

Abstract
Experiments investigated maturation of endothelial function in the postnatal period. Carotid arteries isolated from newborn (postnatal day 1, P1) to P21 mice were assessed in myographs at transmural pressure (PTM) of 20 mmHg (P1 blood pressure, BP). Acetylcholine was ineffective in P1 but powerfully dilated P7 arteries, whereas NO-donor DEA-NONOate caused similar dilation at P1 and P7. Dilation to acetylcholine at P7 was abolished by inhibition of NO synthase (NOS) (l-NAME) or of phosphoinositide-3-kinase (PI3K) (wortmannin, LY294002). Endothelial NOS (eNOS) expression decreased in P7 compared with P1 arteries, although acetylcholine increased PO4-eNOS-Ser1177 in P7 but not in P1 arteries. Endothelial maturation may therefore reflect increased signaling through PI3K, Akt, and eNOS. Systemic BP increases dramatically in the early postnatal period. After exposing P1 arteries to transient increased PTM (50 mmHg, 60 min), acetylcholine caused powerful dilation and increased PO4-eNOS-Ser1177. Pressure-induced rescue of acetylcholine dilation was abolished by PI3K or NOS inhibition. Transient increased PTM did not affect dilation at P7, or dilation to NO-donor in P1 arteries. Width of endothelial adherens junctions (VE-cadherin immunofluorescence) increased significantly from P1 to P7, and in P1 arteries exposed to transient increased PTM. A function-blocking antibody to VE-cadherin reduced the pressure-induced rescue of acetylcholine responses at P1, and the dilation to acetylcholine in P7 arteries. Therefore, maturation of newborn endothelium dilator function may be induced by increasing BP in the postnatal period. Furthermore, this may be mediated by VE-cadherin signaling at adherens junctions. Interruption of this maturation pathway may contribute to developmental and adult vascular diseases.
Keywords: endothelium-dependent dilation, mechanotransduction, arterial blood pressure, developmental biology
the early postnatal period is associated with extraordinary changes in arterial structure and function, including the morphology and functional activity of endothelial cells (8, 19–21, 32, 39). Endothelial cells lining fetal or newborn arteries have reduced activity of endothelial nitric oxide synthase (eNOS) and decreased NO-mediated endothelial dilation compared with more mature arteries (1, 2, 5, 27, 41, 44–46). However, this postnatal increase in endothelial NO activity is not associated with an increase in eNOS expression suggesting that it may reflect posttranslational regulation or signaling of eNOS (2, 5, 44, 45). The mechanisms contributing to the postnatal maturation of arterial endothelium have not been clearly defined. Furthermore, previous studies have mostly focused on the pulmonary circulation (1, 2, 5, 27, 46), which is subject to unique changes during the early postnatal period and may not be representative of the systemic circulation (2, 5).
Increased understanding of arterial maturation might provide novel insight into developmental and adult vascular diseases. Elastic central arteries play a key role in ensuring optimal organ perfusion by stabilizing pulsatile pressure and flow from the heart (36, 37). These elastic arteries expand during systole to store a significant portion of the stroke volume, which they release during diastole to support continuous organ blood flow (36, 37, 43). Remodeling of central arteries during vascular disease causes the arteries to become less elastic and more rigid, resulting in altered pulse wave reflections, increased pulse pressure, and detrimental effects on ventricular load and organ perfusion (14, 25, 30, 36, 37). The early postnatal maturation of arterial endothelium occurs in the face of relatively dramatic increases in blood pressure but with relatively stable blood flows in central arteries (4, 20). Indeed, almost 20 years ago, Kobayashi and Sakai (20) suggested that such postnatal maturation of central arterial endothelium might result from stretch imposed by increasing blood pressure (BP). This intriguing concept has never been further evaluated or considered.
Therefore, the aim of the present study was to investigate the early postnatal maturation of the endothelium on mouse carotid arteries and to determine whether increasing pressure might contribute to this maturation process.
MATERIALS AND METHODS
Use of animals was approved by the Institutional Animal Care and Use Committee of the Johns Hopkins University and complied with the “Guide for the Care and Use of Laboratory Animals” published by the National Institutes of Health. Newborn (postnatal day 1 or P1), 1-wk-old (P7), and 3-wk-old (P21) male and female mice (C57BL6, Jackson Labs) were killed by CO2 asphyxiation. The carotid arteries were rapidly removed and placed in cold Krebs-Ringer bicarbonate solution containing (in mM) 118.3 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, 25.0 NaHCO3, and 11.1 glucose (control solution).
Analysis of vascular responses.
Carotid arteries were cannulated at both ends with glass micropipettes, secured using 12-0 nylon monofilament suture, and placed in a microvascular chamber (Living Systems, Burlington, VT) (3, 34). Unless stated otherwise, the arteries were maintained at a constant transmural pressure (PTM) of 20 mmHg in the absence of flow. This PTM represents the mean arterial BP of P1 mice (17). The chamber was superfused with control solution, maintained at 37°C, pH 7.4, and gassed with 16% O2-5% CO2-balance N2. The chamber was placed on the stage of an inverted microscope (Nikon TMS-F) connected to a video camera (CCTV camera; Panasonic). The vessel image was projected on a video monitor, and the internal diameter was continuously determined by a video dimension analyzer (Living Systems Instrumentation) and was monitored using a BIOPAC (Santa Barbara, CA) data-acquisition system (3, 34).
After a period of equilibration (60–90 min), arterial segments were constricted with the thromboxane receptor agonist U46619, and once the constriction was stable, vasodilatation to the endothelial agonist acetylcholine (10−9 to 10−7 M) or to the NO donor DEA-NONOate (10−9 to 10−7 M) was determined. These concentration-response curves were generated by increasing the agonist concentration in full-log increments once the response to the previous concentration had stabilized. Only one cycle of constriction-vasodilatation was performed on each artery. Concentration-response curves to acetylcholine were determined in paired carotid arteries with one artery analyzed under control conditions and the other artery studied after a pharmacological or biomechanical intervention. When pharmacological agents were studied, the preparations were incubated for 30 min with the drugs before and during exposure of the arteries to the vasodilator stimuli. In some experiments, the arteries were transiently exposed to an elevated PTM of 50 mmHg, which is equivalent to the systolic BP of P7 mice (17), for 60 min before returning PTM to 20 mmHg. Then, 10 min after returning PTM to 20 mmHg, the arteries were constricted with U46619 and vasodilator responses assessed as described above. To determine the role of VE-cadherin clustering at adherens junctions on vasodilator responses, the arteries were exposed to intraluminal administration of a function blocking antibody against mouse VE-cadherin (BV13, 50 μg/ml; eBioscience, CA) or a control antibody (50 μg/ml; eBioscience) for 2.5–3 h before vasodilator responses were analyzed.
Endothelial imaging.
Carotid arteries were mounted in specialized “flipper” chambers (Living Systems) that enabled the blood vessel assembly to be rapidly (∼1 s) transferred from control solution to paraformaldehyde (3%, 4°C, 30 min). Arteries were flipped at a PTM of 20 mmHg and cut open longitudinally during fixation. Preliminary experiments assessed the time course of acetylcholine-induced changes in eNOS phosphorylation in P1 and P7 arteries (1–10 min) and demonstrated that the effect of the agonist was maximal between 5 and 10 min, which is the time point that was used for comparisons in the study. After fixation, arteries were rinsed in PBS (3 × 10 min), permeabilized (Triton X, 0.5%, 15 min), rinsed again (PBS, 3 times) and then incubated in donkey serum (1.5%, 15 min) to reduce nonspecific binding. Arteries were then incubated overnight with primary antibodies: goat polyclonal antibody to VE-cadherin (1:500 dilution, Santa Cruz Biotechnology), mouse monoclonal antibody to eNOS (1:150 dilution, BD Biosciences), and/or a rabbit polyclonal antibody to phospho-eNOS (Ser1177) (1:200 dilution, Cell Signaling). Arteries were rinsed (PBS, 3 × 15 min) then incubated with AlexaFluor 488, AlexaFluor 568, or rhodamine-labeled secondary antibodies (donkey anti-goat, donkey anti-rabbit, donkey anti-mouse) (1:200 dilution, Invitrogen, Carlsbad, CA; or Jackson ImmunoResearch) for 2 h. After rinsing (PBS, 3 × 10 min), arteries were incubated with Draq5 (5 μmol/l, 45 min) (Biostatus, Leicestershire, UK) to label nuclei. Samples were viewed using a Leica AOBS-equipped SP5 laser-scanning microscope. Images (1,024 × 1,024 pixels) were obtained using sequential acquisition, a pinhole of 1 Airy unit, scan speed of 400 Hz, 6 line averaging and an optical zoom of 3.0. For AlexaFluor488, excitation was at 488 nm and emission was captured from 492 to 541 nm; for AlexaFluor 568 or rhodamine, excitation was at 543 nm and emission was captured from 555 to 620 nm; and for DRAQ, excitation was at 633 nm and emission captured from 650 to 750 nm. For direct comparison of fluorescent intensity, arteries were processed using the same approach and the same instrument settings. When comparing fluorescent signals between different groups, the mean of the average signal intensities in the reference (or control) arteries was set as 100%, and the intensity of all control and test images (including acetylcholine-treated arteries) were expressed relative to that value (12). This approach maintains an estimate of variance in the control group, enabling appropriate statistical analyses (12). When analyzing the width of adherens junctions, a region of interest (ROI) grid, comprising four lines 15 μm apart, was placed perpendicular to the long axis of endothelium, which had been stained for VE-cadherin. Particular care was taken to avoid the terminal parts of any cell, which might run parallel to the ROI grid. The width of adherens junctions was determined as the distance in microns that VE-cadherin intensity was above the immediate background level.
Statistical analysis.
Vasomotor responses were expressed as a percentage change in baseline diameter before administrating the agent. Unless stated otherwise, data are expressed as means ± SE for n number of experiments, where n equals the number of animals from which blood vessels were studied. Statistical evaluation of the data was performed by Student's t-test for paired or unpaired observations. When more than two means were compared, analysis of variance was used. If a significant F value was found, Tukey's or Dunnett's tests for multiple comparisons were employed to identify differences among groups. Values were considered to be statistically different when P < 0.05 (NS = not significant).
RESULTS
Maturation of endothelium-dependent vasodilatation in neonatal mouse carotid arteries.
Dilation to the endothelium-dependent agonist, acetylcholine (10−9 to 10−6 M), was minimal in P1 arteries, increased dramatically by P7, and increased further by P21 (Fig. 1A). Maximal dilation to acetylcholine was 23.4 ± 6.9% (n = 11) of the constriction to U46619 in P1 arteries, 75.5 ± 6.1% in P7 arteries (n = 8, P < 0.001 compared with P1), and 102.9 ± 3.1% in P21 arteries (n = 6; P < 0.001 compared with P1, P < 0.05 compared with P7). Between P21 and P100, there was no further significant increase in response to acetylcholine (data not shown). Dilation to the exogenous NO-donor, DEA-NONOate, was similar in P1, P7, and P21 arteries (Fig. 1B). Inhibition of NO synthase (NOS) by l-NAME (10−4 M) abolished dilation to acetylcholine in P1 and P7 arteries and reduced responses to the endothelial agonist in P21 arteries (Fig. 1C). The residual l-NAME-resistant dilation to acetylcholine in P21 arteries was not inhibited by treatment with the cyclooxygenase inhibitor, indomethacin (10−5 M) (data not shown). These results indicate that the endothelial response to acetylcholine increases in postnatal arteries: between P1 and P7, this reflects an increase in NO activity, whereas from P7 to P21 there is an additional increase in an l-NAME/indomethacin-resistant response, likely mediated by endothelium-derived hyperpolarizing factor or EDHF. This EDHF-like response was not analyzed further, and in subsequent experiments, particular attention was focused on the increase in NO activity between P1 and P7.
Fig. 1.

Regulation of carotid arterial dilation in the immediate postnatal period. Arteries were isolated from postnatal day 1 (P1, newborn), P7, and P21 mice, and analyzed in a microperfusion system at a transmural pressure of 20 mmHg. Functional responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as means ± SE. To observe dilation, arteries were initially constricted to ∼80% of baseline diameter with the thromboxane receptor agonist U46619 (U4). For statistical analysis, * symbols indicate significant difference compared with P1; # symbols indicate significant difference between P7 and P21. In each case, 1 symbol = P < 0.05; 2 symbols = P < 0.01; 3 symbols = P < 0.001. A: under control conditions in P1 arteries, acetylcholine caused significant dilation only at the highest dose tested (10−6 M) but caused markedly increased dilation in P7 and P21 arteries [n = 6 (P21), 8(P7), or 11(P1)]. In B, the NO donor DEA-NONOate caused dilation that was similar in P1, P7, and P21 arteries [n = 5 (P1), 7 (P21), or 9 (P7)]. In C, in the presence of the NO synthase inhibitor l-NAME (10−4 M), acetylcholine did not cause significant dilation in P1 or P7 arteries but continued to cause marked dilation in P21 arteries [n = 5 (P21, P1) or 6 (P7)].
Maturation of endothelial signaling in neonatal mouse carotid arteries.
In contrast to the early postnatal increase in dilation to acetylcholine, the expression of endothelial NOS (eNOS, determined by immunofluorescent labeling) decreased significantly during the early postnatal period (Fig. 2).
Fig. 2.
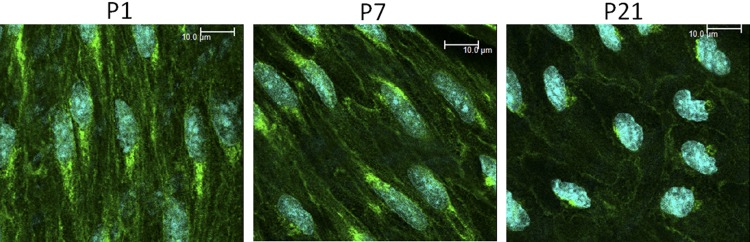
Representative laser scanning microscope (LSM) fluorescent images demonstrating the expression and localization of endothelial NO synthase (eNOS) in endothelial cells of P1 (left), P7 (middle), and P21 (right) arteries. eNOS is represented in green, while nuclei are colored light blue. The white line represents 10 μm. Compared with P1 arteries, eNOS expression decreased by 21.8 ± 2.7% in P7 (P < 0.001, n = 4) and 53.1 ± 2.3% in P21 arteries (P < 0.001, n = 4).
In P7 arteries, the dilation to acetylcholine was markedly reduced following inhibition of phosphoinositide 3-kinase (PI3K) by wortmannin (3 × 10−7 M) or LY294002 (5 × 10−5 M) (Fig. 3). The maximal dilation to acetylcholine in P7 arteries was reduced from 70.8 ± 3.6% of the constriction to U46619 in control arteries (combined controls, n = 14) to 24.2 ± 5.8% after LY294002 (n = 7, P < 0.001) or 35.7 ± 3.1% after wortmannin (n = 7, P < 0.001). The PI3K-Akt signaling pathway can activate eNOS via Akt-mediated phosphorylation of the enzyme at Ser1177. Indeed, in P7 arteries, acetylcholine significantly increased the levels of PO4-eNOS Ser1177 determined by immunofluorescent labeling (to 149.0 ± 5.6% of control, P < 0.001, n = 8) (Fig. 3). Consistent with the markedly diminished dilation to acetylcholine in P1 arteries, the agonist did not significantly increase the level of PO4-eNOS Ser1177 in these newborn arteries (to 100.8 ± 3.3% of control, P = NS, n = 8) (Fig. 3).
Fig. 3.
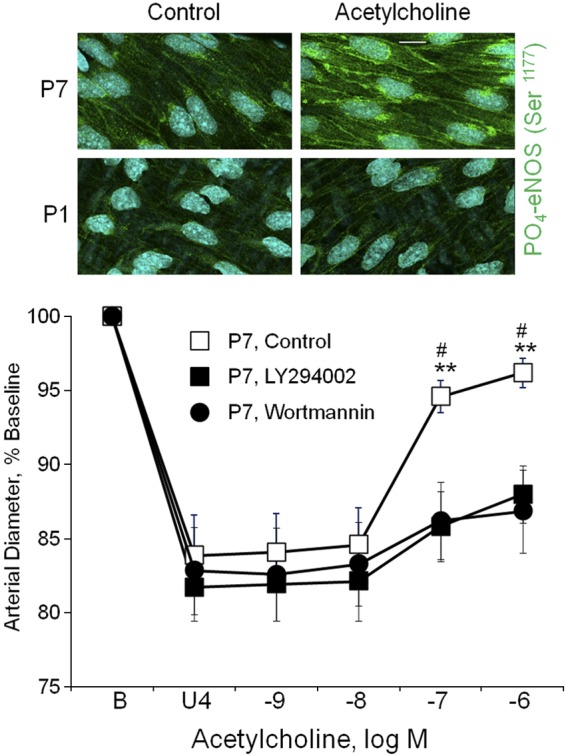
Endothelial signaling in neonatal arteries. Top: representative LSM fluorescent images demonstrating the effect of acetylcholine (10−6 M) on the Ser1177 phosphorylated form of endothelial NO synthase (PO4-eNOS Ser1177) in endothelial cells of P1 and P7 arteries. PO4-eNOS Ser1177 is represented in green, while nuclei are colored light blue. The white line represents 10 μm. Acetylcholine significantly increased levels of PO4-eNOS Ser1177 in endothelial cells of P7 arteries (to 149.0 ± 5.6% of control, P < 0.001, n = 8) but not in P1 arteries (to 100.8 ± 3.3% of control, P = NS, n = 8). There was no significant difference in the basal control levels of PO4-eNOS Ser1177 between P1 and P7 arteries (basal level in P7 arteries was 101.9 ± 7.7% of the level observed in P1 arteries, P = NS, n = 5). Bottom: effects of phosphoinositide 3-kinase (PI3K) inhibition with wortmannin (3 × 10−7 M) or LY294002 (5 × 10−5 M) on dilator responses to acetylcholine in P7 carotid arteries. Paired isolated arteries were analyzed in a microperfusion system at a transmural pressure of 20 mmHg and treated with or without a PI3K inhibitor. To observe dilation, arteries were initially constricted to ∼80% of baseline diameter with the thromboxane receptor agonist U46619 (U4). Functional responses are expressed as a percentage of the baseline diameter (B) and presented as means + SE (n = 7 for each inhibitor). For statistical analysis, * symbols indicate significant difference with arteries treated with wortmannin, and # symbols indicate significant difference with arteries treated with LY294002. In each case, 1 symbol = P < 0.05; 2 symbols = P < 0.01. The inhibitors markedly reduced dilation to acetylcholine.
These results suggest that the increase in endothelial NO activity in P7 compared with P1 arteries reflects an increased ability of acetylcholine to activate the PI3K-Akt-eNOS pathway.
Influence of increased pressure on endothelium of newborn arteries.
The early postnatal period is associated with a marked elevation in BP, increasing from a mean arterial BP of ∼20 mmHg at P1 to >40 mmHg at P7 (17). Transient exposure of P1 arteries to elevated PTM (50 mmHg, 60 min) caused a marked increase in dilation of these newborn arteries to acetylcholine (Fig. 4A), increasing the maximal dilation from 23.8 ± 7.4% of the constriction to U46619 in P1 arteries maintained at 20 mmHg to 73.0 ± 6.2% in P1 arteries transiently exposed to elevated PTM (n = 6, P < 0.01). The transient exposure to elevated PTM did not affect responses to the NO-donor DEA-NONOate in P1 arteries (Fig. 4B). Inhibition of NOS with l-NAME (10−4 M) prevented the pressure-induced amplification of responses to acetylcholine (after l-NAME, no significant response to 10−6 M acetylcholine: 0.6 ± 9.5% of the constriction to U46619, n = 4). In P7 arteries, the transient increase in PTM did not significantly affect response to the endothelial agonist (Fig. 4C). Likewise, in P7 arteries, dilator responses to acetylcholine or to DEA-NONOate were similar in arteries maintained at PTMs of 20 and 50 mmHg (Fig. 5).
Fig. 4.

Effects of a transient increase in transmural pressure (PTM) on dilation of isolated neonatal carotid arteries. Paired arteries were isolated from P1 and P7 mice, and dilation analyzed in a microperfusion system at a transmural pressure (PTM) of 20 mmHg. In one artery of each pair, PTM was transiently increased to 50 mmHg for 1 h then returned to 20 mmHg (transient high PTM), whereas in the other artery PTM was maintained at 20 mmHg (control PTM). To observe dilation, arteries were constricted to 75–85% of baseline diameter with the thromboxane receptor agonist U46619 (U4). Dilation was assessed in response to acetylcholine in P1 arteries (A); the NO donor DEA-NONOate in P1 arteries (B), or acetylcholine in P7 arteries (C). Functional responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as means ± SE; n = 6 (A) or 4 (B, C). For statistical analysis, **Significant difference between responses at control PTM and after exposure to a transient high PTM (P ≤ 0.01). The transient increase in pressure significantly increased dilator responses to acetylcholine in P1 but not P7 arteries and did not significantly affect dilation to DEA-NONOate.
Fig. 5.

Effects of a sustained increase in transmural pressure (PTM) on dilation of isolated P7 carotid arteries. Arteries were isolated from P7 mice, and dilation analyzed in a microperfusion system at a constant transmural pressure (PTM) of 20 mmHg (constant control PTM) or constant PTM of 50 mmHg (constant high PTM). To observe dilation, arteries were constricted to 75–85% of baseline diameter with the thromboxane receptor agonist U46619 (U4). Dilation was assessed in response to acetylcholine (A) or the NO donor DEA-NONOate (B). Functional responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as means ± SE (n = 6) There were no statistically significant differences between dilator responses in arteries maintained at 20 and 50 mmHg.
Pressure-induced amplification of dilation to acetylcholine in P1 arteries was markedly reduced by inhibition of PI3K with wortmannin (3 × 10−7 M) or LY294002 (5 × 10−5 M) (Fig. 6). In P1 arteries transiently exposed to increased PTM, maximal dilation to acetylcholine was significantly reduced from 89.3 ± 13.2% of the U46619-induced constriction in untreated arteries (combined controls, n = 11) to 17.3 ± 8.4% in wortmannin-treated arteries (n = 6, P < 0.01), and to 4.1 ± 8.9% in LY294002-treated arteries (n = 5, P < 0.01). Furthermore, although acetylcholine did not increase PO4-eNOS Ser1177 in P1 arteries maintained at a PTM of 20 mmHg (Fig. 3), after a transient increase in PTM (50 mmHg, 60 min), acetylcholine increased endothelial levels of PO4-eNOS Ser1177 (to 145.5 ± 6.5% of control, P < 0.001, n = 12) (Fig. 6).
Fig. 6.
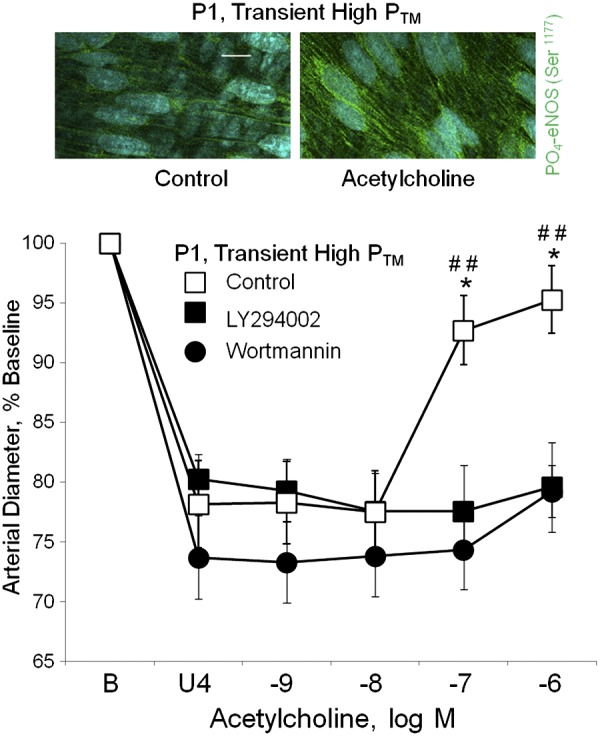
Effects of a transient increase in transmural pressure (PTM) on endothelial signaling in newborn arteries (P1). All arteries were exposed to a transient increase in PTM (to 50 mmHg, 60 min), then PTM was returned to 20 mmHg before assessing responses to acetylcholine. Top: representative LSM fluorescent images demonstrating the effect of acetylcholine (10−6 M) on PO4-eNOS Ser1177 in endothelial cells of P1 arteries that had been transiently exposed to increased PTM (50 mmHg, 60 min). PO4-eNOS Ser1177 is represented in green, while nuclei are colored light blue. Acetylcholine significantly increased levels of PO4-eNOS Ser1177 (to 145.5 ± 6.5% of control, P < 0.001, n = 12). This contrasts with arteries maintained at a PTM of 20 mmHg (Fig. 3), where acetylcholine did not increase PO4-eNOS Ser1177. The white line represents 10 μm. Bottom: effects of phosphoinositide 3-kinase (PI3K) inhibition with wortmannin (3 × 10−7 M) or LY294002 (5 × 10−5 M) on dilator responses to acetylcholine in P1 carotid arteries that had been exposed to increased PTM (50 mmHg, 60 min). Paired isolated arteries were analyzed in a microperfusion system and treated with or without a PI3K inhibitor. After transiently increasing PTM to 50 mmHg, PTM was returned to 20 mmHg for analysis of dilation. Arteries were constricted to ∼75% of baseline diameter with the thromboxane receptor agonist U46619 (U4). Functional responses are expressed as a percentage of the baseline diameter (B) and presented as means + SE (n = 5 for LY294002, n = 6 for wortmannin). For statistical analysis, * symbols indicate significant difference with arteries treated with wortmannin, and # symbols indicate significant difference with arteries treated with LY294002. In each case, 1 symbol = P < 0.05; 2 symbols = P ≤ 0.01. The inhibitors markedly reduced dilation to acetylcholine.
Role of VE-cadherin clustering in the endothelial response to increased pressure.
The early postnatal maturation of arterial endothelium was associated with increased complexity of cellular junctions. Adherens junctions, identified by immunofluorescent labeling of VE-cadherin, were generally narrow and uncomplicated structures in P1 arteries, but the early postnatal period was associated with increased folding and increased width of the junctions (Fig. 7). Indeed, the width of adherens junctions increased from 1.25 ± 0.07 μm in P1 arteries (n = 7 sections from different arteries, comprising 231 junctions) to 1.80 ± 0.05 μm in P7 arteries (n = 8 sections from different arteries, comprising 276 junctions) (P < 0.001). Transient exposure of P1 arteries to elevated pressure (60 min, 50 mmHg) increased the width of adherens junctions from 1.16 ± 0.07 μm in arteries maintained at 20 mmHg (n = 6 sections from different arteries, comprising 197 junctions) to 1.47 ± 0.06 μm after transient exposure to elevated pressure (n = 6 sections from different arteries, comprising 240 junctions) (P < 0.01).
Fig. 7.
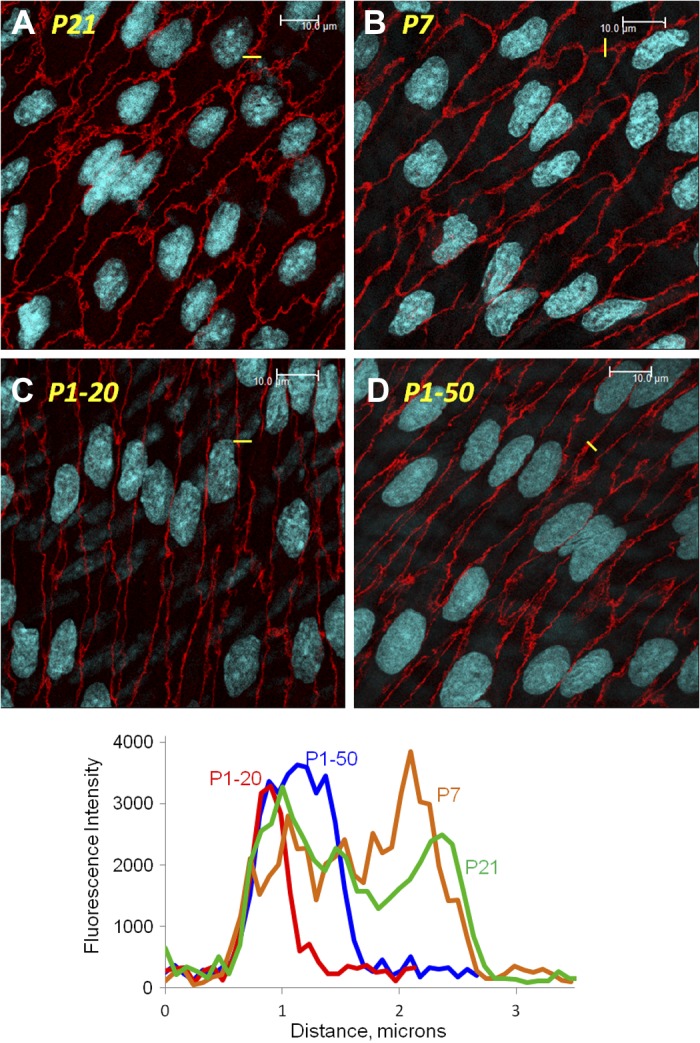
Representative LSM fluorescent images demonstrating the staining of VE-cadherin (red) and the organization of endothelial adherens junctions in P21 (A), P7 (B), and P1 (C, D) arteries. P1 arteries were assessed after they had been maintained at a transmural pressure (PTM) of 20 mmHg (C) or after PTM was transiently increased (50 mmHg, 60 min) before returning to 20 mmHg for analysis (D). The white line represents 10 μm. As described in materials and methods, the width of adherens junctions was determined by constructing region of interest (ROI) grids, comprising 4 lines 15 μm apart, which were placed perpendicular to the long axis of endothelium that had been stained for VE-cadherin. The width of adherens junctions was determined as the distance in μm that VE-cadherin intensity was above the immediate background level. An example of a small component of a ROI grid overlaying a single junction is shown in yellow in each image and the corresponding intensity profiles presented in the bottom graph [red P1–20, P1 arteries, which were maintained at 20 mmHg; blue P1–50, P1 arteries that were exposed to a transient increase in PTM (50 mmHg, 60 min); brown, P7 arteries; green, P21 arteries].
To determine whether clustering of VE-cadherin at adherens junctions contributed to the endothelial response to increasing pressure, neonatal arteries were treated intraluminally with a function-blocking antibody to mouse VE-cadherin (BV13, 50 μg/ml) or a control antibody. As expected, this treatment markedly reduced the localization of VE-cadherin at endothelial cell:cell contacts (not shown). In newborn arteries treated with the function blocking antibody and transiently exposed to an increase in PTM (50 mmHg, 60 min), the pressure-induced amplification of dilator responses to acetylcholine was markedly inhibited (Fig. 8A). In newborn arteries transiently exposed to increased PTM, maximal dilation to acetylcholine was 76.6 ± 9.8% of the constriction to U46619 in control antibody-treated arteries and 30.6 ± 4.1% in VE-cadherin antibody-treated arteries (n = 5, P < 0.05). The function blocking antibody to VE-cadherin also prevented the acetylcholine-induced increase in PO4-eNOS Ser1177 in arteries exposed to a transient increase in PTM (to 105.3 + 4.9% of control, P = NS, n = 8) (Fig. 8). This treatment also reduced the endothelial dilation to acetylcholine in P7 arteries (Fig. 8B) but did not affect the dilation to NO donor DEA-NONOate (P1 arteries, Fig. 8C).
Fig. 8.
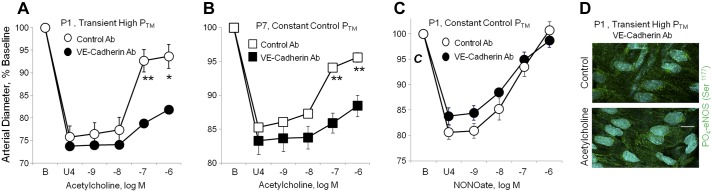
Effects of a VE-cadherin function blocking antibody on responses to acetylcholine or the NO-donor DEA-NONOate in neonatal arteries. In A, B, and C, dilator responses of paired arteries were determined in a microperfusion system (at a PTM of 20 mmHg), after they were constricted to 75–85% of baseline diameter with the thromboxane receptor agonist U46619 (U4). One artery of each pair was treated with a VE-cadherin function blocking antibody (BV13, 50 μg/ml) and the other with a control antibody (50 μg/ml). Dilator responses are expressed as a percentage of the baseline diameter (B), and presented as means + SE for n = 5 (A, C) or 4 (B). For statistical analysis, * symbols indicate a significant difference between responses in control and VE-cadherin antibody-treated arteries (1 symbol = P < 0.05; 2 symbols = P ≤ 0.01). In A, both P1 carotid arteries were exposed to a transient increase in PTM (50 mmHg, 60 min) before assessing dilator responses to acetylcholine (at 20 mmHg). The function blocking antibody to VE-cadherin markedly inhibited the response to acetylcholine. B: in P7 arteries maintained at 20 mmHg, the VE-cadherin function blocking antibody markedly inhibited the response to acetylcholine. C: in P1 arteries maintained at 20 mmHg, the function blocking antibody did not affect dilation to the NO donor DEA-NONOate. D: representative LSM fluorescent images presenting the effect of acetylcholine (10−6 M) on PO4-eNOS Ser1177 in P1 arteries, which were treated with the VE-cadherin function blocking antibody and exposed to a transient increase in PTM (50 mmHg, 60 min). PO4-eNOS Ser1177 is represented in green, while nuclei are colored light blue. Under these conditions, acetylcholine did not significantly increase levels of PO4-eNOS Ser1177 (to 105.3 + 4.9% of control, P = NS, n = 8). This contrasts with untreated P1 arteries exposed to a transient increase in PTM, where acetylcholine significantly increased PO4-eNOS Ser1177 (Fig. 6). The white line represents 10 μm.
DISCUSSION
Numerous studies in different vascular beds have demonstrated that arterial endothelium-dependent relaxation is reduced in the fetal and immediate postnatal period compared with more mature arteries (1, 2, 5, 27, 41, 45). Relaxation mechanisms contributing to this developmental regulation of endothelial function appear to vary within and between different vascular beds and include alterations in the expression and/or activity of eNOS, altered responsiveness of smooth muscle to NO, and alterations in the mediators contributing to endothelium-dependent relaxation (5). Few studies have focused on endothelial-mediated dilation in central arteries during the immediate postnatal period. In the present study, we observed profound depression in dilation to the endothelial agonist acetylcholine in newborn compared with more mature carotid arteries. By P7, endothelium-dependent dilation was dramatically increased, and there was a further small increase to adult levels of response by P21. The dilation to acetylcholine in P7 arteries was abolished by the NOS inhibitor l-NAME, indicating that the emergence of endothelial dilation during the first postnatal week is mediated entirely by NO. Throughout the early postnatal period (P1 to P21), there was no significant difference in dilation to an exogenous NO donor (DEA-NONOate), suggesting that any difference in endothelial NO-mediated dilation likely reflected differences in endothelial activity. As expected, eNOS was localized in a perinuclear compartment (presumably the Golgi) and at the plasma membrane (47). However, although endothelial NO-mediated dilation increased dramatically between P1 and P7, the postnatal period was associated with diminishing expression of endothelial NOS, as determined by immunofluorescent labeling and confocal microscopy, which is consistent with a previous study that assessed eNOS expression in fetal and adult ovine carotid arteries (45).
eNOS expression decreased markedly in P21 compared with P1 or P7 arteries. Interestingly, the profound decrease in eNOS expression at P21 occurred concomitantly with the emergence of an l-NAME/indomethacin-resistant component in the dilation to acetylcholine. Therefore, the activity of an EDHF-like mediator appears to be developmentally regulated in carotid arterial endothelium, albeit on a distinct time course compared with endothelium-derived NO. Although the magnitude of endothelial dilation to acetylcholine changed little after P21, the response was entirely l-NAME/indomethacin-resistant in adult arteries (not shown), suggesting that this switch in balance of endothelial mediators continues after the initial postnatal period. Because of the profound diminution in expression of eNOS and the presence of a prominent NO-independent endothelial dilation in P21 arteries, we focused our subsequent analysis of endothelial responses to the immediate postnatal period, namely P1 and P7 arteries.
The emergence of NO-mediated dilation to acetylcholine in P7 arteries was associated with acetylcholine-induced increase in phosphorylation of eNOS at Ser1177, which is linked to activation of eNOS (10). Although a number of kinases can phosphorylate Ser1177, including AMPK and PKA, an important physiological pathway is via PI3K-Akt signaling (10, 15, 16). Indeed, the endothelial dilation to acetylcholine in P7 arteries was virtually abolished by selective inhibition of PI3K with wortmannin or LY294002. Therefore, the initial postnatal emergence of NO-mediated dilation to acetylcholine in mouse carotid artery appears to reflect increased signaling through PI3K, Akt, and eNOS. Several studies in the developing pulmonary circulation have proposed that pulmonary eNOS is uncoupled in the postnatal period, leading to generation of superoxide (2, 29, 33). However, there were differences with regard to underlying mechanisms (impaired eNOS-HSP90 coupling; BH4 deficiency) and timing of the effect (2, 29, 33). Indeed, Mata-Greenwood et al. (29) observed pulmonary eNOS uncoupling between P3 and P14, but not in the immediate postnatal period (before P3). Furthermore, when the superoxide generating effects of uncoupled eNOS were inhibited (sepiapterin supplementation; cell-permeable SOD mimic MnTMPyP), the existing dilator response to acetylcholine at P3 and P14 was amplified, but the minimal dilation in newborn arteries was unaffected (29). Therefore, although eNOS uncoupling can occur in the pulmonary postnatal circulation, it may not explain the diminished endothelium-dependent dilation to acetylcholine in newborn pulmonary arteries. In mouse carotid arteries, the emergence of endothelial dilation paralleled the ability of acetylcholine to increase phosphorylation of eNOS at Ser1177, which was not the case in the pulmonary circulation (2). Furthermore, the SOD mimic MnTMPyP (50 μM) did not uncover or amplify relaxations to acetylcholine in neonatal mouse carotid arteries (data not shown). Because of the unique changes occurring in the neonatal pulmonary circulation (2, 5), pulmonary endothelial changes may not be representative of the systemic circulation or central arteries.
The postnatal period is characterized by marked changes in hemodynamics. A steady increase in systemic mean arterial blood pressure (MABP) occurs during fetal development (to ∼20 mmHg at P1), then increases to ∼45 mmHg by P7 and to ∼60 mmHg by P21 (17). Vascular endothelial and smooth muscle cells are mechanosensitive, and hemodynamic changes are thought to be important determinants of embryonic vascular development (7, 13, 32). Although arterial BP continually increases during this period, carotid arterial blood flow decreases at birth then gradually increases thereafter (4). The postnatal fall in blood flow is thought to reflect increased arterial O2 content enabling adequate O2 delivery at lower flows (4). Almost 20 years ago, based on a correlation analysis, Kobayashi and Sakai (20) proposed that the postnatal maturation of arterial endothelium resulted from the stretch imposed by increasing BP. This intriguing concept has never been tested until now. When newborn arteries were transiently exposed for 1 h to an increased PTM of 50 mmHg and then returned to their control PTM of 20 mmHg, there was a dramatic emergence of endothelial dilation to acetylcholine. This procedure did not alter the acetylcholine response in P7 arteries. Indeed, the transient increase in PTM appeared to mimic the maturation process occurring in the immediate postnatal period. After this intervention, the magnitude of the acetylcholine dilation was similar in P1 and P7 arteries. Likewise, the mechanism underlying the rescued P1 dilation appeared similar to that occurring in P7 arteries. The pressure-induced rescue of acetylcholine dilation in P1 was dependent entirely on NO activity, was markedly reduced by PI3K inhibitors, was associated with increased phosphorylation of eNOS Ser1177, and occurred without any change in arterial responsiveness to exogenous NO. Therefore, these experiments suggest that pressure-induced stretch of newborn arteries mimicked the early postnatal maturation of endothelium, enabling increased activity of PI3K/Akt signaling, increased PO4-eNOS, and increased endothelium-dependent NO-mediated relaxation.
The effect of increased pressure (to 50 mmHg) on newborn arterial endothelium was long-lasting and was obviously still present after PTM was returned to the control level of 20 mmHg. The present study did not analyze the reversibility of the pressure-induced changes, but acetylcholine-induced dilation remained amplified for at least 60 min after the PTM was returned to the control level. This suggests that once activated, the pressure-induced endothelial signaling pathway diminishes at a relatively slow rate. Slow deactivation of the pressure-induced biomechanical signaling likely explains why P7 arteries had similar dilator responses when studied at a relatively normal arterial pressure (50 mmHg) or a reduced pressure (20 mmHg, control PTM). Future studies remain to be done to assess longer term effects of changes in PTM or arterial BP on endothelial responses of developing arteries.
VE-cadherin proteins located on adjacent endothelial cells interact and cluster at cell:cell borders forming a component of cellular junctions, the adherens junctions. Importantly, VE-cadherin, through its binding partner β-catenin, can amplify PI3K-Akt signaling (22–24). Endothelial adherens junctions were narrow linear structures in P1 arteries and were significantly wider and more complex in more mature arteries. Kobayashi and Sakai (20) originally observed increased organization of endothelial cellular junctions during early postnatal development and, again based on a correlation analysis, proposed that this was mediated by the stretch imposed from developmental increases in BP. Indeed, transient exposure of P1 arteries to increased pressure significantly increased the width of adherens junctions, again mimicking the maturation process. This is consistent with experiments on cultured endothelial cells demonstrating that tugging forces stimulated the growth and stabilization of adherens junctions (28). The clustering of VE-cadherin:β-catenin at adherens junctions directly amplifies PI3K signaling, an effect that is prevented by VE-cadherin function-blocking antibodies (23, 24, 40). To determine whether the pressure and maturational effects on endothelial adherens junctions might contribute to the observed functional effects, we treated isolated neonatal arteries with intraluminal VE-cadherin functioning- blocking antibody or a control antibody. The function-blocking antibody markedly reduced the pressure-induced emergence of dilation and increased PO4-eNOS Ser1177 to acetylcholine occurring in P1 arteries, and the developmental emergence of dilation to the agonist occurring in P7 arteries. Therefore, our results are consistent with a model whereby increased pressure in developing arteries, through stretch-mediated activation of endothelial adherens junctions and VE-cadherin clustering, leads to amplification of PI3K-Akt signaling and emergence of PI3K-Akt-eNOS signaling and dilation.
The “resting” state of normal mature endothelium is an active rather than a passive process (9, 31). In this regard, the potential role of adherens junctions and VE-cadherin clustering in mediating the postnatal maturation of arterial endothelial cells is especially appealing. These structures and VE-cadherin-dependent signaling have previously been proposed as key mediators of vascular stability (9). Impaired protective signaling at endothelial adherens junctions either in developing or mature arteries can disrupt vascular integrity and may be an important trigger for the development of vascular disease (9, 31).
An alternate mode of VE-cadherin:β-catenin signaling has been implicated in endothelial responses to shear stress. Shear stress reportedly utilizes PECAM-1 to trigger transactivation of VEGFR2, which causes increased PI3K-Akt signaling and NO production (11, 18, 35, 42). Within this model, VE-cadherin:β-catenin acts as a scaffolding intermediary facilitating the transactivation of VEGFR2, but VE-cadherin:β-catenin does not respond directly to shear stress (6, 18, 38). Importantly, VE-cadherin does not need to be localized at cell:cell junctions for this role, and therefore responses to shear stress are not inhibited by function blocking antibodies to VE-cadherin (6, 42). This role is therefore distinct from the activity of VE-cadherin in mediating direct amplification of PI3K-Akt signaling (23, 24, 40) and the pressure-induced maturation of endothelial dilation in neonatal arteries. Interestingly, in cultured endothelial cells, although shear stress activates Akt in a VEGFR2-dependent manner, stretch activates Akt independently of VEGFR2 (11, 18, 26, 35, 42). Therefore, stretch and shear stress can activate similar but distinct signaling pathways in endothelial cells.
In conclusion, the maturation of newborn endothelium may be induced by increasing blood pressure in the postnatal period. Furthermore, this may be mediated by stretch-induced activation of VE-cadherin signaling at adherens junctions, resulting in enhanced PI3K-Akt signaling and increased eNOS activity. Retention or reemergence of the newborn endothelial phenotype may contribute to the development of vascular disease by negating the normal protective role of this important cell layer.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Award HL-102715 to N. A. Flavahan.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.F., M.M.M., I.M.L., and N.A.F. conception and design of research; S.F., M.M.M., I.M.L., and N.A.F. performed experiments; S.F., I.M.L., and N.A.F. analyzed data; S.F., I.M.L., and N.A.F. interpreted results of experiments; S.F., M.M.M., I.M.L., and N.A.F. edited and revised manuscript; S.F., M.M.M., I.M.L., and N.A.F. approved final version of manuscript; N.A.F. prepared figures; N.A.F. drafted manuscript.
REFERENCES
- 1. Abman SH, Chatfield BA, Rodman DM, Hall SL, McMurtry IF. Maturational changes in endothelium-derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Physiol Lung Cell Mol Physiol 260: L280–L285, 1991 [DOI] [PubMed] [Google Scholar]
- 2. Aschner JL, Zeng H, Kaplowitz MR, Zhang Y, Slaughter JC, Fike CD. Heat shock protein 90-eNOS interactions mature with postnatal age in the pulmonary circulation of the piglet. Am J Physiol Lung Cell Mol Physiol 296: L555–L564, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bailey SR, Eid AH, Mitra S, Flavahan S, Flavahan NA. Rho kinase mediates cold-induced constriction of cutaneous arteries: role of alpha2C-adrenoceptor translocation. Circ Res 94: 1367–1374, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Bendeck MP, Keeley FW, Langille BL. Perinatal accumulation of arterial wall constituents: relation to hemodynamic changes at birth. Am J Physiol Heart Circ Physiol 267: H2268–H2279, 1994 [DOI] [PubMed] [Google Scholar]
- 5. Boegehold MA. Endothelium-dependent control of vascular tone during early postnatal and juvenile growth. Microcirculation 17: 394–406, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conway D, Schwartz MA. Lessons from the endothelial junctional mechanosensory complex. F1000 Biol Rep 4: 1, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Culver JC, Dickinson ME. The effects of hemodynamic force on embryonic development. Microcirculation 17: 164–178, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis EC. Endothelial cell connecting filaments anchor endothelial cells to the subjacent elastic lamina in the developing aortic intima of the mouse. Cell Tissue Res 272: 211–219, 1993 [DOI] [PubMed] [Google Scholar]
- 9. Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 16: 209–221, 2009 [DOI] [PubMed] [Google Scholar]
- 10. Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflügers Arch 459: 793–806, 2010 [DOI] [PubMed] [Google Scholar]
- 11. Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci 118: 4103–4111, 2005 [DOI] [PubMed] [Google Scholar]
- 12. Goel A, Su B, Flavahan S, Lowenstein CJ, Berkowitz DE, Flavahan NA. Increased endothelial exocytosis and generation of endothelin-1 contributes to constriction of aged arteries. Circ Res 107: 242–251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10: 53–62, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho PC, Melbin J, Nesto RW. Scholarly review of geometry and compliance: biomechanical perspectives on vascular injury and healing. ASAIO J 48: 337–345, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Igarashi J, Bernier SG, Michel T. Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. Differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem 276: 12420–12426, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Igarashi J, Michel T. Sphingosine 1-phosphate and isoform-specific activation of phosphoinositide 3-kinase beta. Evidence for divergence and convergence of receptor-regulated endothelial nitric-oxide synthase signaling pathways. J Biol Chem 276: 36281–36288, 2001 [DOI] [PubMed] [Google Scholar]
- 17. Ishii T, Kuwaki T, Masuda Y, Fukuda Y. Postnatal development of blood pressure and baroreflex in mice. Auton Neurosci 94: 34–41, 2001 [DOI] [PubMed] [Google Scholar]
- 18. Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res 93: 354–363, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Jinguji Y. Developmental stage dependent expression of the endothelial stress fibers and organization of fibronectin fibrils in the aorta of chick embryos. Zoolog Sci 20: 1359–1366, 2003 [DOI] [PubMed] [Google Scholar]
- 20. Kobayashi N, Sakai T. Postnatal reorganization of actin filaments and differentiation of intercellular boundaries in the rat aortic endothelial cells. Cell Tissue Res 278: 471–482, 1994 [DOI] [PubMed] [Google Scholar]
- 21. Kocher O, Skalli O, Cerutti D, Gabbiani F, Gabbiani G. Cytoskeletal features of rat aortic cells during development. An electron microscopic, immunohistochemical, and biochemical study. Circ Res 56: 829–838, 1985 [DOI] [PubMed] [Google Scholar]
- 22. Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72: 463–493, 2010 [DOI] [PubMed] [Google Scholar]
- 23. Lampugnani MG, Dejana E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb Res 120, Suppl 2: S1–S6, 2007 [DOI] [PubMed] [Google Scholar]
- 24. Lampugnani MG, Zanetti A, Breviario F, Balconi G, Orsenigo F, Corada M, Spagnuolo R, Betson M, Braga V, Dejana E. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell 13: 1175–1189, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lemarie CA, Tharaux PL, Lehoux S. Extracellular matrix alterations in hypertensive vascular remodeling. J Mol Cell Cardiol 48: 433–439, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Li M, Chiou KR, Bugayenko A, Irani K, Kass DA. Reduced wall compliance suppresses Akt-dependent apoptosis protection stimulated by pulse perfusion. Circ Res 97: 587–595, 2005 [DOI] [PubMed] [Google Scholar]
- 27. Liu SF, Hislop AA, Haworth SG, Barnes PJ. Developmental changes in endothelium-dependent pulmonary vasodilatation in pigs. Br J Pharmacol 106: 324–330, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci USA 107: 9944–9949, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mata-Greenwood E, Jenkins C, Farrow KN, Konduri GG, Russell JA, Lakshminrusimha S, Black SM, Steinhorn RH. eNOS function is developmentally regulated: uncoupling of eNOS occurs postnatally. Am J Physiol Lung Cell Mol Physiol 290: L232–L241, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol 105: 1652–1660, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest 118: 3355–3366, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakamura H. Electron microscopic study of the prenatal development of the thoracic aorta in the rat. Am J Anat 181: 406–418, 1988 [DOI] [PubMed] [Google Scholar]
- 33. Nandi M, Leiper J, Arrigoni F, Hislop A, Vallance P, Haworth S. Developmental regulation of GTP-CH1 in the porcine lung and its relationship to pulmonary vascular relaxation. Pediatr Res 59: 767–772, 2006 [DOI] [PubMed] [Google Scholar]
- 34. Nowicki PT, Flavahan S, Hassanain H, Mitra S, Holland S, Goldschmidt-Clermont PJ, Flavahan NA. Redox signaling of the arteriolar myogenic response. Circ Res 89: 114–116, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Osawa M, Masuda M, Kusano K, Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? J Cell Biol 158: 773–785, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Safar ME. Hypertension, systolic blood pressure, and large arteries. Med Clin North Am 93: 605–619, Table of Contents, 2009. [DOI] [PubMed] [Google Scholar]
- 37. Safar ME, Levy BI, Struijker-Boudier H. Current perspectives on arterial stiffness and pulse pressure in hypertension and cardiovascular diseases. Circulation 107: 2864–2869, 2003 [DOI] [PubMed] [Google Scholar]
- 38. Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci USA 99: 9462–9467, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sugimoto K, Fujii S, Takemasa T, Yamashita K. Factors inducing codistribution of marginal actin fibers and fibronectin in rat aortic endothelial cells. Am J Physiol Heart Circ Physiol 272: H2188–H2194, 1997 [DOI] [PubMed] [Google Scholar]
- 40. Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 10: 923–934, 2008 [DOI] [PubMed] [Google Scholar]
- 41. Thompson LP, Weiner CP. Acetylcholine relaxation of renal artery and nitric oxide synthase activity of renal cortex increase with fetal and postnatal age. Pediatr Res 40: 192–197, 1996 [DOI] [PubMed] [Google Scholar]
- 42. Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 89: 957–989, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. White CR, Hamade MW, Siami K, Chang MM, Mangalwadi A, Frangos JA, Pearce WJ. Maturation enhances fluid shear-induced activation of eNOS in perfused ovine carotid arteries. Am J Physiol Heart Circ Physiol 289: H2220–H2227, 2005 [DOI] [PubMed] [Google Scholar]
- 45. Williams JM, Hull AD, Pearce WJ. Maturational modulation of endothelium-dependent vasodilatation in ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol 288: R149–R157, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Zellers TM, Vanhoutte PM. Endothelium-dependent relaxations of piglet pulmonary arteries augment with maturation. Pediatr Res 30: 176–180, 1991 [DOI] [PubMed] [Google Scholar]
- 47. Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler Thromb Vasc Biol 26: 1015–1021, 2006 [DOI] [PubMed] [Google Scholar]