

Abstract
Autophagy is an essential cellular mechanism for cell homeostasis and survival by which damaged cellular proteins are sequestered in autophagosomal vesicles and cleared through lysosomal machinery. The autophagy pathway also plays an important role in immunity and inflammation via pathogen clearance mechanisms mediated by immune cells, including macrophages and neutrophils. In particular, recent studies have revealed that autophagic activity is required for the release of neutrophil extracellular traps (NETs), representing a distinct form of active neutrophil death, namely NETosis. Although NET formation is beneficial during host defense against invading pathogens, the mechanisms that promote excessive NETosis under pathological conditions remain ill defined. In the present study, we aimed to characterize the role of the mammalian target of rapamycin (mTOR) in NETosis. As mTOR kinase is known as a key regulator of autophagy in many mammalian cells including neutrophils, we hypothesized that mTOR may play a regulatory role in NET release by regulating autophagic activity. Our data show that the pharmacological inhibition of the mTOR pathway accelerated the rate of NET release following neutrophil stimulation with the bacteria-derived peptide formyl-Met-Leu-Phe (fMLP), while autophagosome formation was enhanced by mTOR inhibitors. This increased mTOR-dependent NET release was sensitive to inhibition of respiratory burst or blockade of cytoskeletal dynamics. Overall, this study demonstrates a pivotal role for the mTOR pathway in coordinating intracellular signaling events downstream of neutrophil activation leading to NETosis.
Keywords: neutrophil, autophagy, neutrophil extracellular traps, mTOR, histones
autophagy plays a primary role in cellular protein homeostasis by regulating turnover of stable and damaged intracellular proteins (24). During autophagy, a double-membrane vesicle containing captured cellular components develops into an autophagosome, followed by fusion with a lysosome to break down the vesicle contents and inner membrane. One of the key regulators of autophagy includes mammalian target of rapamycin (mTOR), a serine/threonine kinase that regulates cell growth, proliferation, and protein synthesis (12, 16, 19). In mammalian cells, mTOR kinase directly interacts with the unc-51-like kinase (ULK) complex, which plays a major role in controlling early steps of autophagosome formation, to inhibit membrane targeting of the ULK complex and other autophagy-related gene (Atg) proteins (16, 19). Inhibition of mTOR activity is known to play an essential step in the initiation of autophagy.
Autophagy has been shown to play a key role in regulating leukocyte responses ranging from phagocytosis of pathogens (12) and cytokine secretion (10) to the formation of neutrophil extracellular traps (NETs; Ref. 26). As the first line of host defense, activated neutrophils can release NETs, which is defined as decondensed chromatin fibers armed with antimicrobial granular cargos to trap and kill pathogens (1, 4). The process of NET release has been defined as “NETosis,” an active form of cell death distinct from apoptosis or necrosis (5). Despite their protective role against infection, increasing evidence has suggested that NETs may also promote pathological outcomes in inflammatory and/or thrombotic conditions (21, 28, 31). However, it is unclear which signaling pathways regulate NET release in response to inflammatory stimuli. The phorbol ester phorbol-myristate-acetate (PMA) is commonly used to induce NETs in vitro, whereas variable results have been reported for the induction of NETs by cytokines, chemokines, and microorganisms (26). For instance, the bacteria-derived peptide, formyl-Met-Leu-Phe (fMLP), which can potently stimulates reactive oxygen species (ROS) production (29), has been shown by Neeli et al. (23) to trigger nuclear chromatin release, while NET formation via fMLP was not observed by other groups (25, 30).
Recently, mTOR has been shown to regulate lipopolysaccharide (LPS)-induced NET release by posttranscriptional control of hypoxia-inducible factor-1α (HIF-1α) protein expression (15). However, the authors did not examine the role of mTOR in the autophagy pathway during NETosis. In this study, we test our hypothesis that the mTOR pathway negatively regulates NET formation via the modulation of autophagic activity in response to fMLP. Our data show that mTOR inhibition with rapamycin accelerated the kinetics of NET formation upon stimulation with fMLP, which was associated with the rapid formation of autophagosomes. These findings are in contrast to the inhibitory effect of rapamycin in LPS-induced NETosis (15). Our data suggest that mTOR activation may prevent neutrophils from undergoing autophagy, thus playing a pivotal role in determining a form of active cell death in activated neutrophils. Moreover, we show that autophagic activity may have a role in histone citrullination, a characteristic histone modification during NETosis and that NET formation requires ROS production and functional cytoskeletal machinery, highlighting multiple layers of regulatory mechanisms underlying NETosis.
MATERIALS AND METHODS
Reagents.
Rapamycin was from LC Laboratories (Woburn, MA). Anti-microtubule-associated protein light chain 3B (LC3B) antibody (no. 2775) was from Cell Signaling (Danvers, MA) and used for autophagosome detection. Anti-neutrophil elastase (NE) antibody (ab21595) and anti-histone H3 (citrulline 2 + 8 + 17) antibody (H3Cit; ab5103) were from Abcam (Cambridge, MA) and used as NET markers. The cell-permeable DNA dye Hoechst 33342, the cell-impermeable DNA dye Sytox green, and the intracellular ROS sensor H2DCFDA were from Invitrogen (Grand Island, NY). All other reagents were from Sigma-Aldrich (St. Louis, MO) or previously named sources (3, 8).
Preparation of human neutrophils.
Human neutrophils were purified as previously described (9, 14). Briefly, human venous blood was drawn in accordance with an Oregon Health & Science University Institutional Review Board-approved protocol from healthy donors into citrate-phosphate-dextrose and layered over an equal volume of Polymorphprep, followed by centrifugation at 500 g for 45 min at 18°C. The lower layer containing neutrophils was collected and washed with HBSS by centrifugation at 400 g for 10 min. To remove contaminating red blood cells, the pellet was resuspended in sterile H2O for 30 s, followed by the immediate addition of 10× PIPES buffer (250 mM PIPES, 1.1 mM NaCl, and 50 mM KCl pH 7.4). After centrifugation at 400 g for 10 min, the pellet was resuspended in PMN buffer (HBSS containing 2 mM CaCl2, 2 mM MgCl2 and 1% wt/vol BSA).
Live-cell imaging.
Purified human neutrophils (2 × 106/ml) were incubated on fibronectin-coated μ-slide eight-well chamber (Ibidi, Verona, WI) at 37°C for 30 min, followed by incubation with indicated inhibitors or vehicle (DMSO) in PMN buffer containing Hoechst 33342 (10 μg/ml) and Sytox green (1 μM). For the detection of intracellular ROS production, adherent neutrophils were loaded with H2DCFDA (20 μM) for 30 min. The cell medium was replaced with PMN buffer containing inhibitor or vehicle (DMSO) as indicated, and cells were further incubated for 30 min, followed by stimulation with fMLP (1 μM) or PMA (10 nM). Upon the addition of agonists, fluorescent signals were detected using a Zeiss Axiovert fluorescent microscope at various time points, and neutrophil morphology was monitored using a differential interference contrast microscope (2). To quantify the kinetics of NET formation, the number of Sytox-positive cells was counted from at least 100 cells per time point for each treatment using ImageJ software.
Immunofluorescence microscopy.
Purified human neutrophils (2×106/ml) were incubated at 37°C for 30 min in the presence of inhibitors or vehicle (DMSO) for 30 min. Subsequently, cells were stimulated with fMLP (1 μM) or PMA (10 nM), followed by fixation with 4% paraformaldehyde for 10 min. For NET detection, fixed cells were blocked with blocking buffer (10% FBS, 5 mg/ml BSA, and 0.1% Triton X-100 in PBS). For autophagosome detection, cells were permeabilized with methanol for 3 min after fixation, followed by incubation with blocking buffer. Cells were then stained with anti-NE (1:100), anti-H3Cit (1:50), or anti-LC3B (1:200) in blocking buffer overnight at 4°C. Secondary goat anti-rabbit IgG antibody conjugated with AlexaFluor 488 (1:500) and Hoechst 33342 (10 μg/ml) in blocking buffer were added and incubated for 2 h in the dark. Coverslips were mounted onto glass slides and visualized with a Zeiss Axiovert fluorescent microscope. For data presentation, the fluorescent intensities of each image were adjusted based on signals detected in neutrophil samples in the absence of primary antibodies. For the quantification of LC3B puncta, the fluorescent signal in all images was adjusted to a fixed threshold, and LC3B-positive particles in the fields of view were counted using a particle analysis function in ImageJ.
Analysis of data.
Data are shown as means ± SE. Statistical analysis was performed using paired Student's t-test. Probability values of P < 0.05 were selected to be statistically significant.
RESULTS
mTOR activity regulates the rate of NETosis.
Neutrophils can respond to various stimuli including cytokines and microbial components such as fMLP to elicit antibacterial functions (1, 4). In neutrophils, fMLP is known to bind the specific G protein-coupled receptors, namely formyl-peptide receptors (FPRs), and activate downstream signaling cascades including the mTOR pathway. The FPR-mTOR signaling axis has been shown to play an important role in neutrophil chemotaxis (13); however, it is unknown whether mTOR regulates NET release induced by fMLP. In this study, we first tested whether mTOR activity plays a role in driving NETosis by using the specific pharmacological inhibitor for mTOR rapamycin and WYE-354. To determine the kinetics of NET release, extracellular release of DNA from stimulated neutrophils was monitored using the cell-impermeable DNA dye Sytox green, along with the cell-permeable DNA dye Hoechst 33342 by direct live-cell imaging. In the presence of fMLP, the percentage of Sytox-positive cells increased in a time-dependent manner (Fig. 1A). Treatment of neutrophils with rapamycin or WYE-354 before the addition of fMLP resulted in a significant increase in the Sytox-positive population compared with treatment with vehicle or fMLP alone (Fig. 1, A and B). In parallel, the protein kinase C (PKC) activator PMA potently induced extracellular DNA release in a time-dependent manner (Fig. 1A). In the absence of agonists, only a minimal increase in the percentage of Sytox-positive cells was observed, even after 180 min (8.1 ± 3.8% after 180 min).
Fig. 1.
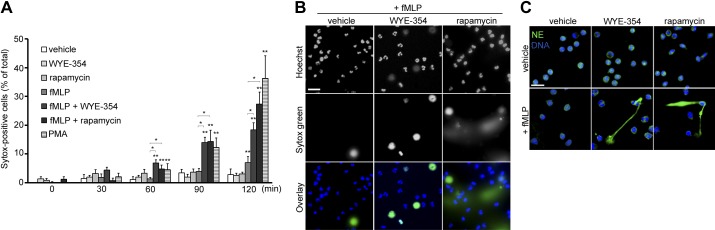
Mammalian target of rapamycin (mTOR) inhibition facilitates the rate of neutrophil extracellular trap (NET) release. A: human neutrophils were treated with vehicle (0.1% DMSO), WYE-354 (30 μM), or rapamycin (100 nM) for 30 min, followed by stimulation with vehicle (HBSS), formyl-Met-Leu-Phe (fMLP; 1 μM), or phorbol-myristate-acetate (PMA; 10 nM). Time-lapse images of intracellular DNA (Hoechst 33342) and extracellular DNA (Sytox green) were obtained at every 30 min using a fluorescence microscope. Results were obtained from 3 independent experiments and presented as the mean percentage of Sytox-positive cells ± SE. *P < 0.05, fMLP-stimulated neutrophils pretreated with vehicle vs. rapamycin; **P < 0.05, compared with unstimulated, vehicle-treated neutrophils. B: representative images of neutrophils stimulated with fMLP in the presence of vehicle, WYE-354, or rapamycin at 2-h time point. C: neutrophils treated with vehicle (0.1% DMSO), WYE-354 (30 μM), or rapamycin (100 nM) were incubated with vehicle (HBSS) or fMLP (1 μM), fixed, and stained for neutrophil elastase (NE; green) and DNA (Hoechst 33342; blue). Scale bar = 20 μm. Images are representative of at least 3 independent experiments.
To next determine whether extracellular DNA release is caused by NETosis as opposed to apoptotic cell death, we examined the localization of neutrophil elastase (NE), a well-known NET protein, in neutrophils under basal and mTOR-inhibited conditions. As shown in Fig. 1C, neutrophils treated with WYE-354 or rapamycin displayed extracellular localization of NE associated with DNA in response to fMLP, suggesting NET formation. Taken together, these results suggest that mTOR activity negatively regulates the early stage of NETosis and controls the kinetics of NET formation.
mTOR inhibition promotes autophagosome formation.
mTOR kinase negatively regulates the translocation of autophagy machinery proteins, such as the ULK complex, to autophagic structures to inhibit the initiation of autophagy (12, 19). Autophagy has been shown to play a critical role in NET formation, as inhibition of autophagy by phosphatidylinositol 3-kinase inhibitors resulted in neutrophil apoptosis (26). Massive cell vacuolization has been described during the early stage of NETosis (5, 26), likely due to active autophagic activity. The facilitation of NET formation by mTOR inhibition led us to question what role the mTOR pathway plays in regulating neutrophil autophagy in response to fMLP. Neutrophils were treated with the phosphatidylinositol 3-kinase inhibitor wortmannin in the presence of rapamycin and stimulated with fMLP to assess whether autophagic activity was required for the acceleration of NETosis. Here, NETs were detected as colocalized immunofluorescence of DNA and cirtullinated histone H3 (H3Cit), which is known to be generated by peptidylarginine deiminase 4 (PAD4) during NET formation. As shown in Fig. 2A, in response to fMLP, a portion of rapamycin-treated neutrophils displayed an increased formation of DNA structures associated with H3Cit, compared with vehicle-treated cells. In contrast, wortmannin treatment abrogated extracellular DNA release as well as histone cirtullination (Fig. 2A). The inhibitory effect of wortmannin on NETosis was verified in PMA-stimulated neutrophils, in which histone citrullination was absent for cells that were pretreated with wortmannin (Fig. 2A).
Fig. 2.
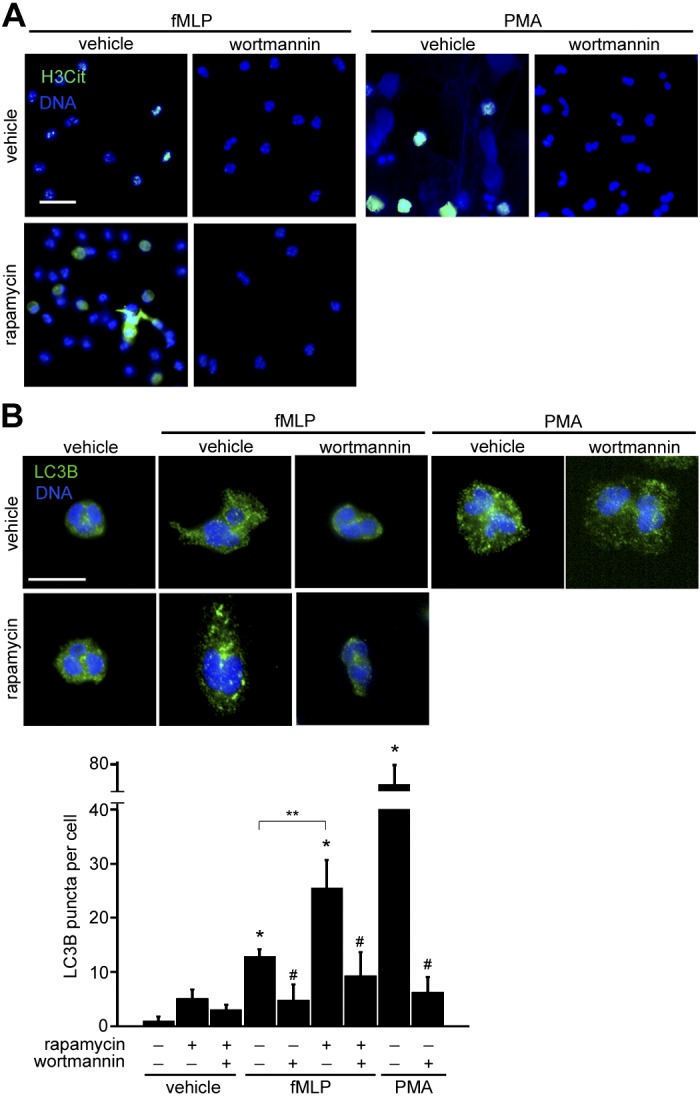
Increased autophagic activity by mTOR inhibition mediates NET release. A: human neutrophils were treated with vehicle (0.1% DMSO) or rapamycin (100 nM) in the presence or absence of the phosphatidylinositol 3-kinase/autophagy inhibitor wortmannin (100 nM) for 30 min, followed by stimulation with fMLP (1 μM) or PMA (10 nM) for 2 h. Cells were then stained for citrullinated histone H3 (histone H3 citrulline 2 + 8 + 17; green) and DNA (blue). B: cells were stimulated with fMLP (1 μM) or PMA (10 nM) for 30 min in the presence of vehicle or rapamycin (100 nM) and/or wortmannin (100 nM) and stained for microtubule-associated protein light chain 3B (LC3B; green) and DNA (blue). LC3B-positive puncta per cell were quantified using ImageJ. Results are shown as the mean LC3B puncta count ± SE. *P < 0.05, compared with unstimulated neutrophils; **P < 0.05, vehicle vs. rapamycin for fMLP-stimulated neutrophils; #P < 0.05, vehicle vs. wortmannin for fMLP- or PMA-stimulated neutrophils. Results were obtained from 3 independent experiments. Scale bar = 20 μm (A) and 10 μm (B).
Next, to examine the effect of rapamycin on autophagic activity, neutrophils were stimulated, fixed, and stained for microtubule-associated LC3B protein. LC3B can be used as a marker of autophagosomes, as it undergoes posttranslational modification and associates with autophagic structures upon the initiation of autophagy (12, 18). Our results show that while LC3B distributed uniformly in the cytosol of neutrophils before the addition of agonists, the immunofluorescence of LC3B was associated with punctate structures after stimulation with fMLP or PMA, suggesting autophagosome formation (Fig. 2B). In the presence of rapamycin, LC3B accumulation was further increased at 30 min postaddition of fMLP (Fig. 2B). The formation of LC3B-associated puncta was inhibited in the presence of wortmannin (Fig. 2B). The blockade of autophagolysosomal fusion by bafilomycin A1 led to an increase in the fluorescent intensity of LC3B in stimulated neutrophils (data not shown), indicating that LC3B accumulation was due to rapamycin-mediated upregulation of autophagic flux, not due to impaired autophagosome degradation. Overall, these data imply that rapamycin facilitates NET release via the control of autophagy influx in response to fMLP and that the mTOR pathway negatively regulates NETosis downstream of FPR signaling.
Cytoskeletal machinery is required for NET formation.
Microtubules, actin, and myosin play an essential role in neutrophil cytoskeletal reorganization. In addition, these cytoskeletal components contribute to the process of autophagosome formation and lysosomal fusion during autophagy in mammalian cells (20). To study the role of cytoskeletal reorganization in regulating the autophagic flux downstream of FPR-mTOR signaling pathway, neutrophils were incubated with rapamycin in combination with inhibitors of actin polymerization (cytochalasin D), microtubule polymerization (nocodazle), or myosin II activity (blebbistatin). While rapamycin increased the extent of LC3B-associated puncta in fMLP-stimulated neutrophils, treatment with cytochalasin D, nocodazole, or blebbistatin attenuated LC3B accumulation (Fig. 3, A and B). We next examined whether cytoskeletal machinery was coupled with mTOR-mediated regulation of NET release. The enhanced formation of DNA meshwork by rapamycin was observed after neutrophil incubation with fMLP for 2 h, compared with cells treated with fMLP alone (Fig. 3C). The presence of cytochalasin D, nocodazole, or blebbistatin abrogated extracellular DNA release in neutrophils activated by fMLP (Fig. 3C). Taken together, these results suggest that cytoskeletal machinery is required for rapamycin-enhanced NET formation, mediated via its regulation of autophagy.
Fig. 3.
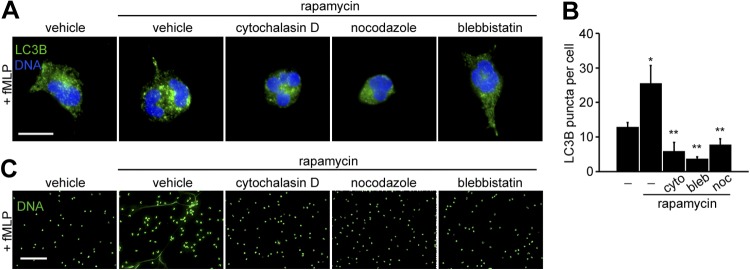
Cytoskeletal dynamics are required for autophagy and NETosis. Human neutrophils were treated with vehicle (0.1% DMSO) or rapamycin (100 nM) in the presence of cytochalasin D (cyto; 10 μM), nocodazole (noc; 10 μM) or blebbistatin (bleb; 100 μM) for 30 min. Cells were stimulated with fMLP (1 μM) for 30 min (A) or 2 h (C). Fixed cells were stained for LC3B (A; green) and DNA (Hoechst 33342; blue) or DNA (C; Sytox green; green). B: LC3B-positive puncta per cell were quantified using ImageJ. Results are shown as the mean LC3B puncta count ± SE. *P < 0.05, compared with vehicle; **P < 0.05, compared with rapamycin-treated neutrophils. Data shown are representative from 3 independent experiments. Scale bar = 10 μm (A) and 100 μm (B).
Establishment of the link between the mTOR axis and ROS production during NETosis.
fMLP, as well as PMA, are also known as potent inducers of respiratory burst, by which neutrophils rapidly produce a large amount of ROS via NADPH oxidase and release ROS to phagosomes or the extracellular environment (29). ROS production shapes a critical component of NETosis, as neutrophils isolated from chronic granulomatous disease patients, who have dysfunctional NADPH oxidase, are unable to form NETs in response to infectious signals (5, 17, 26). In addition, extracellularly added hydrogen peroxide (H2O2) is sufficient to trigger NETs (5, 22). We speculated that altered levels of ROS production are associated with rapid NETosis through mTOR inhibition. Neutrophils were loaded with the ROS sensor H2DCFDA, and intracellular ROS production was monitored using live-cell imaging. Intracellular ROS was increased in neutrophils activated by fMLP or PMA compared with baseline (Fig. 4A). Rapamycin treatment did not alter basal ROS levels, or fMLP-induced ROS production (Fig. 4A). We next assessed whether NADPH oxidase activity or extracellular ROS was required for mTOR-mediated promotion of NETosis. While neutrophil treated with rapamycin and fMLP elicited H3Cit-positive NET structure, the addition of diphenyleneiodonium chloride, an inhibitor of NADPH oxidase, blocked NET release and decreased histone cirtullination to basal levels (Fig. 4B). Catalase, which catalyzes the decomposition of H2O2, did not abrogate histone citrullination (Fig. 4B). Moreover, treatment of neutrophils with SOD, which mediates the dismutation of superoxide anion and produces H2O2, promoted H3Cit-associated NET release (Fig. 4B). Overall, these results demonstrate an indispensable role of ROS production via NADPH oxidase, likely regulated independently of the mTOR pathway, for NETosis.
Fig. 4.
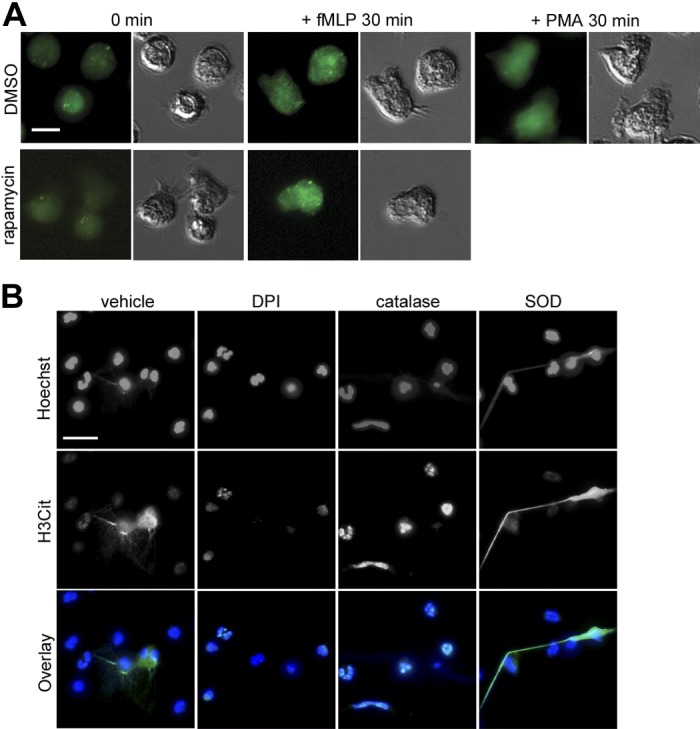
mTOR inhibition does not affect reactive oxygen species (ROS) production. A: human neutrophils were loaded with the ROS sensor H2DCFDA (20 μM), pretreated with vehicle (0.1% DMSO) or rapamycin (100 nM) for 30 min and stimulated with fMLP (1 μM) or PMA (10 nM). Representative H2DCFDA fluorescence images (green) and differential interference contrast images are shown at 0 and 30 min after stimulation, obtained from 3 independent experiments. B: neutrophils were incubated with rapamycin (100 nM) in the presence of vehicle or the NADPH inhibitor diphenyleneiodonium chloride (DPI; 10 μM), the H2O2 scavenger catalase (100 U/ml), or the O2− scavenger SOD (80 U/ml) before stimulation with fMLP (1 μM) for 2 h. Cells were fixed and stained for citrullinated histone H3 (histone H3 citrulline 2 + 8 + 17; green) and DNA (blue). Scale bar = 10 μm (A) and 20 μm (B).
DISCUSSION
In this study, we demonstrate that the mTOR pathway contributes to the regulation of NET formation via autophagy downstream of FPR signaling in human neutrophils. Our current model of mTOR-mediated NET regulation is shown in Fig. 5. As a distinct form of active cell death, NET formation requires both autophagic activity and ROS production by NADPH oxidase; in fact, the lack of either axis leads to neutrophil apoptosis, followed by secondary necrosis (26). The well-known NET inducer PMA can potently activate PKC, leading to robust ROS production via NADPH oxidase (29), while activating autophagy (18, 25). fMLP binding to FPRs can activate not only NADPH oxidase (29) but also the mTOR pathway (13), which may inhibit the autophagy pathway. The present study demonstrates that the blockade of mTOR activity by pharmacological inhibitors increased autophagic activity and drives neutrophils towards NETosis. Interdependently of autophagy, the activity of NADPH oxidase and cytoskeletal machinery plays key roles in regulating NET release.
Fig. 5.

Link between mTOR-mediated autophagy regulation and NET formation in neutrophils. The present study demonstrated a key role of mTOR in NET release. fMLP binds to the specific G protein-coupled receptor and potently activates NADPH oxidase, leading to reactive oxygen species production; in parallel, mTOR activation downstream of fMLP signaling prevents neutrophils from undergoing autophagy. The abrogation of mTOR activity by rapamycin drives autophagy and consequently induces histone citrullination and NET release in response to fMLP. Cytoskeletal components such as actin, myosin, and microtubules are required for autophagy and NET formation.
A critical role for the mTOR pathway has been shown in the early stage of autophagy by inhibiting the docking of autophagy-related proteins to autophagic structures (12, 16). In neutrophils, the mTOR pathway is activated in response to various inflammatory stimuli including fMLP, whereas mTOR activation induced by fMLP contributes to cytoskeletal reorganization during chemotaxis via the control of cAMP production and rear contraction (13). Our results show that mTOR inhibition by rapamycin accelerated NETosis, which paralleled with increased autophagy influx. We propose that this mTOR-dependent regulatory mechanism may explain the inconsistent effects of fMLP on NET release observed in previous studies (23, 25, 30). Contrary to our study, Mclnturff et al. (15) reported that rapamycin inhibited NET release through translational control of HIF-1α. It should be noted that the authors of that study induced NETs using LPS, which is a potent inducer of neutrophil autophagy via Toll-like receptor 4 (7, 18) but is unable to trigger NADPH activity (29). It is not clear whether mTOR activity plays a role in the process of NETosis other than via autophagy regulation. At least in the context of FPR signaling, we show that mTOR activity limits the rate of NETosis via its negative effect on autophagy, while rapamycin treatment does not alter fMLP-induced ROS production. In physiological settings, neutrophils are likely to be exposed to multiple inflammatory signals which would cooperatively activate various cellular functions. Therefore, under certain circumstances, the combination of signal inputs from specific surface receptors including FPR may preferentially drive neutrophils toward an active form of death, NETosis, as opposed to the prolonged survival that allows neutrophils to elicit effective surveillance and clearance.
Histone hypercitrullination by PAD4 has been shown to play a critical role in unfolding chromatin structures downstream of ROS production (22, 23, 27, 32). A recent study has shown that the treatment of a human osteosarcoma cell line with a specific PAD4 inhibitor attenuated cancer cell growth via the downregulation of the mTOR signaling axis, resulting in an increase in autophagy flux (33). In our study, inhibition of autophagy by wortmannin abrogated histone citrullination and NET release induced by cotreatment with rapamycin and fMLP, suggesting an autophagy-dependent mechanism for histone citrullination. It is still unclear whether mTOR can serve to directly regulate PAD4 activity during NETosis. It is possible that the autophagy axis contributes to histone citrullination and subsequent chromatin decondensation by facilitating granule disintegration and PAD4 mobilization in neutrophils during the release of NETs. Notably, actin and microtubule polymerization have been shown to be required for histone citrullination via PAD4 during NET formation (22). In this study, we show that inhibition of actin, myosin, and microtubule dynamics attenuated fMLP-driven NET formation in the presence of rapamycin, which was associated with impaired autophagosome formation. Accordingly, the failure of neutrophils to undergo NETosis in the presence of cytoskeletal inhibitors may be due to disrupted autophagic processes interdependently of histone citrullination. We propose that the accelerated NET release in the presence of mTOR inhibition may depend on cytoskeletal machinery at multiple stages of NETosis.
The pathological roles of NETs have been reported in the contexts of autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis, where many autoantibodies are developed against citrullinated self-antigens (e.g., histones) and/or neutrophil proteins including NE, myeloperoxidase, and proteinase-3 (11, 28). Impairment of NETs degradation has found in SLE patients, suggesting that NETs contribute to the development of autoantibodies by providing autoantigens and damage-associated molecular patterns (6). While autophagy is known to facilitate antigen presentation in macrophages and B-cells during SLE and rheumatoid arthritis (10), our study indicates that dysregulated autophagic activity in neutrophils may promote the exposure of citrullinated histones in these autoimmune diseases.
In summary, the present study reveals a complex set of regulatory mechanisms, including the autophagy pathway, underlying NET formation in activated human neutrophils. As the ability of neutrophils to combat against infection is not limited to NET release (i.e., degranulation, phagocytosis), future studies will be focused on elucidating the missing links explaining how neutrophils have evolved a strategy for pathogen clearance in response to inflammatory stimuli.
GRANTS
This work was supported by the National Institutes of Health (National Heart, Lung, and Blood Institute Grant R01-HL-101972).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.I. and O.J.T.M. conception and design of research; A.I. performed experiments; A.I. analyzed data; A.I. interpreted results of experiments; A.I. prepared figures; A.I. drafted manuscript; A.I. and O.J.T.M. edited and revised manuscript; A.I. and O.J.T.M. approved final version of manuscript.
ACKNOWLEDGMENTS
O. J. T. McCarty is an American Heart Association Established Investigator. A. Itakura is a Vertex Scholar.
REFERENCES
- 1. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30: 459–489, 2012 [DOI] [PubMed] [Google Scholar]
- 2. Aslan JE, Itakura A, Gertz JM, McCarty OJ. Platelet shape change and spreading. Methods Mol Biol 788: 91–100, 2012 [DOI] [PubMed] [Google Scholar]
- 3. Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood 118: 3129–3136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borregaard N. Neutrophils, from marrow to microbes. Immunity 33: 657–670, 2010 [DOI] [PubMed] [Google Scholar]
- 5. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176: 231–241, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 107: 9813–9818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 106: 6226–6231, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Itakura A, Aslan JE, Sinha S, White-Adams TC, Patel IA, Meza-Romero R, Vandenbark AA, Burrows GG, Offner H, McCarty OJ. Characterization of human platelet binding of recombinant T cell receptor ligand. J Neuroinflammation 7: 75, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Itakura A, Verbout NG, Phillips KG, Insall RH, Gailani D, Tucker EI, Gruber A, McCarty OJ. Activated factor XI inhibits chemotaxis of polymorphonuclear leukocytes. J Leukoc Biol 90: 923–927, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones SA, Mills KH, Harris J. Autophagy and inflammatory diseases. Immunol Cell Biol, 2013 [DOI] [PubMed] [Google Scholar]
- 11. Knight JS, Carmona-Rivera C, Kaplan MJ. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front Immunol 3: 380, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 469: 323–335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu L, Das S, Losert W, Parent CA. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell 19: 845–857, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCarty OJ, Tien N, Bochner BS, Konstantopoulos K. Exogenous eosinophil activation converts PSGL-1-dependent binding to CD18-dependent stable adhesion to platelets in shear flow. Am J Physiol Cell Physiol 284: C1223–C1234, 2003 [DOI] [PubMed] [Google Scholar]
- 15. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, Yost CC. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood 120: 3118–3125, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res 20: 748–762, 2010 [DOI] [PubMed] [Google Scholar]
- 17. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, Papayannopoulos V, Zychlinsky A. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117: 953–959, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitroulis I, Kourtzelis I, Kambas K, Rafail S, Chrysanthopoulou A, Speletas M, Ritis K. Regulation of the autophagic machinery in human neutrophils. Eur J Immunol 40: 1461–1472, 2010 [DOI] [PubMed] [Google Scholar]
- 19. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22: 132–139, 2010 [DOI] [PubMed] [Google Scholar]
- 20. Monastyrska I, Rieter E, Klionsky DJ, Reggiori F. Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc 84: 431–448, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol 179: 199–210, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun 1: 194–201, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 180: 1895–1902, 2008 [DOI] [PubMed] [Google Scholar]
- 24. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90: 1383–1435, 2010 [DOI] [PubMed] [Google Scholar]
- 25. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 18: 581–588, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21: 290–304, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol 3: 360, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, Piconese S, Parenza M, Guiducci C, Vitali C, Colombo MP. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 120: 3007–3018, 2012 [DOI] [PubMed] [Google Scholar]
- 29. Sheppard FR, Kelher MR, Moore EE, McLaughlin NJ, Banerjee A, Silliman CC. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J Leukoc Biol 78: 1025–1042, 2005 [DOI] [PubMed] [Google Scholar]
- 30. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5: e1000639, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209: 819–835, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184: 205–213, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Li P, Wang S, Hu J, Chen XA, Wu J, Fisher M, Oshaben K, Zhao N, Gu Y, Wang D, Chen G, Wang Y. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J Biol Chem 287: 25941–25953, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]