold increase and shear stress induced a 1.41 ± 0.25-fold increase. The results suggest that the activity of HDAC, which is normally sequestered at the nuclear lamina, does appear to be hindered in the absence of lamin A/C. On the other hand, HAT activity in both cell types showed decreased fold inductions over control but still followed a similar trend to HDAC activity. Compared with untreated control cells, histone acetylase activity increased by 1.11 ± 0.03-fold following treatment with dexamethasone, and 25 dyn/cm2 shear stress led to a slightly greater 1.17-fold change (both P < 0.05; Fig. 8B). Again, this trend appears to be lost in lamin shRNA cells. Compared with untreated lamin shRNA cells, dexamethasone induced a 1.04 ± 0.04-fold change in histone acetylase activity, and shear stress induced a 0.98 ± 0.06-fold change. Our findings suggest that activities of HDAC and to a less extent HAT are increased by dexamethasone and to a greater extent by shear stress in endothelial cells. However, histone acetylation in the presence of a stimulus such as dexamethasone or shear stress is altered in a lamin-deficient state.
Fig. 8.

Histone acetylase (HAT) and deacetylase (HDAC) proteins show altered function in the absence of lamin A/C. A: HDAC proteins in control shRNA cells showed increasing and significant activity when BAECs were exposed to either 25 μM Dex (2.19 ± 0.35-fold) or 25 dyn/cm2 shear stress (2.95 ± 0.52-fold) compared with static (*P < 0.05, compared with control). Lamin shRNA BAECs showed a lesser and insignificant increase under stimulation (1.68 ± 0.25-fold for Dex, 1.42 ± 0.25-fold for shear stress) compared with static. B: HAT proteins in control shRNA BAECs showed significant increases in activity upon stimulation by Dex (1.11 ± 0.03-fold) or shear stress (1.17 ± 0.04-fold; *P < 0.05, compared with control). Lamin shRNA BAECs failed to show an identical trend, as Dex and shear stress both failed to induce significant increases in HAT activity (1.04 ± 0.04- and 0.99 ± 0.07-fold, respectively).
DISCUSSION
Using the image analysis algorithm that we developed based on Bayesian statistics, we can quantify time-dependent intracellular movement of fluorescently labeled GR. Here, we demonstrate that GFP-GR nuclear translocation after induction by dexamethasone or shear stress is independent of an intact nuclear lamina. We successfully silenced lamin A/C gene using specific shRNA and confirmed lamin deficiency by both Western blot and image analysis (Fig. 1). After treatment with dexamethasone or shear stress, cells with control or lamin A/C gene silencing shRNA demonstrated a similar extent of GFP-GR nuclear localization to what we had reported for non-shRNA-treated cells (57). Our imaging data demonstrate that nuclear lamina is not required for nuclear import of GR after induction with either dexamethasone or shear tress; and in all cases, we have confirmed nuclear localization of endogenous GR via protein analysis.
Lamins participate in linking the nucleus to the cytoskeleton networks through the LINC complex (18) by attaching to SUN proteins within the nuclear envelope, which are further connected to nesprin proteins on the outer nuclear envelope. Nesprins in turn attach to actin and microtubules via dynein and intermediate filaments via plectin (67). We have previously reported that disrupting the endothelial actin and microtubule networks does not prevent nuclear localization of GFP-GR with dexamethasone or shear stress since GR may utilize alternative methods, such as diffusion, to translocate (57). It is not surprising then that silencing lamin proteins also would not interfere with nuclear localization of GR proteins. Nuclear import of GR is independent of the lamin-dependent, nucleus-cytoskeleton link.
Interestingly, for cells with nontargeting control shRNA, we observed elevated GFP-GR nuclear presence under the higher shear stress of 25 dyn/cm2 compared with 10 dyn/cm2. We had previously reported this trend for non-shRNA-treated cells (57). However, this shear-dependent difference is lost in cells lacking nuclear lamina (Fig. 3B and 4B). Nuclear lamina plays an important role in maintaining nuclear structure and mechanics (22, 50). In cells with intact nuclei, we observed a greater degree of GFP-GR nuclear localization under higher shear stress, but this distinction was less obvious in lamin-deficient cells. This could be in part due to increased nuclear stiffness in cells with intact lamina and proper signal transduction cascades. In cells lacking lamin A/C, nuclei are more deformed and fragile with decreased nuclear mechanical stiffness as well as impaired activation of mechanosensitive genes (19, 39). As a result, they are potentially more susceptible to all levels of shear stress. This would also suggest disruption of lamina-dependent signal transduction cascade, for example, the MAPK signaling pathway that phosphorylates GR independent of a ligand (35, 56).
Our data suggest that lower shear stress (5 dyn/cm2) may have a different effect on GR nuclear translocation in cells with intact lamina (Fig. 5). We observed less GFP-GR brightness change in the nucleus and a greater cytoplasmic GR presence, which suggests that GR translocation under athero-genic levels of shear stress is more hindered compared with athero-protective levels (10 or 25 dyn/cm2). This would be consistent with athero-protective property of higher shear stress, with upregulation of anti-inflammatory glucocorticoid-responsive genes. In the absence of lamina, this was not the case as we observed similar GR nuclear localization to cells sheared at higher magnitudes. Again, this may be due to the nuclei becoming more susceptible to mechanical stresses as well as disrupted signaling cascade.
Alternatively, we also examined GR behavior under shear stress in a nonendothelial cell line, specifically wild-type or lamin-deficient MEFs. Both types of cells demonstrated different GR responses to dexamethasone and shear stress compared with endothelial cells. GR content is greater in the cytoplasmic fraction initially and shifts significantly to the nucleus with dexamethasone induction and the cytoplasm with shear induction (Fig. 6). Other groups have found that GR cellular localization varies at each phase of the cell cycle. In HeLa, A549, and HEK cells, GR has been observed to show both cytoplasmic and nuclear localization through interphase, with peak accumulation occurring immediately before cell division. It is excluded from the nucleus upon the initiation of mitosis through early G1. This is likely due to changes in GR phosphorylation in the AF-1 domain by CDK proteins, whose activity changes based on the cell cycle (55). Moreover, these cells have been shown to be resistant to dexamethasone immediately following cell division, and as a result GR failed to translocate (41, 55). Thus GR activity under shear may be cell dependent, which would explain why MEFs failed to show similar nuclear translocation to BAECs. The effects of dexamethasone and shear stress on GR nuclear translocation and transcriptional regulation we observed may be specific to endothelial cells. These results warrant future investigations into ligand independent activation mechanisms of GR among different cell types, specifically regulatory proteins upstream of GR in response to stress signals.
After we confirmed nuclear translocation of endogenous GR in lamin A/C-silenced cells, we examined activation of transcription based on GRE promoters using a GRE dual-luciferase reporter plasmid (Fig. 7). In control shRNA cells with intact lamina, dexamethasone and shear stress induced significant increases in luciferase activity compared with untreated cells (P < 0.05), with dexamethasone being a much more potent inducer of GRE-based transcription activation compared with shear stress (P < 0.05). This finding closely reflects the extent of GFP-GR nuclear localization we assessed quantitatively for control cells and also agrees with our previous report of GRE promoter activation (47) as well as others studies on GRE induction by dexamethasone (6, 35, 38).
In lamin-deficient cells, dexamethasone and shear stress both induced significant increases in luciferase expression compared with untreated control shRNA cells (Fig. 7B). Our data suggest that in the absence of lamin A/C there was increased transcriptional activity and gene expression from our GRE promoter plasmid. Recently, lamin proteins have been shown to be mediators of oxidative stress (54, 62). In the absence of lamin proteins, there is an increase in reactive oxygen species, which causes the 14-3-3 zeta isoform proteins to bind to GR, and Galliher-Beckley (35) showed that in oxidative stress induced by H2O2 more 14-3-3 zeta is recruited to the gene promoter region. Thus there are other mechanisms, such as in during oxidative stress, through which lamin deficiency could affect signal mechanisms, ultimately leading to altered transcription of target genes. Furthermore, some studies have represented the nuclear lamina as a genetically repressive environment (24, 61). The absence of the nuclear lamina would therefore lead to a loss of heterochromatin at the inner nuclear membrane and unregulated gene transcription, a result of certain transcription factors no longer being sequestered there as inactive complexes (39).
To confirm functional transcriptional regulation in cells with and without lamin A/C, under dexamethasone or shear stress induction, we evaluated transcription of NF-κBIA and DUSP1. NF-κBIA functions to inhibit the ubiquitous NF-κB transcription factor by sequestering it in the cytoplasm and plays a significant role in inflammatory responses in low or pulsatile shear stress, for example (12, 25). Glucocorticoids antagonize NF-κB activity by upregulating NF-κBIA expression levels (5, 17). The DUSP family of proteins dephosphorylates MAPK to inhibit inflammatory signaling (8). They inhibit endothelial cell activation and mediate its anti-inflammatory effects under laminar shear stress: DUSP1 was found to reduce both VCAM-1 expression and p38 phosphorylation in response to laminar flow (65). Dexamethasone significantly upregulated DUSP1 expression within 5 min and inhibited both TNF-α-induced p38 MAPK and E-selectin expression (33). Our results in control shRNA BAECs support the anti-inflammatory effects of both NF-κBIA and DUSP1 in endothelial cells: their transcriptions are increased in the presence of either dexamethasone or shear stress. Figure 7, D and E, also correlates the corresponding functional effects of GR activation under dexamethasone and shear stress. Compared with NF-κBIA, DUSP1 showed significantly elevated transcription in lamin-deficient cells (Fig. 7E). This may infer a difference in between NF-κBIA and DUSP1 in their chromatin structures before treatment (25), and the absence of lamin may have further affected the chromatin unfolding as others have reported (23, 36). Similarly, we observed a significant high transcription of NF-κBIA and DUSP1 under shear stress in the absence of lamin A/C, possibly due to altered acetylation of histone proteins in the presence of shear stress (44).
Trichostatin A, a class 1 HDAC inhibitor, has recently been shown to induce chromatin changes and relocate it away from the nuclear periphery, similar to what is observed in lamin A/C-deficient cells (34). TSA treatment of human umbilical vein endothelial cells resulted in suppressed eNOS expression (31). Similarly, our control shRNA BAECs treated with TSA alone also demonstrated reduced GRE expression (data not shown). Subsequent treatment with dexamethasone induced a significant 33-fold increase in luciferase reporter gene expression (Fig. 7C), similar to the increased GRE transcription observed in cells lacking nuclear lamina (Fig. 7B). Thus either inhibiting HDAC or silencing lamin A/C resulted in elevated, dexamethasone-induced transcriptional activity in BAEC. Disrupted lamina in lamin shRNA cells could affect transcription of GRE consensus genes via loss of HDAC functions and/or subsequent increased acetylation of HAT proteins (e.g., histone H3), as was observed for wild-type and lamin-deficient MEFs (34). Along with upregulation of anti-inflammatory genes, glucocorticoids also downregulate expression of inflammation associated genes such as interleuken-11, MAP3K14 (which stimulates NF-κB activity), and CCL2 (participates in the pathogenesis of atherosclerosis) (60). Thus defective nuclear lamina may potentially negatively affect the normal repression of these genes in the presence of GR binding to GRE as well.
Interestingly, TSA had little effect on transcription activation in sheared BAEC; the correlation between HDAC inhibition and lamin deficiency was not observed in sheared cells (Fig. 7, B and C). Lamin deficiency had a much more significant impact on activating GRE transcription than HDAC inhibition under shear, which emphasizes the importance of nuclear lamina in mechanotransduction in response to shear stress. Furthermore, our study suggests that shear-induced transcriptional activation pathways are different and less HDAC dependent than those activated by dexamethasone binding. Activated GR has been shown to interact with class 1 HDACs (HDAC2) upon ligand binding to inhibit NF-κB gene activation (3). Inside the nucleus, after binding to the GRE promoter, HDAC1 is eventually acetylated and gene expression is suppressed (59). Direct interaction of HDAC with the ligand-GR complex would be interrupted by TSA. Inhibiting HDAC could therefore have a more pervasive impact than lamin silencing under dexamethasone, resulting in additional nonregulated transcriptional activity. This could explain the higher increase in GRE activation with dexamethasone in TSA-treated control cells than lamin-deficient cells. In addition, shear stress could be utilizing other mechanosensitive signaling pathways that are independent of GR but are still impacted by the presence of nuclear lamina. In fact, mutations in the lamin gene can lead to increased phosphorylation of MAPK proteins in cardiomyocytes (56), which could increase GR phosphorylation under shear compared with cells with intact lamina. Thus, nuclear lamina, rather than HDAC, is a more significant mediator of shear-induced transcriptional activity.
Histone proteins, which interact with lamins, are responsible for maintaining chromatin organization and architecture. Histone acetylation by HAT results in increased transcription while HDAC remove acetyl groups from histones, resulting in tightened chromatin and suppressed gene expression. Here, we hypothesize that the absence of A-type lamins led to loss of HDAC function when they were no longer sequestered at the nuclear periphery, resulting in open chromatin and increased transcriptional activity (3, 53). To further investigate the effect of dexamethasone and shear stress on histone acetylation, we found that in response to either stimulus, control cells with intact lamina showed increasing HDAC and HAT activity compared with untreated cells (Fig. 8). The greater increase observed in the presence of shear stress is likely due to the greater array of genes and transcription regulators that are involved in endothelial mechanotransduction. On the other hand, dexamethasone would only trigger the GR transcription pathway. HDAC and HAT activity following dexamethasone or shear stress treatment is significantly reduced in the absence of lamina. As previously discussed, the decreased HDAC activity in lamin shRNA cells may be due to it no longer being sequestered at the lamina in its native state, where it interacts with other lamin binding proteins. Transforming chromatin to euchromatin will eventually require deacetylation by HDAC to return it to its heterochromatin state. With respect to HAT, lamin deficiency has been shown to cause increased HAT activity (23, 34). When HAT activity results for both cell types were normalized to that of untreated control shRNA cells, we did indeed see acetylase activity in untreated lamin shRNA cells to be similar to those of sheared cells with intact lamina. These data support the increased GRE reporter gene expression in lamin-deficient cells compared with control cells and further suggest that nuclear lamina is a more significant mediator of transcriptional activity than either biochemical (dexamethasone) or mechanical (shear stress) induction.
In summary, using our novel image analysis algorithm, we have confirmed that nuclear translocation of GFP-GR is independent of nuclear lamina under both dexamethasone and shear stress induction. Both dexamethasone and shear stress induced GRE-based transcription activation to levels that corresponded to the extent of receptor nuclear localization. In the absence of lamin A/C, transcriptional activity increased significantly with dexamethasone or shear stress, and treating control cells with the HDAC inhibitor TSA also significantly increased transcription by dexamethasone. Dexamethasone and shear stress increased HDAC and HAT activities in control cells but not in absence of nuclear lamina. Our data suggest that while nuclear lamin does not affect nuclear import of GR, it plays an essential role in the subsequent transcriptional regulation. Dexamethasone-activated transcription is dependent on HDAC, while shear stress activated transcription is more dependent on the presence of nuclear lamina. We continue to further elucidate the mechanotransduction pathways involved in gene transcription and the role of nuclear proteins. While GR activation under shear stress may be unique to endothelial cells, this study also contributes to our understanding of the disease mechanisms of laminopathies by linking effects of mechanical stress and chromatin regulation at the nuclear lamina.
GRANTS
This study was funded partially by American Heart Association Grant 09SDG2060548 and National Institute on Aging R03-AG-035248-01.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.N. performed experiments; A.N. analyzed data; A.N. interpreted results of experiments; A.N. and J.Y.J. prepared figures; A.N. and J.Y.J. drafted manuscript; A.N. and J.Y.J. edited and revised manuscript; A.N. and J.Y.J. approved final version of manuscript; J.Y.J. conception and design of research.
REFERENCES
- 1. Adcock IM. Glucocorticoid-regulated transcription factors. Pulm Pharmacol Ther 14: 211–219, 2001 [DOI] [PubMed] [Google Scholar]
- 2. Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol 79: 376–384, 2001 [DOI] [PubMed] [Google Scholar]
- 3. Adenuga D, Rahman I. Oxidative stress, histone deacetylase and corticosteroid resistance in severe asthma and COPD. Curr Resp Med Rev 3: 57–68, 2007 [Google Scholar]
- 4. Al-Shali KZ, Hegele RA. Laminopathies and atherosclerosis. Arterioscler Thromb Vasc Biol 24: 1591–1595, 2004 [DOI] [PubMed] [Google Scholar]
- 5. Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol 71: 9–15, 2002 [PubMed] [Google Scholar]
- 6. Avenant C, Kotitschke A, Hapgood JP. Glucocorticoid receptor phosphorylation modulates transcription efficacy through GRIP-1 recruitment. Biochemistry 49: 972–985, 2010 [DOI] [PubMed] [Google Scholar]
- 7. Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 94: 557–572, 1998 [DOI] [PubMed] [Google Scholar]
- 8. Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev 30: 830–882, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 8: 323–327, 1994 [DOI] [PubMed] [Google Scholar]
- 10. Blake G, Ridker P. Inflammatory bio-markers and cardiovascular risk prediction. J Intern Med 252: 283–294, 2002 [DOI] [PubMed] [Google Scholar]
- 11. Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 21: 285–288, 1999 [DOI] [PubMed] [Google Scholar]
- 12. Braddock M, Schwachtgen JL, Houston P, Dickson MC, Lee MJ, Campbell CJ. Fluid shear stress modulation of gene expression in endothelial cells. News Physiol Sci 13: 241–246, 1998 [DOI] [PubMed] [Google Scholar]
- 13. Burke B, Stewart CL. Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol 3: 575–585, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Butin-Israeli V, Adam SA, Goldman AE, Goldman RD. Nuclear lamin functions and disease. Trends Genet 28: 464–471, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 9: 109–112, 2000 [DOI] [PubMed] [Google Scholar]
- 16. Chien S. Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol 292: H1209–H1224, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol 275: 79–97, 2007 [DOI] [PubMed] [Google Scholar]
- 18. Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172: 41–53, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dahl KN, Ribeiro AJ, Lammerding J. Nuclear shape, mechanics, mechanotransduction. Circ Res 102: 1307–1318, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation 109: III27–32, 2004 [DOI] [PubMed] [Google Scholar]
- 21. De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 70: 726–736, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Vos WH, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, Manders EM, Verstraeten VL, van Steensel MA, Marcelis CL, van den Wijngaard A, Vaux DJ, Ramaekers FC, Broers JL. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet 20: 4175–4186, 2011 [DOI] [PubMed] [Google Scholar]
- 23. Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22: 832–853, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Demmerle J, Koch AJ, Holaska JM. The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem 287: 22080–22088, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deroo BJ, Archer TK. Glucocorticoid receptor activation of the I kappa B alpha promoter within chromatin. Mol Biol Cell 12: 3365–3374, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng 103: 177–185, 1981 [DOI] [PubMed] [Google Scholar]
- 27. Diamond MI, Miner JN, Yoshinaga SK, Yamamoto KR. Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 249: 1266–1272, 1990 [DOI] [PubMed] [Google Scholar]
- 28. Eickelberg O, Roth M, Lorx R, Bruce V, Rudiger J, Johnson M, Block LH. Ligand-independent activation of the glucocorticoid receptor by beta2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J Biol Chem 274: 1005–1010, 1999 [DOI] [PubMed] [Google Scholar]
- 29. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423: 293–298, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 341: 1715–1724, 1999 [DOI] [PubMed] [Google Scholar]
- 31. Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D'Abreo C, Marsden PA. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem 280: 24824–24838, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Frangos JA, McIntire LV, Eskin SG. Shear stress induced stimulation of mammalian cell metabolism. Biotechnol Bioeng 32: 1053–1060, 1988 [DOI] [PubMed] [Google Scholar]
- 33. Furst R, Schroeder T, Eilken HM, Bubik MF, Kiemer AK, Zahler S, Vollmar AM. MAPK phosphatase-1 represents a novel anti-inflammatory target of glucocorticoids in the human endothelium. FASEB J 21: 74–80, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Galiova G, Bartova E, Raska I, Krejci J, Kozubek S. Chromatin changes induced by lamin A/C deficiency and the histone deacetylase inhibitor trichostatin A. Eur J Cell Biol 87: 291–303, 2008 [DOI] [PubMed] [Google Scholar]
- 35. Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol 31: 4663–4675, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP. Nuclear lamins: building blocks of nuclear architecture. Genes Dev 16: 533–547, 2002 [DOI] [PubMed] [Google Scholar]
- 37. Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL. The nuclear lamina comes of age. Nat Rev Mol Cell Biol 6: 21–31, 2005 [DOI] [PubMed] [Google Scholar]
- 38. Gupta V, Awasthi N, Wagner BJ. Specific activation of the glucocorticoid receptor and modulation of signal transduction pathways in human lens epithelial cells. Invest Ophthalmol Vis Sci 48: 1724–1734, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ho CY, Lammerding J. Lamins at a glance. J Cell Sci 125: 2087–2093, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Htun H, Barsony J, Renyi I, Gould DL, Hager GL. Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc Natl Acad Sci USA 93: 4845–4850, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu JM, Bodwell JE, Munck A. Cell cycle-dependent glucocorticoid receptor phosphorylation and activity. Mol Endocrinol 8: 1709–1713, 1994 [DOI] [PubMed] [Google Scholar]
- 42. Hutchison CJ. Lamins: building blocks or regulators of gene expression? Nat Rev Mol Cell Biol 3: 848–858, 2002 [DOI] [PubMed] [Google Scholar]
- 43. Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma? Nat Cell Biol 6: 1062–1067, 2004 [DOI] [PubMed] [Google Scholar]
- 44. Illi B, Nanni S, Scopece A, Farsetti A, Biglioli P, Capogrossi MC, Gaetano C. Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ Res 93: 155–161, 2003 [DOI] [PubMed] [Google Scholar]
- 45. Ishida T, Peterson T, Kovach N, Berk B. MAP kinase activation by flow in endothelial cells. Role of beta 1 integrins and tyrosine kinases. Circ Res 79: 310–316, 1996 [DOI] [PubMed] [Google Scholar]
- 46. Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol 16: 2382–2392, 2002 [DOI] [PubMed] [Google Scholar]
- 47. Ji JY, Jing H, Diamond SL. Shear stress causes nuclear localization of endothelial glucocorticoid receptor and expression from the GRE promoter. Circ Res 92: 279–285, 2003 [DOI] [PubMed] [Google Scholar]
- 48. Ku D, Giddens D, Zarins C, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 5: 293–302, 1985 [DOI] [PubMed] [Google Scholar]
- 49. Kuchan M, Frangos J. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am J Physiol Cell Physiol 266: C628–C636, 1994 [DOI] [PubMed] [Google Scholar]
- 50. Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113: 370–378, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Libby P, DiCarli M, Weissleder R. The vascular biology of atherosclerosis and imaging targets. J Nucl Med 51, Suppl 1: 33S–37S, 2010 [DOI] [PubMed] [Google Scholar]
- 52. Lloyd DJ, Trembath RC, Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum Mol Genet 11: 769–777, 2002 [DOI] [PubMed] [Google Scholar]
- 53. Luo Y, Jian W, Stavreva D, Fu X, Hager G, Bungert J, Huang S, Qiu Y. Trans-regulation of histone deacetylase activities through acetylation. J Biol Chem 284: 34901–34910, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Malhas AN, Lee CF, Vaux DJ. Lamin B1 controls oxidative stress responses via Oct-1. J Cell Biol 184: 45–55, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matthews L, Johnson J, Berry A, Trebble P, Cookson A, Spiller D, Rivers C, Norman M, White M, Ray D. Cell cycle phase regulates glucocorticoid receptor function. PloS One 6: e22289, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest 117: 1282–1293, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nayebosadri A, Christopher L, Ji JY. Bayesian image analysis of dexamethasone and shear stress-induced glucocorticoid receptor intracellular movement. Ann Biomed Eng 40: 1508–1519, 2012 [DOI] [PubMed] [Google Scholar]
- 58. Pavalko FM, Gerard RL, Ponik SM, Gallagher PJ, Jin Y, Norvell SM. Fluid shear stress inhibits TNF-alpha-induced apoptosis in osteoblasts: a role for fluid shear stress-induced activation of PI3-kinase and inhibition of caspase-3. J Cell Physiol 194: 194–205, 2003 [DOI] [PubMed] [Google Scholar]
- 59. Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, Hendarwanto A, Martinez ED, Chen Y, Lu H, Adkins NL, Stavreva DA, Wiench M, Georgel PT, Schiltz RL, Hager GL. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol Cell 22: 669–679, 2006 [DOI] [PubMed] [Google Scholar]
- 60. Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19: 2163–2171, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shevelyov YY, Nurminsky DI. The nuclear lamina as a gene-silencing hub. Curr Issues Mol Biol 14: 27–38, 2012 [PubMed] [Google Scholar]
- 62. Sieprath T, Darwiche R, De Vos WH. Lamins as mediators of oxidative stress. Biochem Biophys Res Commun 421: 635–639, 2012 [DOI] [PubMed] [Google Scholar]
- 63. Tseng H, Peterson T, Berk B. Fluid shear stress stimulates mitogen-activated protein kinase in endothelial cells. Circ Res 77: 869–878, 1995 [DOI] [PubMed] [Google Scholar]
- 64. Wilson KL, Berk JM. The nuclear envelope at a glance. J Cell Sci 123: 1973–1978, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zakkar M, Chaudhury H, Sandvik G, Enesa K, Luong le A, Cuhlmann S, Mason JC, Krams R, Clark AR, Haskard DO, Evans PC. Increased endothelial mitogen-activated protein kinase phosphatase-1 expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis. Circ Res 103: 726–732, 2008 [DOI] [PubMed] [Google Scholar]
- 66. Zarins C, Giddens D, Bharadvaj B, Sottiurai V, Mabon R, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res 53: 502–514, 1983 [DOI] [PubMed] [Google Scholar]
- 67. Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet 22: 2335–2349, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]