

Abstract
We used mouse cortical collecting duct principal cells (mpkCCDc14 cell line) as a model to determine whether statins reduce the harmful effects of cyclosporine A (CsA) on the distal nephron. The data showed that treatment of cells with CsA increased transepithelial resistance and that the effect of CsA was abolished by lovastatin. Scanning ion conductance microscopy showed that CsA significantly increased the height of cellular protrusions near tight junctions. In contrast, lovastatin eliminated the protrusions and even caused a modest depression between cells. Western blot analysis and confocal microscopy showed that lovastatin also abolished CsA-induced elevation of both zonula occludens-1 and cholesterol in tight junctions. In contrast, a high concentration of CsA induced apoptosis, which was also attenuated by lovastatin, elevated intracellular ROS via activation of NADPH oxidase, and increased the expression of p47phox. Sustained treatment of cells with lovastatin also induced significant apoptosis, which was attenuated by CsA, but did not elevate intracellular ROS. These results indicate that both CsA and lovastatin are harmful to principal cells of the distal tubule, but via ROS-dependent and ROS-independent apoptotic pathways, respectively, and that they counteract probably via mobilization of cellular cholesterol levels.
Keywords: transepithelial resistance, scanning ion conductance microscopy, confocal microscopy, zonula occludens-1, cholesterol, kidney injury, reactive oxygen species
statins, as inhibitors of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase used to reduce cholesterol synthesis, have been administrated to treat the dyslipidemia that commonly occurs in organ transplant recipients (38). This dyslipidemia may be due to the use of immunosuppressant drugs such as cyclosporine A (CsA) (34). However, there is an ongoing debate as to whether statins can be safely used for these patients to reduce cholesterol. Some clinical case analyses have indicated that statins can induce rhabdomyolysis, in which skeletal muscle breaks down and leads to renal injury in transplant recipients on CsA treatment (1, 20), which implies that a reduced dosage of statins should be used for patients receiving CsA treatment (3). In contrast, there are other data showing that statins are beneficial in outcomes of renal transplant recipients receiving CsA treatment (15, 22). When given in low or modest dosages, statins are quite safe and effective for treatment of hypercholesterolemia in organ transplant recipients (2, 8, 19, 42, 47). In a rat model of kidney transplantation, pravastatin significantly reduced renal interstitial inflammation and fibrosis (21). However, there are few in vitro data showing the molecular mechanism by which statins can reverse side effects of CsA at the cellular level. Our recent in vitro studies have shown that CsA elevates levels of cholesterol in distal nephron cells (45), probably through inhibition of ATP-binding cassette transporter A1 (ABCA1), which is responsible for cholesterol outward transport (46). We assume that locally elevated cholesterol in distal nephron cells may be responsible for some of the side effects of CsA. As HMG-CoA reductase inhibitors, statins might prevent side effects of CsA by reducing cholesterol levels in renal tubular cells.
One side effect of CsA on renal tubular cells is to increase transepithelial resistance, which reflects, at least in part, paracellular permeability (16, 27). Paracellular permeability is mainly controlled by tight junctions, specialized membrane and protein complexes formed between cells. Since tight junctions are raft-like (cholesterol-rich) membrane microdomains (30), CsA may modulate tight junctions and reduce paracellular permeability by elevating levels of cholesterol or tight junction proteins. The possible involvement of cholesterol in cell membranes or intracellular pools encouraged us to hypothesize that lovastatin may attenuate CsA-induced reduction of paracellular permeability. Another side effect of CsA is to cause kidney injury. It has long been known that CsA induces apoptosis of proximal tubule cells (9, 33). A recent study (40) has shown that CsA also induces apoptosis of inner medullary collecting duct cells by elevating osmolality. However, it remains unclear whether CsA can induce apoptosis of cortical collecting duct (CCD) cells. In a rat model, rosuvastatin attenuated CsA-induced inflammation, apoptosis, and fibrosis in the kidney (29). Since CsA-induced renal tubular injury is associated with increased levels of cholesterol ester (48), it is very likely that statins attenuate CsA-induced kidney injury by reducing cholesterol levels in the cell. However, recent studies (7, 10) have suggested that high-potency statins can increase the risk of acute kidney injury. This is not surprising because cholesterol not only maintains normal cell membrane integrity but also allows cell to gain cytoresistance (48). Therefore, we hypothesized that CsA and statins might counteract in the regulation of cell mortality by modulating cellular cholesterol levels.
Here, we report that lovastatin attenuates CsA-decreased paracellular permeability and CsA-induced apoptosis in cultured CCD cells and that, conversely, CsA attenuates lovastatin-induced apoptosis. Therefore, the results of the present study suggest that statins may benefit patients receiving CsA treatment after organ transplantation and that CsA may be used to treat statin-induced kidney injury.
MATERIALS AND METHODS
Cell culture.
A mouse CCD principal cell line (mpkCCDc14 cells) (5) was used in the present study. Cells were cultured on Transwell inserts with permeable polyester membranes, as we have previously reported (12). Briefly, mpkCCDc14 cells were incubated in a 1:1 mix of DMEM-F-12 medium (Invitrogen, Carlsbad, CA) supplemented with 50 nM dexamethasone, 1 nM triiodothyronine, 20 mM HEPES, 2 mM l-glutamine, 0.1% penicillin-streptomycin, and 2% heat-inactivated FBS. Cells were maintained at 37°C with 5% CO2 in air for 2 wk until they formed a confluent monolayer and became fully polarized.
Chemicals and solutions.
The majority of chemicals were obtained from Sigma Chemical. The NaCl bath solution for all experiments contained (in mM) 145 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES (pH was adjusted to 7.4 with NaOH). All concentrations throughout this article represent final concentrations.
Measurement of transepithelial resistance.
After culture on the polyester membrane of Transwell inserts for 2 wk, mpkCCDc14 cells were fully polarized and formed cell monolayers. The resistance across the cell monolayer was measured with electrodes connected to a volt-ohm meter, the EVOM (World Precision Instruments, Sarasota, FL), as we have previously reported (26). Resistance was corrected for the surface area of the polyester membrane (expressed in Ω·cm2).
Scanning ion conductance microscopy.
Scanning ion conductance microscopy (SICM) experiments were performed as previously described (11). SICM images of the apical membrane of mpkCCDc14 cells were obtained with an XE-Bio system (Park Systems, Suwon, Korea) in an adaptive approach-retract-scan mode, as we have previously described (23). Live mpkCCDc14 cells on permeable polyester membranes (Transwell) were mounted on the stage of an inverted Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan). The morphology of the cell surface at nanometer resolution was scanned with the SICM probe, a glass pipette, and by an XY flat scanner attached to the microscopy stage and a Z scanner attached to the SICM head. The glass pipette was fabricated from borosilicate capillaries (inner diameter: 0.6 mm and outer diameter: 1.0 mm) using a CO2-laser-based micropipette puller (P-2000, Sutter Instruments, Novato, CA). The inner and outer diameters of the pipette tip were ∼100 and 200 nm, respectively. Before the experiment, cells were thoroughly washed with NaCl bath solution and transferred into the SICM live cell chamber on an inverted microscopic stage. The glass pipette was filled with electrolyte, NaCl bath solution. The current between the pipette and the cell membrane was set at a constant value by a feedback circuit to keep the pipette tip at a proximal, fixed distance from the cell surface. Images of the apical membrane surface were then made by scanning the cell surface with the pipette. Three-dimensional images were reconstructed, and the heights of protrusions from the apical membrane of mpkCCDc14 cells were calculated with the software attached to the XE-Bio system.
Confocal microscopy.
Confocal microscopy experiments were performed as previously described (26). Briefly, to detect levels of zonula occludens (ZO)-1 in mpkCCDc14 cells, cells were fixed with 4% paraformaldehyde for 10 min, washed twice, permeabilized with 0.1% Triton X-100 in NaCl bath solution for 15 min, and then washed twice. Cells were incubated with rabbit anti-ZO-1 antibody (Invitrogen) for 1 h, washed twice, and incubated with a secondary antibody (Alexa fluor 594, goat anti-rabbit IgG, 5 μg/ml) at room temperature for 1 h. To detect levels of cholesterol, live cells were stained with a membrane-permeable, cholesterol-binding, fluorescent probe, filipin. Before the confocal miscroscopy experiments, cells were washed twice with NaCl bath solution. Immediately after each experimental manipulation, the polyester membrane that supported the mpkCCDc14 cell monolayer was quickly excised and mounted on a glass slide with either a drop of mounting solution for the ZO-1 labeling or a drop of NaCl bath solution to keep the cells alive for the cholesterol labeling. XY or XZ scanning of mpkCCDc14 cells was accomplished with an Olympus FV-1000 confocal microscopy within 3 days for the ZO-1 labeling using fixed cells or within 10 min for the cholesterol labeling using live cells. XY optical sections were performed to provide a flat view of the cells near the apical membrane. XZ optical sections were also performed to provide a lateral view of the cells. In each set of experiments, images were taken using the same parameter settings.
To evaluate apoptosis, mpkCCDc14 cells were stained with both FITC-conjugated annexin V (AV) and propidium iodide (PI), as we have previously described (24). The cell membrane of apoptotic cells was stained with AV because phosphatidylserine, a lipid that has a high binding affinity to AV, is externalized in apoptotic cells. The nuclei of apoptotic cells were stained with PI because the nuclear membrane of apoptotic cells becomes permeable to PI. AV was excited with a 488-nm laser and visualized through a 515-nm emission filter (shown in green). PI was excited with a 488-nm laser and visualized through a 590-nm emission filter (shown in red). To detect levels of intracellular ROS, mpkCCDc14 cells were incubated with 25 μM 2′,7′-dichlorodihydrofluorescein diacetate a membrane-permeable, ROS-sensitive, fluorescent probe for 15 min. Confocal microscopy XY scanning of mpkCCDc14 cells was accomplished within 5–15 min. In each set of experiments, images were taken using the same parameter settings.
Western blot experiments.
To detect levels of ZO-1 and p47phox in mpkCCDc14 cells, cell lysates (100 μg) were loaded and electrophoresed on 10% SDS-PAGE gels for 60–90 min. Gels were blotted onto polyvinylidene fluoride membranes for 1 h at 90 V. After a 1-h block with 5% BSA-PBS-Tween buffer, polyvinylidene fluoride membranes were incubated with primary antibodies (1:1,000 dilution) of either rabbit anti-ZO-1 antibody (lot no. 8831P1, Sigma) or goat anti-NCF1 (p47phox) antibody (lot no. GR84191-5, Abcam), respectively, overnight at 4°C and then respectively incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (1:5,000 dilution, GE Healthcare) for 1 h after four vigorous washes. Finally, blots were visualized with chemiluminescence using an ECL Plus Western Blotting Detection System (GE Healthcare).
Statistical analysis.
ANOVA for multiple comparisons was used for comparison among multiple groups of data. The Holm-Sidak method was used for the posttest of multiple comparisons. Data are shown as means ± SD. P values of <0.05 were considered statistically significant.
RESULTS
Lovastatin abolishes both CsA elevation of transepithelial resistance and CsA modification of cell topographic structure.
Since one side effect of CsA is to reduce the paracellular permeability of renal tubular cells (16, 27), we first examined the effect of CsA on transepithelial resistance across monolayers of a renal principal cell line, mpkCCDc14 cells. As shown in Fig. 1, we found that treatment of cells with 1 μM CsA for 24 h significantly increased transepithelial resistance from 3,978 ± 267 to 5,557 ± 543 Ω·cm2 (n = 12, P < 0.001). Our new finding was that treatment of cells with 25 μM lovastatin, a well-known inhibitor of HMG-CoA reductase that can reduce cholesterol synthesis, decreased transepithelial resistance from the control level (3,978 ± 267 Ω·cm2) to 2,089 ± 243 Ω·cm2 (n = 12, P < 0.001). Since our previous study (45) showed that CsA elevates intracellular cholesterol, we hypothesized that lovastatin might antagonize the effects of CsA. Indeed, resistance remained at control levels (3,776 ± 250 Ω·cm2, n = 12, P > 0.05), when cells were cotreated for 24 h with both 1 μM CsA and 25 μM lovastatin. These data suggest that lovastatin antagonizes the effects of CsA on transepithelial resistance.
Fig. 1.

Effects of cyclosporine A (CsA) and lovastatin on transepithelial resistance. Measurements of transepithelial resistance across mpkCCDc14 cell monolayers showed that CsA increased, but lovastatin decreased, transepithelial resistance. The effect of CsA was abolished by lovastatin. In each group, n = 12. All experiments shown in Figs. 1–4 were performed when cells were either under control conditions or treated for 24 h with 1 μM CsA, 25 μM lovastatin, or 1 μM CsA plus 25 μM lovastatin, respectively.
Since paracellular permeability is generally tightly controlled by tight junctions, we thought that CsA might induce cell membrane topographic changes near tight junctions. SICM allowed us to detect, at nanometer resolution, the difference in height of the cell surface. Therefore, we used SICM to determine whether CsA and lovastatin could modulate apical topographic structures of mpkCCDc14 cells. We found that the apical membrane of mpkCCDc14 cells contained significantly elevated protrusions near tight junctions (Fig. 2, A and B). Treatment of cells with 1 μM CsA for 24 h dramatically increased the height of the protrusions from 251 ± 48 nm (control) to 656 ± 178 nm (n = 10, P < 0.001). Conversely, treatment of cells with 25 μM lovastatin for 24 h reduced tight junction protrusions and caused a depression between cells, as shown by the negative value (−311 ± 121 nm); the changes were statistically significant compared with the height of the protrusions under control conditions (251 ± 48 nm, n = 10, P < 0.001). Interestingly, the height of the protrusions remained unchanged (255 ± 65 nm, n = 10) when cells were cotreated with both 1 μM CsA and 25 μM lovastatin for 24 h. These data suggest that lovastatin reverses CsA-induced topographic changes near tight junctions.
Fig. 2.

Effects of CsA and lovastatin on cell topographic structure. A: representative three-dimensional topographic images of the apical membrane obtained with scanning ion conductance microscopy (SICM). B: SICM imaging analysis of the apical membrane of live mpkCCDc14 cells showing that CsA increased, but lovastatin decreased, the heights of protrusions near tight junctions. The effect of CsA was abolished by lovastatin. In each group, n = 10. Note: CsA also increased the height of cellular microvilli, which serves as an interesting topic for our future studies.
CsA increases, but lovastatin decreases, ZO-1 expression via a cholesterol-dependent mechanism.
The topographic changes near tight junctions implied some remodeling of tight junctions. A previous study (4) has shown that overexpression of ZO-1 increases paracellular permeability. However, upregulation of endogenous ZO-1 expression decreased paracellular permeability, as shown by increased transepithelial resistance (18). Therefore, we determined how CsA would affect levels of ZO-1 in mpkCCDc14 cells. Confocal microscopy data showed that treatment of cells with 1 μM CsA for 24 h significantly increased the amount of ZO-1 in tight junctions (Fig. 3, A and B). In contrast, treatment of cells with 25 μM lovastatin for 24 h to reduce cholesterol synthesis significantly decreased the amount of ZO-1. However, ZO-1 remained approximately unchanged when cells were treated with both 1 μM CsA and 25 μM lovastatin together for 24 h. To confirm the results from confocal microscopy experiments, Western blot experiments were performed using control (untreated) cells and cells treated for 24 h with 1 μM CsA, 25 μM lovastatin, 1 μM CsA plus 25 μM lovastatin, or 30 μg/ml cholesterol. CsA increased, and lovastatin decreased, ZO-1 expression in mpkCCDc14 cells (Fig. 3C). The effect of CsA on ZO-1 expression was abolished by lovastatin and mimicked by cholesterol, indicating that the contrary effects of CsA and lovastatin are probably through a cholesterol-dependent mechanism. We also found that treatment of cells with 30 μg/ml cholesterol significantly increased transepithelial resistance from 3,920 ± 123 to 5,538 ± 328 Ω·cm2 (n = 11, P < 0.001; Fig. 3D), indicating that cholesterol mediates the CsA-induced elevation of transepithelial resistance.
Fig. 3.

Confocal microscopy images of mpkCCDc14 cells showing that CsA increased, but lovastatin decreased, the levels of zonula occludens (ZO)-1. As described in materials and methods, ZO-1 was labeled with a fluorescent antibody. The effect of CsA was abolished by lovastatin. A: representative images from either XY optical sections performed near the apical membrane (top) or XZ optical sections (bottom) when cells were under control conditions or were treated with CsA, lovastatin, or lovastatin plus CsA. All images were taken using the same parameters, including the detector gain. B: summary plots of fluorescence intensity reflecting the levels of ZO-1 under each condition as shown in A. In each group, n = 6. C: Western blots of mpkCCDc14 cells showing that CsA stimulates, and lovastatin inhibits, ZO-1 expression via a cholesterol-dependent mechanism. In these Western blot experiments, cells were either under control conditions or were treated for 24 h with CsA, lovastatin, CsA plus lovastatin, or cholesterol (30 μg/ml). The data represent four separate experiments showing consistent results. D: treatment of cells with cholesterol (30 μg/ml) for 24 h increased transepithelial resistance.
CsA increases, but lovastatin decreases, levels of cholesterol in or near tight junctions.
Since tight junctions are considered to be raft-like (cholesterol-rich) membrane microdomains (30), we also used confocal microscopy to determine the levels of cholesterol in tight junctions by staining cells with a fluorescent, cholesterol-binding compound, filipin. In parallel with the effects on the levels of ZO-1, confocal microscopy showed that treatment of cells with 1 μM CsA for 24 h significantly increased the levels of cholesterol in both tight junctions and the apical membrane of mpkCCDc14 cells (Fig. 4, A and B). In contrast, but as might be expected, treatment of cells with 25 μM lovastatin for 24 h significantly decreased the levels of cholesterol. Cholesterol changed little when cells were treated with both 1 μM CsA and 25 μM lovastatin together for 24 h. These data, together with the data shown in Fig. 3C, suggest that lovastatin antagonizes the effect of CsA on the levels of ZO-1, probably through modification of the levels of cholesterol in the tight junction complex.
Fig. 4.
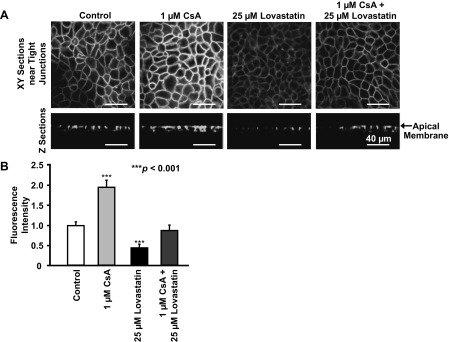
Confocal microscopy images of live mpkCCDc14 cells showing that CsA increases, but lovastatin decreases, the levels of cholesterol in tight junctions. As described in materials and methods, cholesterol was labeled with a fluorescent cholesterol-binding compound, filipin. The effect of CsA is abolished by lovastatin. A: representative images either from XY optical sections performed near the apical membrane (top) or from XZ optical sections (bottom) when cells were under control conditions or were treated with CsA, lovastatin, or lovastatin plus CsA. All images were taken using the same parameters, including the detector gain. To avoid saturating the fluorescence in the area of tight junctions, the detector gain in all experiments was set at a low level; at this level, the cholesterol in other parts of the cell membrane was not visible. B: summary plots of fluorescence intensity reflecting the levels of cholesterol under each condition as shown in A. In each group, n = 7.
A high concentration of CsA induces apoptosis, which is attenuated by lovastatin.
The above experiments showed that CsA at 1 μM caused defective paracellular transport within 24 h. The concentration of CsA can reach 2,629 ng/ml (2.2 μM) in the urine of healthy individuals after a single dose (17). We hypothesized that CsA at a concentration higher than 1 μM and for treatment longer than 24 h may induce apoptosis. Therefore, mpkCCDc14 cells were stained with AV-FITC and PI. Confocal microscopy experiments were performed to detect AV- and PI-positive (apoptotic) mpkCCDc14 cells. Cells were either under control conditions or treated for 72 h with 1 μM CsA, 5 μM CsA, 5 μM CsA plus 5 μM lovastatin, or 5 μM lovastatin. As shown in Fig. 5, A and B, treatment of cells with either 1 μM CsA or 5 μM lovastatin even for 72 h did not affect cell mortality. In contrast, treatment of cells with 5 μM CsA for 72 h caused significant apoptosis of mpkCCDc14 cells, which was significantly attenuated by cotreatment of cells with 5 μM lovastatin. These data suggest that lovastatin can antagonize CsA-induced apoptosis of cultured mpkCCDc14 cells.
Fig. 5.

Lovastatin attenuates CsA-induced apoptosis in mpkCCDc14 cells. A: representative confocal microscopy data showing mpkCCDc14 cells either under control conditions or treated for 72 h with 1 μM CsA, 5 μM CsA, 5 μM CsA plus 5 μM lovastatin, or 5 μM lovastatin. B: summary plots of the average number of apoptotic cells per microscopic field in each experimental condition (calculated from 15 microscopy fields of 3 separate experiments). Apoptotic cells were stained with FITC-conjugated annexin V (AV; shown in green) due to phosphatidylserine externalization and propidium iodide (PI; shown in red) due to a permeable nuclear membrane.
Sustained treatment of mpkCCDc14 cells with lovastatin induces apoptosis, which is attenuated by CsA.
A recent study (10) has indicated that the use of high-potency statins is associated with an increased rate of acute kidney injury. Since the concentration of lovastatin we used in the above experiments to acutely reduce cholesterol synthesis in cells was already relatively high, we hypothesized that sustained treatment of mpkCCDc14 cells with the same concentration of lovastatin might induce apoptosis. Confocal microscopy experiments were performed in mpkCCDc14 cells either under control conditions or treated with 25 μM lovastatin for 24 h, 48 h, or 72 h. As shown in Fig. 6, A and B, treatment of cells for 24 and 48 h did not significantly affect cell mortality. In contrast, treatment of cells for 72 h caused significant apoptosis. Interestingly, the apoptosis induced by sustained treatment with lovastatin was significantly attenuated by cotreatment of cells with 1 μM CsA. These data suggest that CsA can antagonize lovastatin-induced apoptosis of cultured mpkCCDc14 cells.
Fig. 6.

CsA attenuates lovastatin-induced apoptosis in mpkCCDc14 cells. A: representative confocal microscopy data showing mpkCCDc14 cells either under control conditions or treated with 25 μM lovastatin for 24, 48, or 72 h (in the absence or presence of 1 μM CsA). B: summary plots of the average number of apoptotic cells per microscopic field in each experimental condition (calculated from 15 microscopy fields of 3 separate experiments).
A high concentration of CsA, but not lovastatin, elevates ROS levels in mpkCCDc14 cells via activation of NADPH oxidase.
Recent studies (9, 31) have suggested that CsA elevates ROS in mesangial cells and induces apoptosis in renal epithelial LLC-PK1 cells. To examine whether a high concentration of CsA elevates ROS in distal tubular cells, mpkCCDc14 cells were stained with a ROS probe. The data showed that treatment of the cells for 24 h with 5 μM CsA, but not 1 μM CsA, significantly elevated intracellular ROS in mpkCCDc14 cells under control conditions but not in cells cotreated with 50 μM apocynin, a NADPH oxidase inhibitor (Fig. 7, A and C). In parallel, Western blot data showed that treatment of cells for 24 h with 5 μM CsA, but not 1 μM CsA, significantly elevated the expression of p47phox, a regulatory subunit of NADPH oxidase (Fig. 8). Although treatment of cells with 25 μM lovastatin for 72 h caused apoptosis, as shown in Fig. 6, the treatment did not elevate intracellular ROS (Fig. 7, B and D). These data suggest that a high concentration of CsA elevates ROS by activation of NADPH oxidase, probably via stimulation of p47phox expression.
Fig. 7.
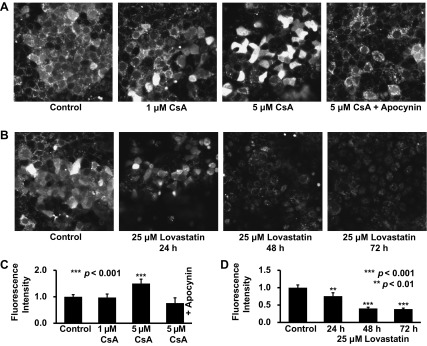
A high concentration of CsA elevates intracellular ROS via activation of NADPH oxidase in mpkCCDc14 cells. A: representative confocal microscopy data showing mpkCCDc14 cells either under control conditions or treated for 24 h with 1 μM CsA, 5 μM CsA, or 5 μM CsA plus 50 μM apocynin. B: representative confocal microscopy data showing mpkCCDc14 cells either under control conditions or treated with 25 μM lovastatin for 24, 48, or 72 h. C and D: summary plots of fluorescence intensity in each experimental condition (calculated from 10 microscopy fields of 4 separate experiments). Intracellular ROS were detected with a membrane-permeable, ROS-sensitive, fluorescent probe, 2′,7′-dichlorodihydrofluorescein diacetate.
Fig. 8.

A high concentration of CsA elevates p47phox expression in mpkCCDc14 cells. A: representative Western blot data showing p47phox expression in mpkCCDc14 cells either under control conditions or treated for 24 h with 1 or 5 μM CsA. B: summary plots of relative expression levels of p47phox in each experimental condition from three separate experiments.
DISCUSSION
Immunosuppresive drugs such as CsA and tacrolimus are extensively used for reducing allograft rejections, and they suppresses immune responses via inhibition of calcineurin, a protein phosphatase (32). However, both CsA and tacrolimus have side effects such as inducing nephrotoxicity (28) and causing hypertension (14). Although CsA- and tacrolimus-induced nephrotoxicity is severe, so far, there are no other drugs that can completely replace their important role in treating the rejection of organ transplants. Therefore, reducing their side effects becomes very critical for improving the management of allograft rejections. A recent study (13) has shown that tacrolimus causes hypertension by stimulating the renal NaCl cotransporter in the distal convoluted tubule via a calcinerin-dependent pathway. Since CsA induces greater hypertension than tacrolimus (36), besides via inhibition of calcineurin, CsA may cause hypertension also via another mechanism. Indeed, CsA, but not tacrolimus, also inhibits the ABCA1 transporter (25), which is responsible for Cho transport out of the cells (46). Our recent study (45) suggested that CsA causes hypertension by also stimulating the epithelial Na+ channel in distal nephron cells via ABCA1-dependent elevation of cholesterol. This study indicates that modulation of cholesterol levels in cell membranes or membrane pools may serve as a signaling pathway to mediate side effects of CsA. Indeed, the results of the present study showed that CsA-induced elevation of both ZO-1 and cholesterol levels in or near tight junctions appears to account for CsA-reduced paracellular permeability.
Using SICM nanotechnology, for the first time, we showed that CsA and lovastatin have contrary effects on topographic structures of the specialized membranes between CCD cells where tight junctions are located. Under basal conditions, the membranes between two cells seem to integrate into one membrane, which forms protrusions on the top of tight junctions; CsA enhances the protrusions, whereas lovastatin eliminates the protrusions. These topographic changes induced by CsA and lovastatin dramatically parallel the changes in the levels of ZO-1 and cholesterol. The concentration of CsA we used (1 μM) to induce these changes is less than the maximum CsA concentration (2,629 ng/ml = 2.2 μM) found in the urine of healthy individuals after a single dose (17). A recent study (6) has shown that pharmacological concentrations of CsA (10–40 μg/ml = 8–33 μM) decrease tight junction proteins, including ZO-1, in proximal tubular cells. Since CsA also causes apoptosis at these concentrations (6), the decreases in tight junction protein levels caused by pharmacological concentrations of CsA may be due to nonspecific protein degradation commonly observed under apoptotic conditions. Our finding that CsA increases, but lovastatin decreases, the levels of cholesterol in tight junctions is significant because these results suggest that cholesterol can be locally synthesized in renal epithelial cells and that its levels in tight junctions can also be locally controlled. Our recent study (45) has shown that CsA promotes exogenous cholesterol accumulation in distal nephron cells, indicating that CsA elevates levels of cholesterol by reducing cholesterol loss from the cell. Since treatment of cells either with lovastatin to inhibit cholesterol synthesis or with exogenous cholesterol can antagonize or mimic, respectively, the effect of CsA on ZO-1 expression, we argue that CsA may enhance ZO-1 expression by elevating cholesterol in the cells. We propose that statins may improve the CsA-induced defects in paracellular transport.
In addition to inducing defective paracellular transport, a high concentration of CsA also causes apoptosis in renal epithelial LLC-PK1 cells (9). The present study showed that CsA at 5 μM can cause apoptosis of cultured mpkCCDc14 cells. Since the CsA concentration in the urine of healthy individuals even after a single dose can reach 2.2 μM (17), we argue that the urinary CsA levels in patients on CsA therapy with reduced kidney function may be even higher than the concentrations we used in the present study. Consistent with the evidence obtained in mesangial cells (31), we showed that CsA also causes oxidative stress in mpkCCDc14 cells. CsA elevates intracellular ROS by activating NADPH oxidase and promoting the expression of the regulatory subunit of NADPH oxidase, p47phox. As an inhibitor of the cholesterol transporter ABCA1, CsA can cause the accumulation of cholesterol in distal nephron cells (45). It has been reported that the assembly and activity of NADPH oxidase are dependent on cholesterol-rich membrane microdomains, lipid rafts (37, 44). Therefore, we argue that the CsA-induced accumulation of cholesterol in mpkCCDc14 cells may be responsible for the CsA-induced elevation of intracellular ROS by activation of NADPH oxidase. Consistent with a previous report (43), we showed that CsA at 1 μM did not affect p47phox expression. However, our new finding is that CsA at 5 μM significantly stimulated p47phox expression, elevated intracellular ROS, and induced apoptosis of mpkCCDc14 cells. Since urinary CsA levels almost can reach the concentration we used in the in vitro experiments, CsA may be harmful to the distal tubule in patients on high doses of CsA. More importantly, CsA-induced apoptosis is attenuated by lovastatin. These data provide important information for further determining whether lovastatin can be used to improve CsA-induced nephrotoxicity.
It is well documented that statins, when given in low or modest dosages, are quite safe and effective for the treatment of hypercholesterolemia in organ transplant recipients (2, 8, 19, 42, 47). Even at a high dose (80 mg), if it is a single dose, atorvastatin can improve acute kidney injury (35). The present in vitro study supports previous findings by showing that treatment of mpkCCDc14 cells with lovastatin even at 25 μM did not induce significant apoptosis within 48 h. However, recent studies (7, 10) have indicated that high-potency statins can increase the risk of acute kidney injury. Indeed, we showed that sustained treatment of mpkCCDc14 cells with lovastatin caused significant apoptosis. Since such a treatment did not elevate intracellular ROS, the mechanism by which lovastatin induces apoptosis remains to be further determined. Our exciting finding is that lovastatin-induced apoptosis can be attenuated by 1 μM CsA. At this concentration, CsA does not induce apoptosis but only reduces paracellular permeability, which is abolished by lovastatin. These results provide useful information for further determining whether CsA can be used to improve acute kidney injury induced by high-potency statins. Although the plasma concentrations of lovastatin in human subjects receiving lovastatin are usually <20 ng/ml (50 nM) (39), a recent study (41) has suggested that simvastatin can be safely used for chemotherapy at a relatively high dose of 15 mg·kg−1·day−1. Assuming the average blood volume is 5,000 ml, this high dose would result in a calculated plasma concentration of simvastatin is ∼7 μM. Therefore, the pharmacological concentrations of lovastatin we used in the present in vitro study provide useful information to control statin nephrotoxicity and to guide the use of statins in cancer chemotherapy.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant 5R01-DK-067110 (to H.-P. Ma) and by National Natural Science Foundation of China Project 81130028 (to B.-Z. Shen).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.-C.L., X.S., X.-Y.L., and S.-P.W. performed experiments; B.-C.L. and C.F. analyzed data; B.-C.L. prepared figures; A.A.A., D.C.E., B.-Z.S., X.-Q.L., and H.-P.M. approved final version of manuscript; H.-P.M. conception and design of research; H.-P.M. interpreted results of experiments; H.-P.M. drafted manuscript; H.-P.M. edited and revised manuscript.
REFERENCES
- 1. Alejandro DS, Petersen J. Myoglobinuric acute renal failure in a cardiac transplant patient taking lovastatin and cyclosporine. J Am Soc Nephrol 5: 153–160, 1994 [DOI] [PubMed] [Google Scholar]
- 2. Arnadottir M, Eriksson LO, Germershausen JI, Thysell H. Low-dose simvastatin is a well-tolerated and efficacious cholesterol-lowering agent in ciclosporin-treated kidney transplant recipients: double-blind, randomized, placebo-controlled study in 40 patients. Nephron 68: 57–62, 1994 [DOI] [PubMed] [Google Scholar]
- 3. Arnadottir M, Eriksson LO, Thysell H, Karkas JD. Plasma concentration profiles of simvastatin 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitory activity in kidney transplant recipients with and without ciclosporin. Nephron 65: 410–413, 1993 [DOI] [PubMed] [Google Scholar]
- 4. Balda MS, Matter K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J 19: 2024–2033, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol 10: 923–934, 1999 [DOI] [PubMed] [Google Scholar]
- 6. Berzal S, Alique M, Ruiz-Ortega M, Egido J, Ortiz A, Ramos AM. GSK3, snail, and adhesion molecule regulation by cyclosporine A in renal tubular cells. Toxicol Sci 127: 425–437, 2012 [DOI] [PubMed] [Google Scholar]
- 7. Carney EF. Acute kidney injury: high-potency statin therapy and risk of acute kidney injury. Nat Rev Nephrol 9: 309, 2013 [DOI] [PubMed] [Google Scholar]
- 8. Cheung AK, DeVault GA, Jr, Gregory MC. A prospective study on treatment of hypercholesterolemia with lovastatin in renal transplant patients receiving cyclosporine. J Am Soc Nephrol 3: 1884–1891, 1993 [DOI] [PubMed] [Google Scholar]
- 9. de Arriba G, Calvino M, Benito S, Parra T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol Lett 218: 30–38, 2013 [DOI] [PubMed] [Google Scholar]
- 10. Dormuth CR, Hemmelgarn BR, Paterson JM, James MT, Teare GF, Raymond CB, Lafrance JP, Levy A, Garg AX, Ernst P. Use of high potency statins and rates of admission for acute kidney injury: multicenter, retrospective observational analysis of administrative databases. Br Med J 346: f880, 2013 [DOI] [PubMed] [Google Scholar]
- 11. Gorelik J, Zhang Y, Shevchuk AI, Frolenkov GI, Sanchez D, Lab MJ, Vodyanoy I, Edwards CR, Klenerman D, Korchev YE. The use of scanning ion conductance microscopy to image A6 cells. Mol Cell Endocrinol 217: 101–108, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Helms MN, Liu L, Liang YY, Al-Khalili O, Vandewalle A, Saxena S, Eaton DC, Ma HP. Phosphatidylinositol 3,4,5-trisphosphate mediates aldosterone stimulation of epithelial sodium channel (ENaC) and interacts with γ-ENaC. J Biol Chem 280: 40885–40891, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Hoorn EJ, Walsh SB, McCormick JA, Furstenberg A, Yang CL, Roeschel T, Paliege A, Howie AJ, Conley J, Bachmann S, Unwin RJ, Ellison DH. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 17: 1304–1309, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoorn EJ, Walsh SB, McCormick JA, Zietse R, Unwin RJ, Ellison DH. Pathogenesis of calcineurin inhibitor-induced hypertension. J Nephrol 25: 269–275, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imamura R, Ichimaru N, Moriyama T, Shi Y, Namba Y, Nonomura N, Matsumiya K, Toki K, Takahara S, Okuyama A. Long term efficacy of simvastatin in renal transplant recipients treated with cyclosporine or tacrolimus. Clin Transplant 19: 616–621, 2005 [DOI] [PubMed] [Google Scholar]
- 16. Kiely B, Feldman G, Ryan MP. Modulation of renal epithelial barrier function by mitogen-activated protein kinases (MAPKs): mechanism of cyclosporine A-induced increase in transepithelial resistance. Kidney Int 63: 908–916, 2003 [DOI] [PubMed] [Google Scholar]
- 17. Klawitter J, Haschke M, Kahle C, Dingmann C, Klawitter J, Leibfritz D, Christians U. Toxicodynamic effects of ciclosporin are reflected by metabolite profiles in the urine of healthy individuals after a single dose. Br J Clin Pharmacol 70: 241–251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ko JA, Murata S, Nishida T. Up-regulation of the tight-junction protein ZO-1 by substance P and IGF-1 in A431 cells. Cell Biochem Funct 27: 388–394, 2009 [DOI] [PubMed] [Google Scholar]
- 19. Kuo PC, Kirshenbaum JM, Gordon J, Laffel G, Young P, DiSesa VJ, Mudge GH, Jr, Vaughan DE. Lovastatin therapy for hypercholesterolemia in cardiac transplant recipients. Am J Cardiol 64: 631–635, 1989 [DOI] [PubMed] [Google Scholar]
- 20. Lasocki A, Vote B, Fassett R, Zamir E. Simvastatin-induced rhabdomyolysis following cyclosporine treatment for uveitis. Ocul Immunol Inflamm 15: 345–346, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Li C, Yang CW, Park JH, Lim SW, Sun BK, Jung JY, Kim SB, Kim YS, Kim J, Bang BK. Pravastatin treatment attenuates interstitial inflammation and fibrosis in a rat model of chronic cyclosporine-induced nephropathy. Am J Physiol Renal Physiol 286: F46–F57, 2004 [DOI] [PubMed] [Google Scholar]
- 22. Lisik W, Schoenberg L, Lasky RE, Kahan BD. Statins benefit outcomes of renal transplant recipients on a sirolimus-cyclosporine regimen. Transplant Proc 39: 3086–3092, 2007 [DOI] [PubMed] [Google Scholar]
- 23. Liu BC, Lu XY, Song X, Lei KY, Alli AA, Bao HF, Eaton DC, Ma HP. Scanning ion conductance microscopy: a nanotechnology for biological studies in live cells. Front Physiol 3: 483, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu BC, Song X, Lu XY, Li DT, Eaton DC, Shen BZ, Li XQ, Ma HP. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim Biophys Acta 1833: 1434–1442, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Llaudo I, Cassis L, Torras J, Bestard O, Franquesa M, Cruzado JM, Cerezo G, Castano E, Petriz J, Herrero-Fresneda I, Grinyo JM, Lloberas N. Impact of small molecules immunosuppressants on P-glycoprotein activity and T-cell function. J Pharm Pharm Sci 15: 407–419, 2012 [DOI] [PubMed] [Google Scholar]
- 26. Ma HP. Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. J Biol Chem 286: 32444–32453, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin-Martin N, Slattery C, McMorrow T, Ryan MP. TGF-β1 mediates sirolimus and cyclosporine A-induced alteration of barrier function in renal epithelial cells via a noncanonical ERK1/2 signaling pathway. Am J Physiol Renal Physiol 301: F1281–F1292, 2011 [DOI] [PubMed] [Google Scholar]
- 28. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009 [DOI] [PubMed] [Google Scholar]
- 29. Nam HK, Lee SJ, Kim MH, Rho JH, Son YK, Lee SM, Kim SE, Kim KH, An WS. Rosuvastatin attenuates inflammation, apoptosis and fibrosis in a rat model of cyclosporine-induced nephropathy. Am J Nephrol 37: 7–15, 2013 [DOI] [PubMed] [Google Scholar]
- 30. Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci 113: 1771–1781, 2000 [DOI] [PubMed] [Google Scholar]
- 31. O'Connell S, Tuite N, Slattery C, Ryan MP, McMorrow T. Cyclosporine A-induced oxidative stress in human renal mesangial cells: a role for ERK 1/2 MAPK signaling. Toxicol Sci 126: 101–113, 2012 [DOI] [PubMed] [Google Scholar]
- 32. O'Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O'Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature 357: 692–694, 1992 [DOI] [PubMed] [Google Scholar]
- 33. Ortiz A, Lorz C, Catalan M, Ortiz A, Coca S, Egido J. Cyclosporine A induces apoptosis in murine tubular epithelial cells: role of caspases. Kidney Int Suppl 68: S25–S29, 1998 [DOI] [PubMed] [Google Scholar]
- 34. Pereira MJ, Palming J, Rizell M, Aureliano M, Carvalho E, Svensson MK, Eriksson JW. The immunosuppressive agents rapamycin, cyclosporin A and tacrolimus increase lipolysis, inhibit lipid storage and alter expression of genes involved in lipid metabolism in human adipose tissue. Mol Cell Endocrinol 365: 260–269, 2013 [DOI] [PubMed] [Google Scholar]
- 35. Quintavalle C, Fiore D, De MF, Visconti G, Focaccio A, Golia B, Ricciardelli B, Donnarumma E, Bianco A, Zabatta MA, Troncone G, Colombo A, Briguori C, Condorelli G. Impact of a high loading dose of atorvastatin on contrast-induced acute kidney injury. Circulation 126: 3008–3016, 2012 [DOI] [PubMed] [Google Scholar]
- 36. Radermacher J, Meiners M, Bramlage C, Kliem V, Behrend M, Schlitt HJ, Pichlmayr R, Koch KM, Brunkhorst R. Pronounced renal vasoconstriction and systemic hypertension in renal transplant patients treated with cyclosporin A versus FK 506. Transpl Int 11: 3–10, 1998 [DOI] [PubMed] [Google Scholar]
- 37. Rao MR, Raghu H, Rao JS. Regulation of NADPH oxidase (Nox2) by lipid rafts in breast carcinoma cells. Int J Oncol 37: 1483–1493, 2010 [DOI] [PubMed] [Google Scholar]
- 38. Riella LV, Gabardi S, Chandraker A. Dyslipidemia and its therapeutic challenges in renal transplantation. Am J Transplant 12: 1975–1982, 2012 [DOI] [PubMed] [Google Scholar]
- 39. Rogers JD, Zhao J, Liu L, Amin RD, Gagliano KD, Porras AG, Blum RA, Wilson MF, Stepanavage M, Vega JM. Grapefruit juice has minimal effects on plasma concentrations of lovastatin-derived 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Clin Pharmacol Ther 66: 358–366, 1999 [DOI] [PubMed] [Google Scholar]
- 40. Schenk LK, Rinschen MM, Klokkers J, Kurian SM, Neugebauer U, Salomon DR, Pavenstaedt H, Schlatter E, Edemir B. Cyclosporin-A induced toxicity in rat renal collecting duct cells: interference with enhanced hypertonicity induced apoptosis. Cell Physiol Biochem 26: 887–900, 2010 [DOI] [PubMed] [Google Scholar]
- 41. van der Spek E, Bloem AC, van de Donk NW, Bogers LH, van der Griend R, Kramer MH, de WO, Wittebol S, Lokhorst HM. Dose-finding study of high-dose simvastatin combined with standard chemotherapy in patients with relapsed or refractory myeloma or lymphoma. Haematologica 91: 542–545, 2006 [PubMed] [Google Scholar]
- 42. Vanhaecke J, Van CJ, Van LJ, Daenen W, De GH. Safety and efficacy of low dose simvastatin in cardiac transplant recipients treated with cyclosporine. Transplantation 58: 42–45, 1994 [PubMed] [Google Scholar]
- 43. Vetter M, Chen ZJ, Chang GD, Che D, Liu S, Chang CH. Cyclosporin A disrupts bradykinin signaling through superoxide. Hypertension 41: 1136–1142, 2003 [DOI] [PubMed] [Google Scholar]
- 44. Vilhardt F, van DB. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J 23: 739–748, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang J, Zhang ZR, Chou CF, Liang YY, Gu Y, Ma HP. Cyclosporine stimulates the renal epithelial sodium channel by elevating cholesterol. Am J Physiol Renal Physiol 296: F284–F290, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang N, Silver DL, Thiele C, Tall AR. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J Biol Chem 276: 23742–23747, 2001 [DOI] [PubMed] [Google Scholar]
- 47. Yoshimura N, Oka T, Okamoto M, Ohmori Y. The effects of pravastatin on hyperlipidemia in renal transplant recipients. Transplantation 53: 94–99, 1992 [DOI] [PubMed] [Google Scholar]
- 48. Zager RA, Andoh T, Bennett WM. Renal cholesterol accumulation: a durable response after acute and subacute renal insults. Am J Pathol 159: 743–752, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]