

Abstract
The activity of glucose-6-phosphate dehydrogenase (G6PD) appears to control a vascular smooth muscle relaxing mechanism regulated through cytosolic NADPH oxidation. Since our recent studies suggest that thiol oxidation-elicited dimerization of the 1α form of protein kinase G (PKG1α) contributes to the relaxation of isolated endothelium-removed bovine pulmonary arteries (BPA) to peroxide and responses to hypoxia, we investigated whether cytosolic NADPH oxidation promoted relaxation by PKG1α dimerization. Relaxation of BPA to G6PD inhibitors 6-aminonicotinamide (6-AN) and epiandrosterone (studied under hypoxia to minimize basal levels of NADPH oxidation and PKG1α dimerization) was associated with increased PKG1α dimerization and PKG-mediated vasodilator-stimulated phosphoprotein (VASP) phosphorylation. Depletion of PKG1α by small inhibitory RNA (siRNA) inhibited relaxation of BPA to 6-AN and attenuated the increase in VASP phosphorylation. Relaxation to 6-AN did not appear to be altered by depletion of soluble guanylate cyclase (sGC). Depletion of G6PD, thioredoxin-1 (Trx-1), and Trx reductase-1 (TrxR-1) in BPA with siRNA increased PKG1α dimerization and VASP phosphorylation and inhibited force generation under aerobic and hypoxic conditions. Depletion of TrxR-1 with siRNA inhibited the effects of 6-AN and enhanced similar responses to peroxide. Peroxiredoxin-1 depletion by siRNA inhibited PKG dimerization to peroxide, but it did not alter PKG dimerization under hypoxia or the stimulation of dimerization by 6-AN. Thus regulation of cytosolic NADPH redox by G6PD appears to control PKG1α dimerization in BPA through its influence on Trx-1 redox regulation by the NADPH dependence of TrxR-1. NADPH regulation of PKG dimerization may contribute to vascular responses to hypoxia that are associated with changes in NADPH redox.
Keywords: glucose-6-phosphate dehydrogenase, hypoxia, thioredoxin
our laboratory has previously reported evidence for the existence of a relaxing mechanism coordinated by the oxidation of cytosolic NADPH generated by the pentose phosphate pathway (9) that appeared to participate in the responses of bovine coronary arteries and bovine pulmonary arteries (BPA) to hypoxia (10–12). Initial studies investigating the mechanism of relaxation to 6-aminonicotinamide (6-AN) and epiandrosterone (Epi), inhibitors of glucose-6-phosphate dehydrogenase (G6PD), in bovine coronary arteries and BPA detected evidence that cytosolic NADPH oxidation appeared to coordinate multiple processes decreasing the levels and actions of intracellular calcium (9, 11, 12). The inhibition of G6PD by these agents also appeared to lower the detection of reactive oxygen species (ROS) derived from Nox oxidases by decreasing the levels of cytosolic NADPH needed to support ROS generation (9, 10). A novel mechanism of relaxation to peroxide by a cGMP-independent thiol oxidation-elicited dimerization-activation of the 1α form of protein kinase G (PKG1α) was first reported in the aorta and coronary circulation of rats (2). In addition, recent studies, including the use of mice lacking PKG1α or possessing a knockin form of PKG1α that cannot dimerize, provide evidence that this form of PKG is a key mediator of relaxation to peroxide in systemic arteries, with properties of it functioning as an endothelium-derived hyperpolarizing factor (16, 21, 25). We found that the peroxide-elicited PKG dimerization mechanism was also present in BPA (18) and that it participated in responses to hypoxia in both BPA and coronary arteries (20). Since PKG1α dimerization and its potential influence on changes in force elicited by hypoxia occurred under conditions in which the oxidation of NADPH was observed (11, 12, 17, 18), we hypothesized that PKG1α dimerization could be an important factor in the regulation of vascular function affected by changes in cytosolic NADPH redox. As decreased biosynthesis of peroxide also appeared to participate in PKG-regulated responses of BPA to hypoxia (5, 20), it remains to be determined whether cytosolic NADPH is merely changing as a result of altering its consumption by processes generating and/or metabolizing peroxide (e.g., via Nox oxidases and/or glutathione peroxidases + reductases, respectively) or whether changes in NADPH redox are actually participating in controlling the redox status of thiols potentially regulating PKG activation through dimerization.
Systems controlling protein thiol redox in the cytosolic region such as thioredoxin-1 (Trx-1) and glutaredoxin could be hypothesized to have a role in promoting PKG1α dimerization under circumstances in which cytosolic NADPH oxidizes because thioredoxin reductase-1 and glutathione reductase utilize NADPH to maintain Trx-1 and glutathione in their reduced forms (6, 8, 17). Evidence has recently been reported, based on the actions of an inhibitor of thioredoxin reductase, that thioredoxin and thioredoxin reductase function to redox cycle the dimerized form of PKG1α back to the reduced monomeric form of PKG (4, 22). Since thioredoxin reductase-1 utilizes NADPH to maintain Trx-1 in its reduced state (8), we hypothesize that the activities of systems controlling cytosolic NADPH redox, such as G6PD, could also function as an additional physiological mechanism regulating PKG dimerization. The objective of the present study was to examine the potential roles of cytosolic NADPH redox and Trx-1 systems in regulating dimerization-activation of PKG1α in BPA. Most experiments were conducted under hypoxia to minimize the activation of PKG in BPA by peroxide stimulation of cGMP generation by soluble guanylate cyclase (sGC) and by peroxide-elicited PKG1α dimerization that potentially occur under aerobic conditions (20). Gene-targeted small inhibitory RNA (siRNA)-transfected BPA were used to deplete enzymes such as G6PD, Trx-1, and thioredoxin reductase-1 to define their roles in the redox control of PKG1α dimerization, whereas siRNA depletion of PKG1α was used to examine the role of PKG1α in the relaxation of BPA by an inhibitor of G6PD. Organoid culture of BPA with 10 μM 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) for 48 h was used to deplete sGC (18). Changes in both the cGMP-dependent and cGMP-independent mechanisms of PKG activation were evaluated based on changes in PKG-mediated phosphorylation on serine 239 of vasodilator-stimulated phosphoprotein (VASP) (24). Relationships between thiol oxidation-elicited subunit dimerization of PKG1α and changes in force generation under hypoxia were examined by detecting the shift in molecular mass of PKG1α in Western blot analysis resulting from subunit dimerization. The depletion of peroxiredoxin-1 by siRNA was examined to determine whether peroxide metabolism by this protein participated in the dimerization of PKG. Since peroxiredoxin-1 was observed to participate in the actions of peroxide, the effects of depletion of this protein were subsequently used to evaluate whether peroxide contributed to the PKG dimerization elicited by promoting NADPH oxidation by inhibiting G6PD. NADPH levels and NADP/NADPH redox were measured to help evaluate its role in the actions of inhibition of G6PD and peroxide on PKG dimerization.
MATERIALS AND METHODS
Materials.
All physiological buffers were prepared with analytical grade reagent salts purchased from J. T. Baker Chemical, and all other chemicals were obtained from Sigma Chemical unless stated otherwise. PKG1α and Trx-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). VASP and thioredoxin reductase-1 antibodies were purchased from Cell Signaling (Beverly, MA). G6PD antibody was purchased from Abcam (Cambridge, MA). Peroxiredoxin-1 antibody (LF-PA0086) was obtained from Abfrontier. Transfection reagents and siRNAs were purchased from Qiagen (Valencia, CA) and Ambion (Austin, TX). All gases were obtained from Tech Air (White Plains, NY).
Tissue preparations.
Bovine lungs were obtained from a slaughterhouse in ice-cold physiological buffered saline. The second and third branches of main lobar pulmonary artery were used for BPA studies (18). Pulmonary arteries were cleaned of their connective tissue and cut into rings of 2- to 3-mm diameter and width. Endothelium was removed by rubbing the lumen. Fresh and organoid-cultured blood vessel rings were used in studies for vascular reactivity and Western blot protein analysis. Organoid cultures were performed (18) with BPA rings in the absence and presence of 10 μM ODQ with Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and 1% antibiotics (penicillin, streptomycin, amphotericin B) for 48 h at 37°C with 5% CO2.
siRNA transfection.
The transfection protocol used in our previous study (20) was slightly adapted to incubate 0.3 pg of siRNA for the first 24 h in DMEM at 37°C with 5% CO2 with the Qiagen TransMessenger Transfection Reagent Kit with a ratio of enhancer R, siRNA, and TransMessenger transfection reagent of 1:3:2.5. Each ring was transfected in a separate well of a sterile 96-well plate with a final volume of 250 μl of the transfection medium. After 24 h the rings were moved to another plate containing DMEM with 1 g/l glucose, 10% fetal bovine serum, and antibiotics (penicillin, streptomycin, Fungizone) for a further 24 h at 37°C with 5% CO2.
Sequences of siRNA were as follows: PKG1α siRNA: sense 5′-GGACAGGACUUAUCAAGCAtt-3′, antisense 3′-UGCUUGAUAAGUCCUGUCCtc-5′; G6PD siRNA: sense 5′-CGCACAUCUUUAUCAUCAUtt-3′, antisense 3′-AUGAUGAUAAAGAUGUGCGta-5′; Trx-1 siRNA: sense 5′-GCCUUUACCUAGUUUUCAAtt-3′, antisense 3′-UUGAAAACUAGGUAAAGGCat-5′; thioredoxin reductase-1 siRNA: sense 5′-GGAUUAAGGCAACAAAUAAtt-3′, antisense: 5′-UUAUUUGUUGCCUUAAUCCta-3′; peroxiredoxin-1 siRNA: sense 5′-CCUUCGACAGAUCACCAUAtt-3′, antisense 5′-UAUGGUGAUCUGUCGAAGGat-3′; scrambled siRNA: sense 5′-UUCUCCGAACGUGUCACGUtt-3′, antisense: 3′-ACGUGACACGUUCGGAGAAtt-5′.
Vascular reactivity.
Endothelium-removed artery rings were mounted on Grass (FT-03) or Coulborne Instruments force displacement transducers for recording isometric force development through the Powerlab data acquisition system from ADInstruments, as previously described (18). Arterial rings were incubated in Krebs-bicarbonate buffer (in mM: 118 NaCl, 4.7 KCl, 1.5 CaCl2, 25 NaHCO3, 1.1 MgSO4, 1.2 KH2PO4, and 5.6 glucose) for 1 h under resting tension of 5 g. In all studies, arterial rings were depolarized with 123 mM KCl-containing Krebs-bicarbonate buffer and then the rings were equilibrated with Krebs-bicarbonate buffer for 30 min. In the initial studies investigating vascular reactivity to G6PD inhibitors 6-AN and Epi, fresh BPA were precontracted with 20 mM potassium (20K) under aerobic conditions. Rings were subsequently exposed to hypoxia by changing the gassing from 21% O2-5% CO2-74% N2 to 5% CO2-95% N2 (Po2 ∼8–10 Torr), and 1 mM 6-AN or 0.5 mM Epi was added once force reached a steady state. For PKG1α, peroxiredoxin-1, and Trx-1 siRNA-transfected or chronic ODQ-treated BPA, arterial rings were precontracted with 25 mM KCl under aerobic conditions before switching to hypoxia, and 1 mM 6-AN or 0.1 mM H2O2 was added. For G6PD, peroxiredoxin-1, Trx-1, and thioredoxin reductase-1 siRNA-transfected BPA, arterial rings were precontracted with 25 mM KCl under aerobic conditions before switching to hypoxia. Rings were flash frozen during vascular reactivity studies for Western blot analysis with liquid nitrogen under hypoxic conditions.
Western blot analysis.
Frozen arterial rings were pulverized and then homogenized in lysis buffer containing protease and phosphatase inhibitors, as previously described (18). Maleimide (100 mM) was included in the lysis buffer to alkylate the thiols to avoid artifactual disulfide bond formation during homogenization (2). A protein quantification assay was performed with the Bradford method, and samples were prepared for gel electrophoresis. Proteins were separated with 10% SDS-polyacrylamide gel. Thiol-reducing conditions were avoided in the samples analyzed for PKG1α dimer. Gels were transferred to PVDF membranes, the membranes were blocked with Tris-buffered saline with Tween 20 + 5% milk for 1 h, and then the membranes were exposed to primary and secondary antibodies. Protein bands were visualized with an enhanced chemiluminescence kit (Pierce) on X-OMAT autoradiography paper (Kodak). Relative changes in PKG1α monomer and dimer forms are reported as percentage of the total PKG1α. Changes in PKG1α monomer and dimer expression were quantified after normalization to β-actin, and phosphorylated VASP was normalized to total VASP in each individual artery studied because the interventions used did not alter total VASP expression. Changes in VASP phosphorylation are reported as the percentage of a specific control condition for each experimental protocol that is defined as 100%. Protein levels were measured by densitometry analysis with UN-SCAN-IT gel software by Silk Scientific. Molecular masses of PKG monomer and dimer are 75 kDa and 150 kDa, respectively.
NADPH and NADP-to-NADPH ratios.
A NADP/NADPH Quantitation Kit (catalog no. K347-100) obtained from Biovision was used to measure NADPH and NADP-to-NADPH ratio (NADP/NADPH) in frozen pulverized arterial rings. The arterial rings were flash frozen during vascular reactivity studies under the conditions described in results.
Statistical analysis.
Data values are means ± SE of the number of arterial segments (n) from different animals. Statistical analyses between two groups were performed with paired and unpaired Student's t-test, and a one-way ANOVA with Newman-Keuls correction was used for comparison between multiple groups. A value of P < 0.05 was used to establish statistical significance.
RESULTS
Inhibitors of G6PD promote relaxation of BPA associated with increased dimerization and PKG1α activity.
BPA were precontracted with 20 mM potassium under aerobic conditions before exposure to hypoxia by changing the gassing in the tissue baths from 21% O2-5% CO2-74% N2 to 5% CO2-95% N2 (Po2 ∼8–10 Torr). Under these hypoxic conditions, 1 mM 6-AN (Fig. 1A) and 0.5 mM Epi (Fig. 2A) promoted 60–75% relaxation of the force generated by 20 mM KCl. 6-AN and Epi caused increased disulfide dimerization of PKG1α (Fig. 1B, Fig. 2B), and phosphorylation of VASP was also increased in the presence of 6-AN and Epi (Fig. 1C, Fig. 2C).
Fig. 1.
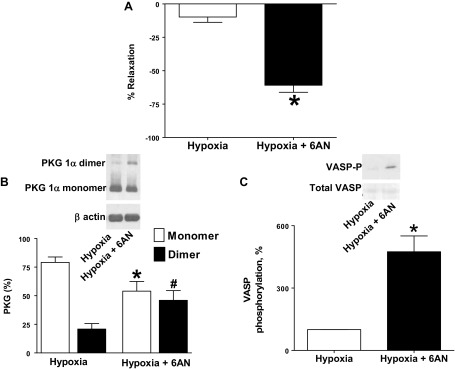
Treatment of bovine pulmonary arteries (BPA) with glucose-6-phosphate dehydrogenase (G6PD) inhibitor 6-aminonicotinamide (6-AN) promotes relaxation, disulfide-mediated dimerization of protein kinase G (PKG)1α, and vasodilator-stimulated phosphoprotein (VASP) phosphorylation. A: 1 mM 6-AN elicits relaxation of BPA precontracted with 20 mM KCl under hypoxia. Data are reported for the change in force under hypoxia in the absence and presence of 6-AN as % relaxation of total force generated by 20 mM KCl under hypoxia (n = 10). B: typical Western blot analysis and summary data showing 1 mM 6-AN-mediated dimerization of PKG1α (n = 10). C: summary data showing that 1 mM 6-AN promoted increased VASP phosphorylation by PKG under the hypoxic conditions studied (n = 7). *P < 0.05 vs. hypoxia in absence of 6-AN; #P < 0.05 vs. hypoxia-dimer.
Fig. 2.
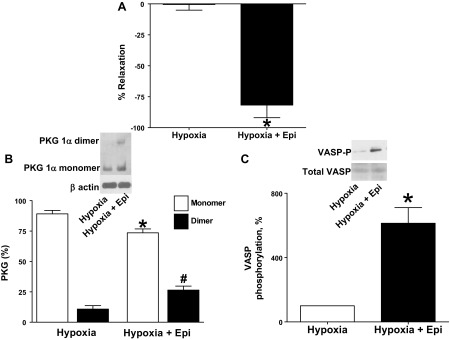
Treatment of BPA with G6PD inhibitor epiandrosterone (Epi) promotes relaxation, disulfide-mediated dimerization of PKG1α, and VASP phosphorylation. A: 0.5 mM Epi elicits relaxation of BPA contracted with 20 mM KCl under hypoxia. Data are reported for the change in force under hypoxia in the absence and presence of Epi as % relaxation of total force generated by 20 mM KCl under hypoxia (n = 10). B: typical Western blot analysis and summary data showing 0.5 mM Epi-mediated dimerization of PKG1α (n = 12). C: summary data showing that 0.5 mM Epi promoted increased VASP phosphorylation by PKG under the hypoxic conditions studied (n = 10). *P < 0.05 vs. hypoxia in absence of Epi; #P < 0.05 vs. hypoxia-dimer.
Effects of siRNA knockdown of PKG1α in BPA on relaxation and alterations in PKG1α dimerization and PKG activity elicited by G6PD inhibitor 6-AN.
Transfection of BPA for 48 h with siRNA for PKG1α resulted in decreased PKG1α monomer and dimer protein expression (20). PKG1α siRNA-transfected BPA were precontracted with 25 mM potassium under aerobic conditions before exposure to hypoxia by changing the gassing in the tissue baths from 21% O2-5% CO2-74% N2 to 5% CO2-95% N2 (Po2 ∼8–10 Torr), and 1 mM 6-AN was added. PKG1α siRNA-transfected BPA demonstrated decreased relaxation to 6-AN (Fig. 3C). PKG1α dimerization (Fig. 3A) and VASP phosphorylation (Fig. 3B) were decreased in PKG1α siRNA-transfected BPA compared with scrambled siRNA control. Relaxation to 6-AN did not differ in BPA organoid cultured in the absence and presence of 10 μM ODQ under conditions previously documented (12) to deplete sGC by ∼85% (Fig. 3D).
Fig. 3.
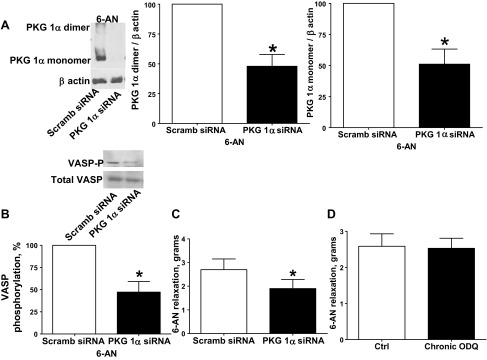
Effects of small inhibitory RNA (siRNA) knockdown of PKG1α in BPA on 1 mM 6-AN-elicited levels of PKG1α dimer and monomer and VASP phosphorylation. A and B: Western blot analysis showing that PKG1α siRNA transfection for 48 h decreased expression of PKG1α monomer and dimer subunits in the presence of 6-AN (n = 6; A) and VASP phosphorylation is decreased by PKG1α siRNA transfection compared with scrambled siRNA controls (B). C: with PKG1α siRNA transfection (n = 6), relaxation to 6-AN is attenuated in BPA precontracted with 25 mM KCl (n = 8). D: soluble guanylate cyclase (sGC) depletion by organoid culture with 10 μM 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) did not alter the relaxation to 6-AN (n = 7). *P < 0.05 vs. scrambled siRNA control response.
siRNA knockdown of G6PD in BPA decreased force generation to 25 mM KCl and increased PKG1α dimerization and PKG activity under hypoxia.
G6PD siRNA transfection of BPA for 48 h resulted in decreased G6PD protein expression (Fig. 4A). G6PD siRNA-transfected BPA demonstrated decreased force generation to 25 mM KCl under both aerobic and hypoxic conditions (Fig. 4B) compared with the scrambled siRNA control. PKG1α dimerization (Fig. 4C) and VASP phosphorylation (Fig. 4D) were increased under hypoxia in BPA transfected with siRNA for G6PD.
Fig. 4.
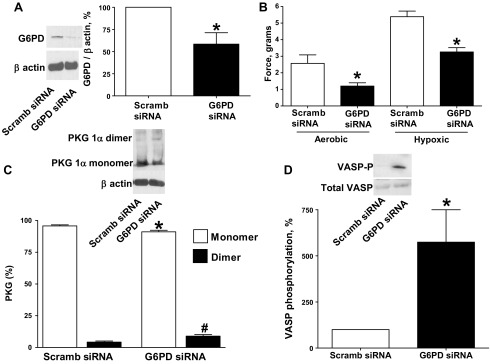
Effects of siRNA knockdown of G6PD in BPA on force generation to 25 mM KCl under aerobic and hypoxic conditions and PKG1α dimerization and VASP phosphorylation under hypoxia. A: Western blot analysis showing that G6PD siRNA transfection for 48 h decreased expression of G6PD (n = 6). B: with G6PD siRNA transfection, contraction to 25 mM potassium under aerobic and hypoxic conditions is attenuated (n = 7) compared with scrambled siRNA controls. C and D: Western blot analysis showing that G6PD siRNA transfection for 48 h increased the observed level of PKG1α dimerization (n = 7; C) and VASP phosphorylation (n = 7; D) under hypoxia compared with scrambled siRNA controls. *P < 0.05 vs. scrambled siRNA control response; #P < 0.05 vs. scrambled siRNA control dimerization response.
siRNA knockdown of Trx-1 in BPA decreased force generation to 25 mM KCl and increased PKG1α dimerization and PKG activity under hypoxia.
Trx-1 siRNA transfection of BPA for 48 h resulted in decreased Trx-1 protein expression (Fig. 5A). Trx-1 siRNA-transfected BPA demonstrated decreased force generation to 25 mM KCl under both aerobic and hypoxic conditions (Fig. 5B) compared with the scrambled siRNA control. PKG1α dimerization (Fig. 5C) and VASP phosphorylation (Fig. 5D) were increased in BPA transfected with siRNA for Trx-1.
Fig. 5.
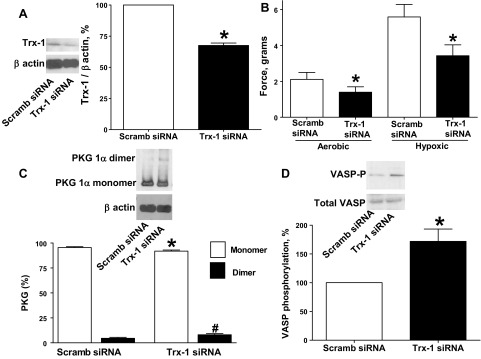
Effects of siRNA knockdown of thioredoxin-1 (Trx-1) in BPA on force generation to 25 mM KCl under aerobic and hypoxic conditions and PKG1α dimerization and VASP phosphorylation under hypoxia. A: Western blot analysis showing that Trx-1 siRNA transfection for 48 h decreased expression of Trx-1 (n = 5). B: with Trx-1 siRNA transfection, contraction to 25 mM potassium under aerobic and hypoxic conditions is attenuated (n = 7) compared with scrambled siRNA controls. C and D: Western blot analysis showing that Trx-1 siRNA transfection for 48 h increased the observed level of PKG1α dimerization (n = 7; C) and VASP phosphorylation (n = 7; D) under hypoxia compared with scrambled siRNA controls. *P < 0.05 vs. scrambled siRNA control response; #P < 0.05 vs. scrambled siRNA control dimerization response.
siRNA knockdown of thioredoxin reductase-1 in BPA decreased force generation to 25 mM KCl and increased PKG1α dimerization and PKG activity under hypoxia.
Thioredoxin reductase-1 siRNA transfection of BPA for 48 h resulted in decreased thioredoxin reductase-1 protein expression (Fig. 6A). Thioredoxin reductase-1 siRNA-transfected BPA demonstrated decreased force generation to 25 mM KCl under both aerobic and hypoxic conditions (Fig. 6B) compared with the scrambled siRNA control. PKG1α dimerization (Fig. 6C) and VASP phosphorylation (Fig. 6D) were increased in BPA transfected with siRNA for thioredoxin reductase-1.
Fig. 6.
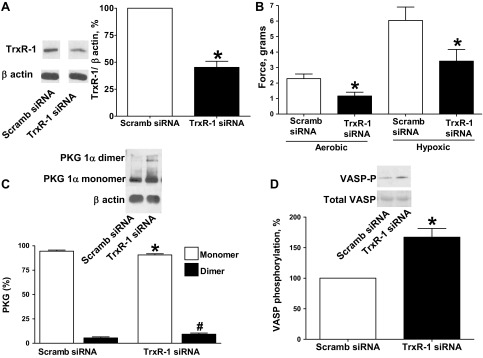
Effects of siRNA knockdown of thioredoxin reductase-1 (TrxR-1) in BPA on force generation to 25 mM KCl under aerobic and hypoxic conditions and PKG1α dimerization and VASP phosphorylation under hypoxia. A: Western blot analysis showing that TrxR-1 siRNA transfection for 48 h decreased expression of TrxR-1 (blot on right) compared with scrambled siRNA controls (blot on left from the same Western blot and animal) (n = 7). B: with TrxR-1 siRNA transfection, contraction to 25 mM potassium under aerobic and hypoxic conditions is attenuated (n = 7) compared with scrambled siRNA controls. C and D: Western blot analysis showing that TrxR-1 siRNA transfection for 48 h increased the observed level of PKG1α dimerization (n = 8; C) and VASP phosphorylation (n = 8; D) under hypoxia compared with scrambled siRNA controls. *P < 0.05 vs. scrambled siRNA control response; #P < 0.05 vs. scrambled siRNA control dimerization response.
Effects of siRNA knockdown of thioredoxin reductase-1 in BPA on relaxation and alterations in PKG1α dimerization and PKG activity elicited by G6PD inhibitor 6-AN and H2O2.
Thioredoxin reductase-1 siRNA-transfected BPA were precontracted with 25 mM potassium under aerobic conditions before exposure to hypoxia by changing the gassing in the tissue baths from 21% O2-5% CO2-74% N2 to 5% CO2-95% N2 (Po2 ∼8–10 Torr) and the subsequent addition of 1 mM 6-AN or 0.1 mM H2O2. Thioredoxin reductase-1 siRNA-transfected BPA showed an attenuation of the observed relaxation to 6-AN (Fig. 7A). PKG1α dimerization (Fig. 7B) and VASP phosphorylation (Fig. 7C) in the presence of 6-AN were also attenuated in thioredoxin reductase-1 siRNA-transfected BPA compared with the scrambled siRNA control. In contrast, siRNA-transfected BPA showed an enhancement of the observed relaxation to H2O2 (Fig. 8A). PKG1α dimerization (Fig. 8B) and VASP phosphorylation (Fig. 8C) in the presence of peroxide were also enhanced in thioredoxin reductase-1 siRNA-transfected BPA compared with the scrambled siRNA control.
Fig. 7.
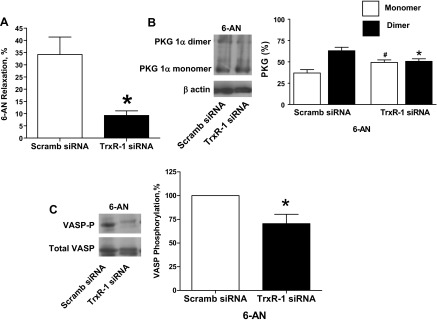
Effects of siRNA knockdown of TrxR-1 in BPA on 1 mM 6-AN-elicited relaxation (A), levels of PKG1α dimer and monomer (B), and VASP phosphorylation (C). A: TrxR-1 siRNA transfection for 48 h attenuated the relaxation to 6-AN (n = 12) compared with 6-AN-treated scrambled siRNA controls. B and C: Western blot analysis showing that the 6-AN-elevated levels of PKG1α dimerization (n = 7; B) and VASP phosphorylation (n = 6; C) observed in Fig. 1 are also attenuated by siRNA depletion of TrxR-1 compared with 6-AN treated scrambled siRNA controls. *P < 0.05 vs. 6-AN-treated scrambled siRNA control response; #P < 0.05 vs. scrambled siRNA control PKG monomer.
Fig. 8.
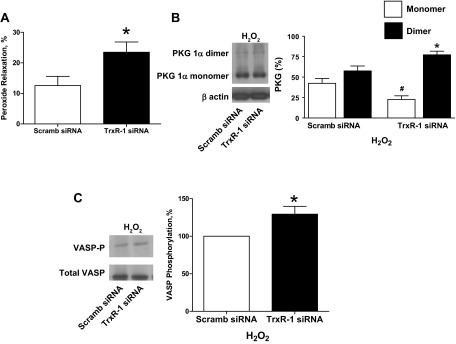
Effects of siRNA knockdown of TrxR-1 in BPA on 0.1 mM H2O2-elicited relaxation and levels of PKG1α dimer and monomer and VASP phosphorylation under hypoxia. A: TrxR-1 siRNA transfection for 48 h enhanced the relaxation to H2O2 (n = 6) compared with H2O2-treated scrambled siRNA controls. B and C: Western blot analysis showing that the increased levels of PKG1α dimerization (n = 6; B) and elevated levels of VASP phosphorylation (n = 6; C) observed with H2O2 (18) are further increased by siRNA depletion of TrxR-1 compared with H2O2-treated scrambled siRNA controls. *P < 0.05 vs. H2O2-treated scrambled siRNA control response; #P < 0.05 vs. scrambled H2O2-treated siRNA control PKG monomer.
siRNA knockdown of peroxiredoxin-1 in BPA did not alter force generation to 25 mM KCl, PKG1α dimerization, and PKG activity under hypoxia.
Peroxiredoxin-1 siRNA transfection of BPA for 48 h resulted in decreased peroxiredoxin-1 protein expression (Fig. 9A). Peroxiredoxin-1 siRNA-transfected BPA did not show altered force generation to 25 mM KCl under both aerobic and hypoxic conditions (Fig. 9B) compared with the scrambled siRNA control. PKG1α dimerization (Fig. 9C) and VASP phosphorylation (Fig. 9D) were not altered in BPA transfected with siRNA for peroxiredoxin-1.
Fig. 9.
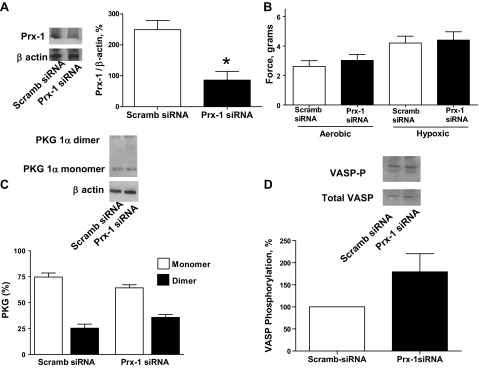
Effects of siRNA knockdown of peroxiredoxin-1 (Prx-1) in BPA on force generation to 25 mM KCl under aerobic and hypoxic conditions and PKG1α dimerization and VASP phosphorylation under hypoxia. A: Western blot analysis showing that Prx-1 siRNA transfection for 48 h decreased expression of Prx-1 (n = 7). B: with Prx-1 siRNA transfection, contraction to 25 mM potassium under aerobic and hypoxic conditions is not attenuated (n = 12) compared with scrambled siRNA controls. C and D: Western blot analysis showing that Prx-1 siRNA transfection for 48 h increased the observed level of PKG1α dimerization (n = 7; C) and VASP phosphorylation (n = 8; D) under hypoxia compared with scrambled siRNA controls. *P < 0.05 vs. scrambled siRNA control response.
Effects of siRNA knockdown of peroxiredoxin-1 in BPA on relaxation and alterations in PKG1α dimerization and PKG activity elicited by G6PD inhibitor 6-AN and H2O2.
Peroxiredoxin-1 siRNA-transfected BPA were precontracted with 25 mM potassium under aerobic conditions before exposure to hypoxia by changing the gassing in the tissue baths from 21% O2-5% CO2-74% N2 to 5% CO2-95% N2 (Po2 ∼8–10 Torr) and the subsequent addition of 1 mM 6-AN or 0.1 mM H2O2. Peroxiredoxin-1 siRNA-transfected BPA did not show an alteration in the observed relaxation to 6-AN (Fig. 10A). PKG1α dimerization (Fig. 10B) and VASP phosphorylation (Fig. 10C) in the presence of 6-AN were also not altered in peroxiredoxin-1 siRNA-transfected BPA compared with the scrambled siRNA control. In contrast, siRNA-transfected BPA showed an inhibition of the observed relaxation to H2O2 (Fig. 11A). PKG1α dimerization (Fig. 11B) and VASP phosphorylation (Fig. 11C) in the presence of H2O2 were also decreased in peroxiredoxin-1 siRNA-transfected BPA compared with the scrambled siRNA control.
Fig. 10.
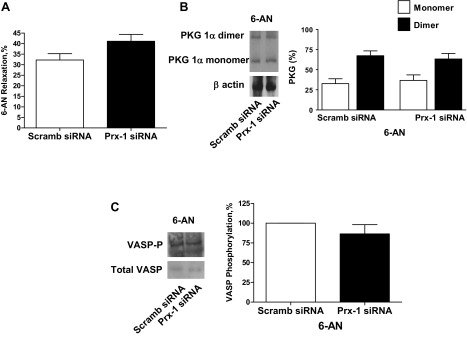
Effects of siRNA knockdown of Prx-1 in BPA on 1 mM 6-AN-elicited relaxation (A), levels of PKG1α dimer and monomer (B), and VASP phosphorylation (C). A: Prx-1 siRNA transfection for 48 h did not alter the relaxation to 6-AN (n = 12) compared with 6-AN-treated scrambled siRNA controls. B and C: Western blot analysis showing that the increases in PKG1α dimerization (n = 7; B) and VASP phosphorylation (n = 6; C) observed in Fig. 1 with 6-AN are also not altered compared with scrambled siRNA controls treated with 6-AN.
Fig. 11.
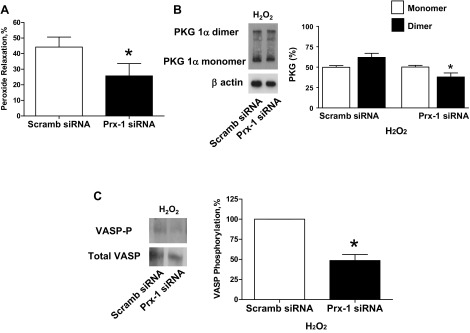
Effects of siRNA knockdown of Prx-1 in BPA on 0.1 mM H2O2-elicited relaxation (A), levels of PKG1α dimer and monomer (B), and VASP phosphorylation (C) under hypoxia. A: Prx-1 siRNA transfection for 48 h inhibited the relaxation to H2O2 (n = 8) compared with H2O2-treated scrambled siRNA controls. B and C: Western blot analysis showing that the increased levels of PKG1α dimerization (n = 7; B) and VASP phosphorylation (n = 7; C) observed with H2O2 (18) are also attenuated compared with H2O2 treated scrambled siRNA controls. *P < 0.05 vs. H2O2-treated scrambled siRNA control response.
Effects of 6-AN and H2O2 on NADPH levels and NADP-to-NADPH ratios in BPA under hypoxia.
NADPH levels and NADP/NADPH were measured to document how they are altered by inhibition of G6PD with 1 mM 6-AN and by 0.1 mM H2O2 under hypoxic conditions. As shown in Fig. 12A, hypoxia increases NADPH compared with aerobic conditions and both 6-AN and H2O2 decrease NADPH under the hypoxic conditions studied. The data in Fig. 12B show that hypoxia caused a decrease in NADP/NADPH, and under these hypoxic conditions both 6-AN and H2O2 caused an increased NADP/NADPH or an oxidation of this pyridine nucleotide.
Fig. 12.

Effect of hypoxia and 1 mM 6-AN and 0.1 mM H2O2 under hypoxia on NADPH levels (A) and NADP-to-NADPH ratios (B) in BPA. *P < 0.05 vs. aerobic control levels; #P < 0.05 vs. hypoxia (n = 8).
DISCUSSION
The present study provides evidence that NADPH redox and the Trx-1 system shown in the model in Fig. 13 appear to participate in the regulation of thiol oxidation-elicited PKG1α dimerization of BPA under the hypoxic conditions examined here. Hypoxic conditions were selected to maximize the levels of cytosolic NADPH in BPA (Ref. 11; Fig. 12) and to minimize the basal levels of PKG1α dimerization and VASP phosphorylation (20). Decreases in cytosolic NADPH resulting from inhibition or depletion of G6PD (Refs. 9, 11; Fig. 12) and siRNA knockdown of Trx-1 and thioredoxin reductase-1 were observed in the present study to promote dimerization of PKG1α associated with increased VASP phosphorylation. Depleting thioredoxin-1 reductase also markedly attenuated relaxation and PKG dimerization and activation caused by further promoting NADPH oxidation by inhibiting G6PD with 6-AN (Fig. 7). These observations suggest that cytosolic NADPH redox can control thiol redox mechanisms influencing PKG1α dimerization through the NADPH dependence of thioredoxin reductase-1 and its regulation of Trx-1 redox. Data in this study did not reveal evidence for the NADPH oxidation mechanism regulating PKG dimerization depending on peroxiredoxin-1 or peroxide metabolism by this enzyme because siRNA depletion of peroxiredoxin-1 did not alter 6-AN-elicited relaxation and PKG dimerization of VASP phosphorylation. In contrast, siRNA depletion of peroxiredoxin-1 attenuated relaxation and PKG dimerization of VASP phosphorylation elicited by H2O2, suggesting that peroxiredoxin-1 has a role in the pathway promoting PKG1α dimerization by exogenous peroxide. In addition, activation of sGC did not appear to participate in the relaxation elicited by inhibiting G6PD with 6-AN because depletion of sGC did not appear to alter this response. Interestingly, as shown by recently observed properties of immunoprecipitation, PKG, G6PD, and thioredoxin reductase may colocalize with each other in BPA (7). Since depletion of PKG1α by siRNA attenuated the relaxing effect of inhibitors of G6PD, activation of PKG1α appears to be a major contributor to the mechanisms involved in relaxation of BPA elicited by promoting cytosolic NADPH oxidation under the conditions examined.
Fig. 13.

Model showing how oxidation of cytosolic NADPH redox resulting from the inhibition of G6PD is hypothesized to utilize the NADPH dependence of TrxR-1 to potentially control thiol oxidation-elicited subunit dimerization-activation of PKG in BPA under the hypoxic conditions examined in the present study. Observations on differences in how PKG dimerization by peroxide appears to depend on Prx-1 vs. inhibition of G6PD depending on TrxR-1 are consistent with both H2O2 and cytosolic NADPH redox having distinct roles in regulating PKG dimerization that are hypothesized to be mediated through their control of Trx-1 redox. The model also illustrates how the hypoxic conditions used in this study potentially decrease basal PKG activity by a combination of increasing cytosolic NADPH by stimulating G6PD activity (11) and decreasing the availability of oxygen needed for a source of intracellular H2O2 generation that appears to stimulate both sGC and PKG dimerization (5, 20) in the cytosolic region. Thus, under these hypoxic conditions, the suppression of cytosolic NADPH generation from NADP+ by inhibitors of G6PD (6-AN and Epi) or depletion of G6PD appears to allow cytosolic NADPH oxidation to promote PKG1α dimerization. The model also contains the potential ways in which H2O2 can promote PKG dimerization considered in discussion, including via its metabolism by Prx-1 and via promoting NADPH oxidation. These are likely to be more important factors under aerobic conditions. e−, NADPH serves as an electron donor for superoxide (O2·−) production by Nox oxidase; VASP-Pi, phosphorylated form of vasodilator-associated protein, an indicator of increased PKG activity.
The findings in the present study are generally consistent with our previous observations in BPA precontracted with KCl suggesting that hypoxia influences vascular function both through decreasing peroxide stimulation of PKG by sGC-derived cGMP (5, 17) and by promoting increases in cytosolic NADPH (11), which could be two factors coordinating the decrease in basal PKG activation that is observed under hypoxia (20). The maintenance of low cGMP levels under the hypoxic conditions studied (5) also minimized the recently identified potential for an attenuation of PKG1α dimerization by cGMP (4). The absence of an effect of siRNA depletion of peroxiredoxin-1 on contraction to 25 mM KCl under aerobic conditions suggests that peroxide metabolism through peroxiredoxin-1 potentially stimulating PKG could not be detected as having a role in the previously reported (5, 20) suppression of force under aerobic conditions that is potentially removed by hypoxia. Thus other factors such as modulation of cytosolic NADPH through its consumption by Nox oxidases for peroxide generation and peroxide metabolism via enzymes such as glutathione peroxidase and reductase and the stimulation of G6PD by hypoxia (11) could contribute to how hypoxia is causing the observed increase in NADPH (Fig. 12) that is associated with the previously reported (20) decrease in PKG1α dimerization. The attenuation of force under hypoxia by inhibition of G6PD and by siRNA knockdown of Trx-1 and thioredoxin reductase-1 and G6PD are observations consistent with the high ratio of cytosolic NADPH to NADP and its control of thioredoxin reductase-1 having a major physiological role in maintaining the high levels of force observed under hypoxic conditions in the pulmonary arteries studied. However, the magnitude of changes in VASP phosphorylation and force generation under hypoxic conditions seemed to vary with the enzyme that was depleted. Thus alterations of systems in addition to PKG could be contributing factors in regulating force when enzymes potentially influencing multiple redox-regulated systems, such as Trx-1 and thioredoxin reductase-1 and G6PD, are depleted.
Peroxiredoxins have been described as a sensors of peroxide in the low micromolar concentration range, because peroxides do not seem to directly react rapidly enough with signaling-associated protein thiols (3, 6). It is possible that peroxiredoxin-1 becomes more important in regulating PKG dimerization when peroxide levels are elevated above the concentrations present under basal aerobic conditions, which have been estimated to be in the nanomolar range in the pulmonary arteries studied (5). Interestingly, early observations of peroxiredoxins forming disulfide complexes with other proteins when cardiac muscle is exposed to oxidant conditions (1, 23) have evolved into recent evidence that peroxiredoxin-1 potentially promotes signaling-associated thiol oxidations by forming transient disulfides with the regulated protein (14). Thus peroxiredoxin-1 could be hypothesized to have a role in directly promoting PKG dimerization once it has been oxidized by peroxide.
The observation that peroxide oxidizes NADPH suggests that NADPH oxidation could be a factor in peroxide promoting PKG dimerization. This could occur through peroxiredoxin-thioredoxin-thioredoxin reductase and/or glutathione peroxidase-glutathione reductase pathways of peroxide metabolism consuming NADPH. While the actual mechanism through which peroxide promotes PKG dimerization is not known, recent reports of the enhancement of this process by an inhibitor of thioredoxin reductase are consistent with thioredoxin and thioredoxin reductase normally functioning to reduce the dimerized form of PKG1α back to its monomeric form (4, 22). The observed enhancement of peroxide responses in BPA depleted of thioredoxin reductase-1 (Fig. 8) is consistent with these previous observations. Since regulation of protein thiol redox by thioredoxin is thought to be a reversible process (6, 8), the oxidation of thioredoxin when thioredoxin reductase or NADPH is depleted could be a factor in promoting PKG dimerization. The observation that thioredoxin-1 depletion, but not peroxiredoxin-1 depletion, regulated basal PKG dimerization suggests that an oxidant process other than peroxide metabolism through peroxiredoxin-1 could be a more dominant factor in promoting thioredoxin oxidation and/or PKG1α dimerization under the hypoxic conditions examined in the present study. If the reversibility of the reactions catalyzed by thioredoxin and thioredoxin reductase is efficient in the region of PKG1α, it may allow the dimerization to behave as if it is in equilibrium with the ratio of cytosolic NADP to NADPH.
Data in the present study show that when cytosolic NADPH was decreased by inhibiting or depleting G6PD activity (9, 11) PKG1α dimerization and VASP phosphorylation were increased under hypoxic conditions where relaxation was observed in BPA precontracted with KCl. Relaxation to G6PD inhibitors was attenuated in BPA depleted of PKG1α by siRNA knockdown, demonstrating that PKG1α-coordinated signaling is involved in the observed relaxation. In addition, siRNA knockdown of G6PD also produced decreased force under hypoxia that was associated with increased PKG1α dimerization and VASP phosphorylation. The depletion of thioredoxin-1 and thioredoxin reductase-1 also decreased aerobic and hypoxic force generation, associated with increases in PKG1α dimerization and VASP phosphorylation. The data in this study show some variability in relationships between changes in force, PKG1α dimerization, and VASP phosphorylation under the conditions studied. Other factors such as cGMP inhibition of PKG dimerization (4), subcellular localization or compartmentalization of signaling (7), observations that small increases in PKG dimerization have been associated with substantial vasodilator responses (22), and alternative actions of peroxide and/or perturbations of the redox systems studied (3, 9) could be contributing factors to the relationships that were observed under baseline hypoxic conditions in the present study. However, the prominent attenuation by siRNA depletion of thioredoxin reductase-1 on relaxation and PKG activation promoted by inhibition of G6PD (Fig. 7) provides evidence that the cytosolic NADPH dependence of thioredoxin reductase-1 has a major role in controlling vascular regulation by PKG dimerization. Overall, the data in the present study together with recent observations of others on the consequences of thioredoxin reductase inhibition (4, 22) support the potential importance of cytosolic NADPH redox in controlling thiol redox mechanisms regulating PKG1α dimerization through its influence on NADPH-dependent thioredoxin reductase-1 activity and thioredoxin in the control of force generation by BPA under hypoxia.
This study provides novel evidence for our hypothesis that cGMP-independent mechanisms promoting PKG1α activation by dimerization are potentially an important factor in the control of contractile function by changes in cytosolic NADPH redox. PKG is a well-established coordinator of relaxation through multiple mechanisms that decrease the levels and actions of intracellular calcium (13, 15). Previous studies document many similarities in mechanisms contributing to vascular relaxation by stimuli of PKG (13, 15) and cytosolic NADPH oxidation (9, 11, 12), including attenuation of rho kinase-mediated inhibition of myosin light chain phosphatase, opening of plasma membrane potassium channels, stimulation of calcium uptake by the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) pump, attenuation of calcium influx mechanisms, and lowering of intracellular calcium. Since hypoxia elicits NADPH increases in BPA (11) and decreases in bovine coronary arteries, changes in cytosolic NADPH redox could be a key factor in controlling the role of regulation by PKG1α dimerization in the vascular oxygen-sensing mechanisms observed in these vascular segments (20) and other systems where hypoxia regulates cytosolic NADPH redox.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL-31069, HL-43023, and HL-66331.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.H.N. and M.S.W. conception and design of research; B.H.N., D.P., and S.K. performed experiments; B.H.N., D.P., S.K., and M.S.W. analyzed data; B.H.N., D.P., S.K., and M.S.W. interpreted results of experiments; B.H.N., D.P., S.K., and M.S.W. prepared figures; B.H.N., S.K., and M.S.W. drafted manuscript; B.H.N., D.P., S.K., and M.S.W. edited and revised manuscript; B.H.N., D.P., S.K., and M.S.W. approved final version of manuscript.
ACKNOWLEDGMENTS
Portions of this study were presented at the 2010 Experimental Biology Meeting in Anaheim, CA (19).
REFERENCES
- 1. Brennan JP, Wait R, Begum S, Bell JR, Dunn MJ, Eaton P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J Biol Chem 279: 41352–41360, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schröder E, Browning DD, Eaton P. Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science 317: 1393–1397, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res 111: 1091–1106, 2012 [DOI] [PubMed] [Google Scholar]
- 4. Burgoyne JR, Prysyazhna O, Rudyk O, Eaton P. cGMP-dependent activation of protein kinase G precludes disulfide activation: implications for blood pressure control. Hypertension 60: 1301–1308, 2012 [DOI] [PubMed] [Google Scholar]
- 5. Burke-Wolin TM, Wolin MS. H2O2 and cGMP may function as an O2 sensor in the pulmonary artery. J Appl Physiol 66: 167–170, 1989 [DOI] [PubMed] [Google Scholar]
- 6. Chae HZ, Oubrahim H, Park JW, Rhee SC, Chock PB. Protein glutathionylation in the regulation of peroxiredoxins: a family of thiol-specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antioxid Redox Signal 16: 506–523, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chettimada S, Rawat DK, Dey N, Kobelja R, Simms Z, Wolin MS, Lincoln TM, Gupte SA. Glc-6-PDH and PKG contribute to hypoxia-induced decrease in contractile phenotype proteins and pulmonary artery contraction. Am J Physiol Lung Cell Mol Physiol 303: L64–L74, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol 287: C246–C256, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G, Koller A, Wolin MS. Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol 285: H2316–H2326, 2003 [DOI] [PubMed] [Google Scholar]
- 10. Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol 288: H13–H21, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Gupte RS, Rawat DK, Chettimada S, Cioffi DL, Wolin MS, Gerthoffer WT, McMurtry IF, Gupte SA. Activation of glucose-6-phosphate dehydrogenase promotes acute hypoxic pulmonary artery contraction. J Biol Chem 285: 19561–19571, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupte SA, Wolin MS. Hypoxia promotes relaxation of bovine coronary arteries through lowering cytosolic NADPH. Am J Physiol Heart Circ Physiol 290: H2228–H2238, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev 86: 1–23, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Jarvis RM, Hughes SM, Ledgerwood EC. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med 53: 1522–1530, 2012 [DOI] [PubMed] [Google Scholar]
- 15. Lincoln TM, Dey N, Sellak H. cGMP-dependent protein kinase signaling mechanisms in smooth muscle: from the regulation of tone to gene expression. J Appl Physiol 91: 1421–1430, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Muller PM, Gnugge R, Dhayade S, Thunemann M, Krippeit-Drews P, Drews G, Feil R. H2O2 lowers the cytosolic Ca2+ concentration via activation of cGMP-dependent protein kinase Iα. Free Radic Biol Med 53: 1574–1583, 2012 [DOI] [PubMed] [Google Scholar]
- 17. Neo BH, Kandhi S, Ahmad M, Wolin MS. Redox regulation of guanylate cyclase and protein kinase G in vascular responses to hypoxia. Respir Physiol Neurobiol 174: 259–264, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neo BH, Kandhi S, Wolin MS. Roles for soluble guanylate cyclase and a thiol oxidation-elicited subunit dimerization of protein kinase G in pulmonary artery relaxation to hydrogen peroxide. Am J Physiol Heart Circ Physiol 299: H1235–H1241, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Neo BH, Kandhi S, Wolin MS. Potential role of NADPH redox in regulating thiol oxidation-elicited subunit dimerization activation of protein kinase G in the relaxation of bovine pulmonary arteries to pentose phosphate pathway inhibitors (Abstract). FASEB J 24: 795.5, 2010 [Google Scholar]
- 20. Neo BH, Kandhi S, Wolin MS. Roles for redox mechanisms controlling protein kinase G in pulmonary and coronary artery responses to hypoxia. Am J Physiol Heart Circ Physiol 301: H2295–H2304, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 18: 286–290, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rudyk O, Prysyazhna O, Burgoyne JR, Eaton P. Nitroglycerin fails to lower blood pressure in redox-dead Cys42Ser PKG1α knock-in mouse. Circulation 126: 287–295, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schröder E, Brennan JP, Eaton P. Cardiac peroxiredoxins undergo complex modifications during cardiac oxidant stress. Am J Physiol Heart Circ Physiol 295: H425–H433, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smolenski A, Bachmann C, Reinhard K, Hönig-Liedl P, Jarchau T, Hoschuetzky H, Walter U. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem 273: 20029–20035, 1998 [DOI] [PubMed] [Google Scholar]
- 25. Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res 110: 471–480, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]