

Abstract
Accumulating evidence indicates that GABA acts beyond inhibitory synaptic transmission and regulates the development of inhibitory synapses in the vertebrate brain, but the underlying cellular mechanism is not well understood. We have combined live imaging of cortical GABAergic axons across time scales from minutes to days with single-cell genetic manipulation of GABA release to examine its role in distinct steps of inhibitory synapse formation in the mouse neocortex. We have shown previously, by genetic knockdown of GABA synthesis in developing interneurons, that GABA signaling promotes the maturation of inhibitory synapses and axons. Here we found that a complete blockade of GABA release in basket interneurons resulted in an opposite effect, a cell-autonomous increase in axon and bouton density with apparently normal synapse structures. These results not only demonstrate that GABA is unnecessary for synapse formation per se but also uncover a novel facet of GABA in regulating synapse elimination and axon pruning. Live imaging revealed that developing GABAergic axons form a large number of transient boutons, but only a subset was stabilized. Release blockade led to significantly increased bouton stability and filopodia density, increased axon branch extension, and decreased branch retraction. Our results suggest that a major component of GABA function in synapse development is transmission-mediated elimination of subsets of nascent contacts. Therefore, GABA may regulate activity-dependent inhibitory synapse formation by coordinately eliminating certain nascent contacts while promoting the maturation of other nascent synapses.
Introduction
Synaptic innervation by GABAergic interneurons in the neocortex is often characterized by specificity and robustness (Huang et al., 2007). For example, basket interneurons selectively target the soma and proximal dendrite of pyramidal neurons, and a single basket cell innervates hundreds of pyramidal neurons with tens of synapses on each target cell (Tamas et al., 1997; Somogyi et al., 1998). Such perisomatic inhibitory innervation contributes to the effective control of the output and synchrony of pyramidal neurons (Bartos et al., 2007; Doischer et al., 2008), but the underlying developmental mechanisms are primarily unknown. During early postnatal weeks, the developing rodent neocortex consists of highly diverse and intermingled cell types. A growing basket interneuron axon therefore explores a highly complex cellular milieu with many potential synaptic partners, yet eventually achieves characteristic perisomatic innervation by early adolescence. How molecular recognition and synaptic activity cooperate to guide GABAergic synapse formation remains poorly understood. At developing excitatory synapses, glutamate transmission coordinates many aspects of synapse development, from activity-dependent validation and maturation to competition and plasticity (Zheng et al., 1994; Wong and Wong, 2001; Zhang and Poo, 2001; Tashiro et al., 2003). Accumulating evidence in the past few years suggests that, similar to glutamate at developing excitatory synapses, GABA may coordinate presynaptic and postsynaptic maturation at inhibitory synapses (Huang, 2009), but the underlying cellular mechanism remains unclear.
A logical approach to study the role of GABA in inhibitory synapse development is to block GABA transmission and examine the consequence. However, such experiments have not been performed, mainly because conventional pharmacological (e.g., GABA receptor antagonists) or genetic (e.g., germline deletion of GABA synthetic enzymes or receptors) (Ji et al., 1999; Patrizi et al., 2008) approaches to block GABA transmission grossly alter circuit activity levels, thus their effects on activity-dependent synapse development would be difficult to interpret. Here we adopted a genetic strategy to completely block either GABA synthesis or release in single GABAergic neurons. GABA is synthesized by two isoforms of glutamic acid decarboxylase (GAD), enzymes GAD67 and GAD65 (Soghomonian and Martin, 1998). In rodent neocortex, GAD67 contributes to ∼90% of the GABA synthesis, whereas GAD65 is responsible for the remaining ∼10% and is primarily localized to a presynaptic terminal (Pinal and Tobin, 1998). GABA is loaded into synaptic vesicles by a single vesicular transporter (vGAT) (McIntire et al., 1997). Using the Cre/loxP system, we deleted either both isoforms of GAD or vGAT in single basket interneurons. These manipulations in sparse neurons should have rather minimal impact on circuit activity levels. In addition, we restricted genetic deletion to specific stages of inhibitory axon and synapse development, thereby circumventing any earlier developmental consequence of GABA blockade (GABAblock). Furthermore, by labeling of GABAblock interneurons and high-resolution imaging, we examined the acute effects of GABA blockade on synapse morphogenesis, the chronic effects on axon extension and the retraction process, and the outcome on synapse density and axon arbor.
Materials and Methods
Mice and DNA constructs.
Gad67flx/flx mice were a gift from Dr. R. Palmiter (University of Washington, Seattle, WA). Gad65+/− mice were a gift from Dr. S. Baekkeskov (University of California, San Francisco, CA). vGATflx/flx mice were a gift from Dr. B. Lowell (Harvard Medical School, Boston, MA). To create a conditional GABA knock-out, Gad67flx/flx and Gad65+/− mice were bred together to generate mice of genotype Gad67flx/flx:Gad65+/−. These animals were then used as breeders to generate Gad67flx/flx:Gad65+/+ and Gad67flx/flx:Gad65−/− animals in the same litter.
PG67-GFP, Cre, Syn-GFP, and tdTomato were described previously (Chattopadhyaya et al., 2007). The Neurexin 1β-SEP construct (Fu and Huang, 2010) was cloned into the PG67 construct.
Slice culture and biolistic transfection.
Slice culture and biolistic transfection were performed as described previously (Chattopadhyaya et al., 2004). Single-cell deletion of both GADs was done in organotypic cultures made from Gad67flx/flx:Gad65−/− mice. Basket cells were biolistically cotransfected with PG67-GFP and PG67-Cre to knockout both GADs by Cre-mediated recombination and label the cell with GFP at the same time. Single-cell deletion of vGAT was done in organotypic cultures made from vGATflx/flx mice using the same method.
Cortical injection of adeno-associated virus and tissue processing.
Postnatal day 18 (P18) mice (of either sex) were anesthetized with an intraperitoneal injection of ketamine/xylazine mixture (0.13 mg/g, 0.01 mg/g body weight). A small hole in the skull was made using a dental drill (Henry Schein), 1 mm anterior from lambda and 4 mm from the midline. The dura was slightly punctured, and virus was delivered by pressure injection using a glass micropipette (tip size, ∼10 μm) attached to a Picospritzer (General Valve). The glass micropipette was lowered to 0.5 mm below the pia surface. To inject virus into the brain, air puffs were delivered (25 psi, 10 ms duration) at a frequency of 0.2–0.4 Hz to inject a virus volume of 0.5–1 μl. The pipette was then held in place for ∼5 min before completely retracting it from the brain. Adeno-associated virus (AAV)-GFP was used for control animals, and AAV-GFP-ires-Cre was used for GABAblock animals.
At P26, AAV-injected mice were anesthetized (sodium pentobarbitone, 6 mg/100 g body weight) and perfused transcardially with 4% paraformaldehyde in phosphate buffer, pH 7.4. Coronal sections (80 μm) were cut from visual cortex using a Vibratome (VT100; Leica). Brain sections were blocked in 10% normal goat serum and 1% Triton X-100. Slices were then immunostained with anti-NeuN (monoclonal, 1:400; Millipore Corporation) and anti-parvalbumin (Pv) antibody (rabbit, 1:1000; Sigma-Aldrich) for analysis of bouton size or anti-GABA (rabbit, 1:1000; Sigma-Aldrich) and Pv (mouse, 1:1000; Sigma-Aldrich) for analysis of GABA levels, followed by Alexa594-conjugated anti-rabbit IgG and Alexa633-conjugated anti-mouse IgG (1: 400; Invitrogen); they were mounted in Vectashield mounting medium (Vector Laboratories).
Immunohistochemistry for synaptic markers.
Organotypic cultures were fixed overnight at 4°C in 4% paraformaldehyde in phosphate buffer, pH 7.4, frozen and thawed in 30% sucrose in PBS, and subjected to immunofluorescence as described above for brain sections. Freeze-thaw is known to increase exposure of antigen and sensitivity of immunostaining in brain tissue. Primary antibodies are used at the following concentrations: vGAT (rabbit antibody to vGAT, 1:200; Synaptic Systems), gephyrin (mouse antibody to gephyrin, 1:400; Synaptic Systems), GAD65 (mouse antibody to Gad65, 1:1000; Millipore Corporation).
Confocal image acquisition and analysis.
Images from confocal planes were acquired with a 63× oil-immersion objective (NA 1.4; Zeiss) using a confocal microscope (LSM 510 or 710; Zeiss). Scans from each channel were collected in multiple track mode and subsequently merged. Care was taken to use the lowest laser power, and no bleedthrough was visible between Alexa594 and Alexa488 channels. For each cell, one to three image stacks in Zoom 1 (134.95 × 134.95 μm) were taken. Images were acquired using the same acquisition parameters for all the samples.
To quantify axon density, we perform a maximum intensity Z-projection and draw a polygon connecting the outermost tips of neurites, creating an outline of the axon arbor in the Z-projection. Axon density is defined as the percentage of GFP+ pixels inside this polygon.
We determined bouton density by measuring the total length of axon and counting the total number of varicosities. Axons were traced, and varicosities were labeled using NeuroLucida software (MicroBrightField). Bouton size was measured by the diameter of a bouton perpendicular to the basket axon using Zeiss LSM software. All quantifications were done blind to the genotype.
Two-photon laser-scanning microscopy.
Live slice cultures were imaged using a custom-built two-photon laser-scanning microscope based on an Olympus laser-scanning microscope (Olympus America). We used a 60× objective (NA 0.9; Olympus) and the Ti-sapphire laser (Chameleon Ultra; Coherent) as the light source. All imaging experiments were performed with 910 nm unless noted otherwise. Fluorescence was detected in whole-field detection mode with a photomultiplier tube (Hamamatsu Corporation). Laser power was adjusted so that additional power failed to reveal previously undetected boutons. Optical sections were collected at 0.5 μm spacing. Slices were kept in a transparent chamber with ACSF (in mm: 2.5 KCl, 1 MgSO4, 2 CaCl2, 25 NaHCO3, 1.25 NaH2PO4, 126 NaCl, and 14 glucose) saturated with 95% O2/5% CO2 and maintained at 35°C (Inline heater; Warner Instruments).
Two-photon microscopy data acquisition and analysis.
Image acquisition was by Fluoview (Olympus). Several time-lapse protocols were used. For observing Syn-GFP formation at equivalent to postnatal day 18 (EP18; P3 plus 15 d in vitro), a two-photon Z-stack was taken every 1 min for 60 min. For examining Syn-GFP puncta dynamics at EP24, a two-photon Z-stack was taken every 15 min for 2 h. For examining axonal filopodia dynamics, a two-photon Z-stack was taken every 10 min for 3 h. Typically, one to two Z-stacks were imaged of each cell (512 × 512, 47 × 47 μm, Z depth of 30–50 μm). The imaged axon areas were 70–150 μm away from the cell body.
Puncta were analyzed by custom-made Matlab applications. Briefly, a projection of 20–30 μm Z-stack of puncta was made for subsequent analysis individually. A look-up table was set by taking into account of the 10% brightest pixels and 10% dimmest pixels. Puncta were thresholded (cut off at 5 times the background using the mean thresholding method) and binerated. The fate of each punctum was tracked in all the time series. Puncta intensity was measured by masking the binerated punctum area back to the grayscale image.
Filopodia were analyzed by tracing the axon arbor and calculating the number of filopodium sites. Filopodia were defined as 0.3–1.5 μm protrusions coming out perpendicular from the axon.
Long-term time-lapse confocol microscopy and analysis.
Images were acquired with a 20× long working distance objective (NA 0.7; Zeiss) on a confocal microscope (LSM 710; Zeiss). Slices were kept in a heated chamber (35°C) with 5% CO2, which was the same condition as in the incubator. Right before the 3 d experiment, slices were transferred into the glass-bottom Petri dishes (number 1.5 glass; MatTek). Scans were collected at a Z interval of 3 μm from the tdTomato channel (Zen software; Zeiss). Care was taken to use as little laser power as possible to minimize the phototoxicity effect. For each cell, every imaging session was <2 min. Only one cell was imaged in one slice. Images were acquired every day for 3 consecutive days. Images were saved as TIFF files.
Images were analyzed with ImageJ. Projections were made, and the StackReg Plug-in (EPFL) was used to align the images so as to identify the dynamics of axon terminals over days. The change of branch length (in micrometers) was measured by the Simple Neurite Tracer Plug-in of ImageJ. An Excel spreadsheet was generated to record each dynamic axon terminal's status over 3 d.
Immunoelectron microscopy.
P26 mice were anesthetized with sodium pentobarbitone (6 mg/100 g body weight, i.p.) and perfused immediately with 100 ml of 0.2% glutaraldehyde and 2% PFA in 0.1 m phosphate buffer, pH7.4. One hour after perfusion, the brains were removed and 50 μm Vibratome (VT100; Leica) sections were cut coronally from the visual cortex. Sections were cryoprotected in 2% glycerol and 20% DMSO in 0.1 m PBS, for 15 min, and freeze-thawed twice in liquid nitrogen. These were then incubated overnight in the primary antibody (1:600 GFP; Millipore Corporation) in PBS at 4°C and for 2 h at room temperature in a biotinylated secondary antibody (1:500 goat anti-rabbit Fab fragment; Jackson ImmunoResearch Laboratories). To reveal the labeling, avidin–biotin–peroxidase complex (ABC Elite; Vector Laboratories) was used for 2 h and incubated in 3,3′-diaminobenzidine tetrachloride (Fluka) and 0.015% hydrogen peroxide. After enhancement, the sections were then washed, postfixed in osmium tetroxide, and embedded in Epon resin (Fluka). Once cured, the regions of interest were localized with the light microscope, and this area was cut away from the section. This was then attached to a blank resin block and serially thin sectioned at a thickness of between 50 and 60 nm. Images were acquired with a Philips CM12 electron microscope with a filament voltage of 80 kV using a digital camera (Megaview 3; SIS). Image series of serial sections were aligned and registered with TrakEM2 (Cardona et al., 2010). Serially reconstructed boutons were rendered in Blender software.
Electrophysiology recording.
Single whole-cell recordings of fluorescent-labeled cells (basket cells) were made with Axopatch 200B or 700B amplifiers (Molecular Devices), using an upright microscope (BX51; Olympus) equipped with infrared-differential interference contrast (IR-DIC) optics and an fluorescence excitation source. Both the IR-DIC image and fluorescence image were captured with a digital camera (Microfire; Optronics). Organotypic slice culture was rapidly moved into the recording chamber and perfused with oxygenated ACSF (in mm: 124 NaCl, 2.5 KCl, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 11 d-glucose, pH 7.35, ∼300 mOsm) at 32–34°C. The internal solution of the recording pipette contained (in mm) 130 K-gluconate, 15 KCl, 10 Na-phosphocreatine, 10 HEPES, 4 ATP-Mg, 0.3 GTP, and 0.3 EGTA, adjusted to pH 7.3 with KOH and to ∼300 mOsm. The pipette resistance was 3–5 MΩ. Signals were recorded and filtered at 2 kHz, digitalized at 10 or 20 kHz (DIGIDATA 1322A; Molecular Devices), and further analyzed using the pClamp 9.0 software (Molecular Devices) for intrinsic properties.
Statistical analysis.
Differences among three groups were assessed with one-way ANOVA on ranks with Dunn's post hoc test for not normally distributed data. Differences between two groups were assessed with Student's t test.
Results
Blockade of GABA release in developing basket interneurons results in higher axon and bouton density
We have developed a cortical organotypic culture system, which recapitulates many aspect of basket interneuron development and allows single-cell labeling and genetic manipulations (Chattopadhyaya et al., 2004). Between EP14 and EP24, basket interneurons undergo extensive axon and synapse growth and begin to achieve their characteristic perisomatic innervation pattern (Chattopadhyaya et al., 2004). We have systematically examined the effects of a progressive reduction of GABA synthesis on axon development by combining germline deletion of Gad65 and conditional deletion of Gad67. Previously, we have shown that germline deletion of Gad65 (i.e., Gad67+/+;Gad65−/−) did not affect axon development (Chattopadhyaya et al., 2007) (Fig. 1a,b,e; p > 0.05; n = 5 for each genotype), but deletion of one or two alleles of Gad67 in single basket interneurons resulted in simpler axon arbor and smaller boutons (Chattopadhyaya et al., 2007). Here we found that a further reduction of GABA levels by removing a Gad65 allele in GAD67−/− basket cells (i.e., Gad67−/−;Gad65+/−) resulted in, similar to Gad67−/−;Gad65+/+, simple axon arbors (Fig. 1c,e; p < 0.05; n = 5 for each genotype). However, a total depletion of GABA in basket cells by deleting both enzymes (i.e., Gad67−/−;Gad65−/− or Gad−/−; Fig. 1d,e) resulted in the opposite phenotype, a twofold increase in axon density over wild-type (WT; i.e., Gad67+/+;Gad65+/+) cells. Indeed, GAD−/− neurons showed exceptionally dense axon arbors as well as substantially higher bouton density (Fig. 1g,j; p < 0.001; n = 5 for each genotype). These phenotypes were unlikely to have resulted from a pathological change of cell identity attributable to GABA depletion because Gad−/− neurons showed normal biophysical properties identical to WT fast-spiking basket interneurons (Fig. 2).
Figure 1.
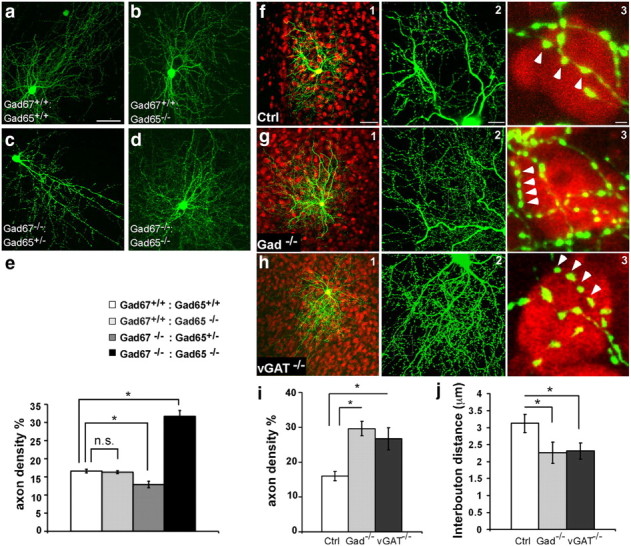
Blockade of GABA release induces basket cell axons and bouton overgrowth. a–d, Comparison of axon arbors in basket cells with different GABA levels at EP24. Basket cells were transfected at EP18. a, A control (Ctrl, Gad67+/+;Gad65+/+) cell with a complex axon arbor. b, A Gad67+/+;Gad65−/− cell showed an axon arbor comparable to control. c, A Gad67−/−;Gad65+/− cell showed a simple axon arbor. d, A Gad67−/−: Gad65−/− cell showed an over exuberant axon arbor. Scale bar, 100 μm. e, Quantification of local axon density in basket cells with different GABA levels. Compared with Ctrl cells, there was no difference in Gad67+/+;Gad65−/− cells (p > 0.05, Mann–Whitney test), a significant decrease in Gad67−/−:Gad65+/+ cells (p < 0.05, Mann–Whitney test), and a highly significant increase in Gad67−/−:Gad65−/− cells (p < 0.001, one-way ANOVA, post hoc Dunn's test; n = 5 cells for each group). f1, A control basket cell (green) at EP24 with an extensive axon arbor that innervates hundreds of nearby pyramidal cells. f2, Axon branches carry prominent presynaptic boutons. f3, Terminal branches innervate pyramidal cell soma with numerous clustered boutons (arrowheads; red, NeuN immunostaining). Scale bars: f1, 100 μm; f2, 10 μm; f3, 2 μm. g, A Gad−/− basket cell shows overall similar arbor size (g1) but increased axon and bouton density (g2) around pyramidal cell soma (g3). h, vGAT−/− basket cells display a similar phenotype as Gad−/− cells. i, Axon density is significantly increased in Gad−/− and vGAT−/− cells compared with controls (n = 10 basket cells for each group; one-way ANOVA, post hoc Dunn's test, p < 0.001). j, Interbouton distances are significantly reduced in Gad−/− and vGAT−/− cells compared with controls (n = 10 basket cells for each group; one-way ANOVA, post hoc Dunn's test, p < 0.001). Asterisks indicate p < 0.05.
Figure 2.

GABA depletion does not alter intrinsic biophysical properties of basket cells. a, A basket cell was identified by GFP expression for patch-clamp recording. b, Protocol for analysis of input resistance. Ra current and Rinput current are measured as shown and Rm = Rinput − Ra. c, Protocol for analysis of action potential (AP). The half-width of the first AP is measured as shown. d, Analysis of fast-spiking adaptation index. Adaptation = last interval/fourth interval. e, Control and Gad−/− basket cells are not significantly different in intrinsic property parameters: Rm, action potential half-width, and fast-spiking adaptation (FS) (n = 6 cells for each group; numbers present mean ± SD; t test, p > 0.05).
To rule out the possibility that the overgrowth of axons and boutons resulted from a nonspecific effect of abolished GABA synthesis (e.g., more accumulation of glutamate), we blocked GABA release in basket cells without disrupting GABA synthesis using slice cultures from the vGATflx/flx mice (Tong et al., 2008). The phenotypes of vGAT−/− and Gad−/− basket cells were nearly identical (Fig. 1h): axon density increased significantly from 16.0 ± 1.3% in the control (Ctrl) cells to 29.6 ± 2.1% in Gad−/− cells and 26.7 ± 3.2% in vGAT−/− cells (Fig. 1i; mean ± SD; p < 0.001; n = 10 cells for each genotype); the average interbouton distance is decreased from 3.1 ± 0.3 μm in the control cells to 2.3 ± 0.4 μm in the Gad−/− cells and 2.4 ± 0.2 μm in the vGAT−/− cells (Fig. 1j; mean ± SD; n = 10 cells for each genotype).
Blockade of GABA release results in smaller and more homogenously sized boutons
In addition to an increase in bouton density, blockade of GABA release resulted in smaller and more similar-sized boutons. The average bouton diameter decreased from 0.806 ± 0.006 μm in controls to 0.568 ± 0.003 μm in Gad−/− and 0.624 ± 0.004 μm in vGAT−/− neurons (Fig. 3a; mean ± SEM; p < 0.001; n = 1000, 100 boutons from 10 cells for each phenotype). The cumulative distribution of bouton size was almost identical between Gad−/− and vGAT−/− but was different from controls (Fig. 3b; Kolmogorov–Smirnov test, p > 0.05 between Gad−/− and vGAT−/−; p < 0.001 between Gad−/− and Ctrl or vGAT−/− and Ctrl; n = 1000, 100 boutons from 10 cells for each phenotype). Notably, the size of GABABlock boutons had a much narrower distribution with a significantly smaller coefficient of variation (Fig. 3c; p < 0.001; n = 1000, 100 boutons from 10 cells for each phenotype).
Figure 3.

Blockade of GABA release in developing basket interneurons results in smaller and more homogenously sized boutons. a, In organotypic cultures, perisomatic bouton size is significantly reduced in Gad−/− and vGAT−/− cells compared with controls (10 cells for each group and 100 boutons from each cell; one-way ANOVA, post hoc Dunn‘s test, p < 0.001). Bar represents mean. Error bars represent SEM. b, The cumulative distribution of bouton size in mutant cells is shifted toward the smaller size (Kolmogorov–Smirnov test, p < 0.001). c, The variance of bouton size is significantly smaller in mutant cells (one-way ANOVA, post hoc Dunn's test, p < 0.001). d, In visual cortex, Gad67flx/flx;Gad65−/− mice and AAV-GFP-ires-Cre-infected basket interneurons (arrowheads; blue, Pv immunostaining) show no GABA immunostaining (red) 7 d after injection compared with neighboring untransfected basket cells (arrows). Scale bar, 20 μm. e, Gad−/− and vGAT−/− cells (in vGATflx/flx mice) in the visual cortex have smaller perisomatic boutons (arrows) compared with control cells. Pyramidal cell somata are labeled by NeuN (red). Note that the interval between boutons appears reduced in these mutant cells. Scale bar, 2 μm. f, Bouton size is significantly reduced in infected Gad−/− and vGAT−/− cells compared with controls (3 animals for each group, 50 boutons from each animal; one-way ANOVA, post hoc Dunn's test, p < 0.001). Bars represent the mean. Error bars represent SEM. g, The cumulative distribution of bouton size in mutant cells is shifted toward the smaller size (Kolmogorov–Smirnov test, p < 0.001). Asterisks indicate p < 0.05.
We extended these studies to adolescent visual cortex in vivo. We used an adeno-associated virus that expresses Cre and GFP (AAV-GFP-ires-Cre) to inactivate either GAD or vGAT by injecting it into the visual cortex of Gad67flx/flx;Gad65−/− mice or vGATflx/flx mice, respectively. AAV-GFP-ires-Cre injected at P18 efficiently deleted the floxed alleles assayed at P26. For example, in the visual cortex of the Gad67flx/flx;Gad65−/− mouse, basket cells (identified by parvalbumin immunoreactivity) no longer contained GABA, whereas basket cells in nearby uninfected visual cortex area expressed GABA immunoreactivity (Fig. 3d). We analyzed perisomatic boutons around pyramidal cells because these boutons were formed primarily by basket cells. In control mice, GFP-labeled basket axon terminals surround NeuN-positive pyramidal cell somata with distinct boutons separated by regular interbouton intervals. Boutons in Gad67flx/flx;Gad65−/− mice and vGATflx/flx mice are smaller and less distinct (Fig. 3e). In the visual cortex of Gad67flx/flx;Gad65−/− mice and vGATflx/flx mice infected with AAV-GFP-ires-Cre, perisomatic boutons are significantly smaller (Fig. 3f; p < 0.001; n = 150 from three animals for each genotype) and more homogeneous in size (Fig. 3g; Kolmogorov–Smirnov test, p < 0.001; n = 150 from three animals for each genotype), similar to organotypic cultures.
GABA transmission is not necessary for inhibitory synapse formation per se
We used several assays and criteria to evaluate whether the supra-numerous boutons in GABAblock basket cells represented synaptic contacts. For presynaptic markers, we examined vGAT in Gad−/− cells and GAD65 in vGAT−/− cells. In control cells, 90.0 ± 4.3% of GFP-labeled boutons colocalized with vGAT(Fig. 4a), and 80.4 ± 3.8% of GFP-labeled boutons colocalized with GAD65 (data not shown). GABAblock cells showed similar colocalization: 84.7 ± 4.4% of boutons in Gad−/− cells contained vGAT (Fig. 4b), and 80.9 ± 6.5% of boutons in vGAT−/− cells contained GAD65 (data not shown) (mean ± SD; p > 0.05; n = 3 cells in each genotype). Analysis of the postsynaptic markers gephyrin and β2 subunit of GABAA receptors shows that both are similarly localized opposed to GABAblock and control boutons. In control cells, 98.8 ± 0.8% of boutons were opposite to gephyrin clusters (Fig. 4c), and 73.9 ± 1.8% of GFP-labeled boutons were opposite to GABAA receptor β2 (Fig. 4e). GABAblock cells showed similar ratios: 99.4 ± 0.4% of boutons in vGAT−/− cells were opposite to gephyrin (Fig. 4d), and 71.7 ± 3.5% of boutons in vGAT−/− cells were opposite to GABAA receptor β2 (Fig. 4f; mean ± SD; p > 0.05; n = 3 cells in each genotype). These results suggest that presynaptic and postsynaptic components are recruited to contact sites independent of GABA release. In addition, we examined the effect of release blockade on the localization of the essential synaptic adhesion molecule neurexin1β. We recently developed a method to detect the membrane-inserted form of neurexin1 and showed that neurexin1β is specifically localized to presynaptic boutons of basket interneurons(Fu and Huang, 2010). In control cells, Nrx1β-SEP (a neurexin1β-synaptopHlorin fusion protein) was often concentrated to a restricted area within a bouton with variable sizes among different boutons (Fig. 4g). In Gad−/− cells, Nrx1β-SEP retained specific localization to axonal boutons but invariably occupied a very small area (Fig. 4h). Indeed, these Nrx1β-SEP patches were significantly smaller and much more homogenous in size (size of 0.025 ± 0.002 versus 0.0168 ± 0.001; mean ± SEM; coefficient of variance, 0.48 ± 0.03 versus 0.35 ± 0.02; mean ± SEM; Student's t test, p < 0.001 for both Nrx1β-SEP size and coefficient of variance; n = 150 from three cells of each genotype). This phenotype is highly concordant with the small and homogenous bouton morphology in Gad−/− cells and suggests that without GABA release, GABAergic boutons retain key synaptic adhesion molecule and weak presynaptic and postsynaptic interactions.
Figure 4.

Release blocked GABAergic boutons represent bona fide synapses. a, Control basket cell presynaptic boutons (green, arrowheads) around a pyramidal cell soma (red) colocalize with the presynaptic marker vGAT (blue). Scale bar, 5 μm. b, Perisomatic boutons (green) from Gad−/− basket cells remain colocalized with vGAT (blue). c, Control presynaptic boutons (circled) are juxtaposed to the postsynaptic marker Gephyrin (red). Scale bar, 1 μm. d, vGAT−/− presynaptic boutons (circled), although much smaller in size, remain juxtaposed to gephyrin. e, Control boutons (green) are juxtaposed (white arrowheads) to postsynaptic GABAA R β2 (red). Scale bar, 1 μm. f, vGAT−/− boutons (green), although much smaller in size, remain juxtaposed to postsynaptic GABAA R β2. g, Expression pattern of neurexin1β-SEP in control axon together with TdTomato. Neurexin1β-SEP puncta localized to subregions within boutons (arrowheads), with substantial variations in puncta size independent of bouton size revealed by tdTomato signals. Scale bar, 1 μm. h, In a Gad−/− axon, neurexin1β-SEP puncta remain localized to boutons (arrowheads) but appear homogenous and tiny in size. i, Syn-GFP puncta are colocalized to presynaptic marker vGAT. Scale bar, 2 μm. j, k, Syn-GFP puncta localize to axonal boutons in a control basket cell (j) (labeled by tdTomato) and in a Gad−/− basket cell (k). Scale bar, 5 μm. Arrowheads point to examples of Syn-GFP puncta that are colocalized with tdTomato-labeled boutons. l, Local reconstruction of control and Gad−/− basket cell axons (red lines) with Syn-GFP puncta (green dots). The average interpuncta distances of Syn-GFP puncta in Gad−/− and vGAT−/− cells are significantly decreased compared with controls (n = 5 cells for each group; p < 0.05, Mann–Whitney test). Scale bar, 5 μm. Asterisks indicate p < 0.05. m, n, Serial EM sections showing a control (m) and Gad−/− (n) basket cell bouton, which forms a symmetric synapse with a postsynaptic cell soma in P26 visual cortex. GFP labeling was conferred by AAV-GFPires-Cre injection into Gad67flx/flx;Gad65−/− mouse cortex from P18 to P26. Both unlabeled (Ctrl) and labeled (Gad−/−, arrow, labeled by dark precipitate from immunogold against GFP) boutons contain misshaped vesicles, typical of GABAergic synapses, which cluster around the active zone (arrow). The synaptic cleft appears typically narrower than those of asymmetric synapses (arrowheads) that are found on nearby dendrites. Scale bar, 500 nm. 3D reconstructions from serial EM images of the same WT and Gad−/− axonal boutons are shown at the bottom, respectively.
Furthermore, we examined the localization of synaptic vesicle components using synaptophysin-GFP (Syn-GFP) fusion protein (Wiedenmann and Franke, 1985; Sudhof et al., 1987; Nakata et al., 1998). Syn-GFP puncta colocalize with inhibitory presynaptic markers such as vGAT (Fig. 4i). Syn-GFP puncta colocalize with GABAblock boutons, identical to their localization in control cells (Fig. 4j,k). Consistent with the bouton density result, the density of Syn-GFP was significantly higher in GABAblock cells (Fig. 4l; one-way ANOVA, post hoc Dunn's test, p < 0.001; n = 5 cells from each genotype). These results indicate that most, if not all, GABAblock boutons contain synaptic vesicles and release machinery.
Finally, we examined the ultrastructure of GABAblock boutons in visual cortex in vivo using electron microscopy. The visual cortex of Gad67flx/flx;Gad65−/− mice or vGATflx/flx mice was infected with AAV-GFP-ires-Cre from P18 to P26. GFP immunoprecipitates (manifested by the darker color) were used to identify GABAblock perisomatic boutons, and nearby GFP-negative perisomatic boutons served as controls. All GFP-positive (i.e., GABAblock) boutons that we observed formed symmetric contacts with typical features of perisomatic inhibitory synapses such as an active zone, synaptic vesicles, and mitochondria (Fig. 4n; 12 boutons from three animals). These GABAblock boutons showed ultrastructural morphology similar to the nearby control boutons (Fig. 4m; 15 boutons from three animals). Together, our results suggest that inhibitory synapse formation per se is independent of GABA transmission.
Increased stability of nascent synaptic boutons after blockade of GABA release
Our experimental system allowed us to examine the acute effect on synapse morphogenesis in the time scale of minutes to hours as well as chronic effects on axon development in the course of several days. Because our genetic blockade of GABA release involved several steps including expression of Cre recombinase (which takes at least 6–12 h), deletion of the Gad67 or vGAT genes, and clearance of existing GAD67 or vGAT proteins [half-life, 4–14 h (Christgau et al., 1991; Rasmussen et al., 2002)], we estimated that it might take 2–3 d from biolistic transfection until GABA blockade to exert an effect on axon and synapse overgrowth.
To examine the acute impact of GABA signaling on synaptogenesis, we transfected basket cells in Gad67flx/flx;Gad65−/− or vGATflx/flx slice cultures with PG67-Cre and PG67 Syn-GFP together at EP16 and imaged the dynamics of Syn-GFP puncta at EP19, a period of extensive axon growth and synapse proliferation (Chattopadhyaya et al., 2004). Because synaptophysin is an integral component of synaptic vesicle, Syn-GFP puncta are thought to represent transmitter release sites; their size and stability likely reflect the maturation status of presynaptic terminals. As the time course of synapse assembly is on the order of tens of minutes (Ruthazer et al., 2006; Sabo et al., 2006), we repeatedly imaged Syn-GFP puncta every minute for 60 min. In control basket cells, we observed frequent and substantial Syn-GFP puncta dynamics (Fig. 5a): puncta appeared, disappeared, and translocated along the axon; their intensity fluctuated significantly over tens of minutes. Although many transient puncta appeared and disappeared during the 1 h imaging session, there was only 1.1 ± 1.8% (mean ± SD; n = 5 cells) puncta net gain at the end of the session. Consistent with this observation, the average interpuncta distance (IPD), calculated by dividing the length of an axon stretch with the total number of puncta along the stretch, was not significantly different between t0 (3.5 ± 0.2 μm) and t60 (3.4 ± 0.2 μm) (Student's t test, p > 0.05; mean ± SEM; n = 5 cells). In control basket cells, Syn-GFP puncta accumulated at many sites throughout axon length but frequently disassembled. It is possible that transient puncta represent weak release sites associated with labile contacts by which developing basket axons explore postsynaptic targets, and the more stable puncta may represent more lasting contacts that would more likely mature into synapses. In Gad−/− cells, on the other hand, Syn-GFP puncta were more stable (Fig. 5b). Puncta detected at t0 mostly persisted through the imaging session, new puncta that appeared often persisted, and the peak intensity of puncta remained stable during the session. There was a 4.4 ± 3.9% (mean ± SD; n = 5 cells) net gain of puncta at the end of the imaging session. The average interpuncta distance, either at t0 (2.5 ± 0.2 μm) or t60 (2.4 ± 0.2 μm), was significantly smaller than that of control cells (Fig. 5d; Student's t test, p < 0.001; n = 5 for each genotype).
Figure 5.

Increased stability of Syn-GFP puncta after blockade of GABA release. a, Dynamics of Syn-GFP puncta along a stretch of control basket cell axon at EP19. Note that puncta appear (green arrows) and disappear (red arrows) over a 60 min period. The profile plot shows that the intensity of signals fluctuates (areas shaded in red), reflecting puncta dynamics. b, Dynamics of Syn-GFP puncta along a stretch of vGAT−/− axon at EP19. Puncta are more stable, and a new one appeared (t40, green arrows). The profile plot along the axon shows that the intensities are relatively stable (shaded in blue), reflecting the more static nature of vGAT−/− puncta. c, The mIPDs in Ctrl, Gad−/−, and vGAT−/− axons are not significantly different (one-way ANOVA, post hoc Dunn's test, p > 0.05). d, The average interpuncta distances are smaller in Gad−/− and vGAT−/− axons than in Ctrl axons at both t0 and t60 (one-way ANOVA, post hoc Dunn's test, p < 0.05). Note that, at EP19, most synGFP puncta are small regardless of genotypes, compared with those of EP24 control cells (Fig. 1f); in fact, synGFP puncta in E19 Gad−/− cells appear larger than in control cells, likely because these puncta are more stable in Gad−/− cells than those that are more mobile in control cells. Asterisks indicate p < 0.05.
To further characterize and compare the dynamic properties of transient puncta between control and Gad−/− cells, we defined a parameter called minimum average interpuncta distance (mIPD). This was calculated first by dividing the length of an axon segment (range, 7–15 μm) with the maximum number of Syn-GFP puncta (i.e., including all transient puncta) that appeared along that segment in a 60 min period; values from different segments were then averaged. Interestingly, mIPD in control (2.7 ± 0.2 μm), Gad−/− (2.5 ± 0.2 μm), and vGAT−/− (2.5 ± 0.2 μm) cells were not significantly different (Fig. 5c; one-way ANOVA, post hoc Dunn's test, p > 0.05; n = 5 for each genotype). This result suggests that axons in each group formed transient contacts at a similar density, which was likely to be an intrinsic property of developing basket axons independent of GABA transmission. Importantly, at either t0 or t60, the average IPD in GABAblock cells, although significantly smaller than that of control cells, was not different from their mIPD (one-way ANOVA, post hoc Dunn's test, p > 0.05; n = 5 for each genotype). These data indicate that the higher Syn-GFP puncta density in GABAblock axons resulted from an enhanced stability of transient puncta at the nascent contact sites. Together, these results suggest that a basket cell axon, while exploring potential postsynaptic partners, forms a large number of transient contacts that recruit synaptic vesicles that release GABA. Many of these transient release sites are quickly disassembled, possibly because of the unfavorable nature of the axon–target interactions. Importantly, GABA transmission is likely necessary to distinguish favorable and unfavorable contacts.
Blockade of GABA transmission suppresses disassembly of Syn-GFP puncta
We also examined the effect of blocking transmission at a later developmental stage when a characteristic pattern of perisomatic innervation begins to emerge (approximately EP24). We transfected basket cells in vGATflx/flx slice cultures with Cre and Syn-GFP at EP21 and imaged the dynamics of puncta at EP24. Two-photon image stacks were collected at a 1 μm Z-interval every 15 min during a 2 h session. Images were projected, thresholded, and binerated to outline and track individual punctum over time. In control cells, 93.0 ± 4.8% of the puncta remained stable over 2 h (Fig. 6a). Whereas 3.0 ± 0.9% new puncta were added, 8.8 ± 4.8% were eliminated (mean ± SD; n = 5 cells, 533 boutons). In vGAT−/− cells, on the other hand, 97.6 ± 1.0% of Syn-GFP puncta were stable (Fig. 6b). Importantly, whereas 8.5 ± 2.0% new puncta were added, only 2.4 ± 1.0% were lost (mean ± SD; n = 5 cells, 684 boutons). Quantification shows that control cells show a higher percentage of loss, whereas vGAT−/− cells have a higher gain of puncta (Student's t test, p < 0.001). Interestingly, both lost and newly formed puncta had lower Syn-GFP intensity compared with more stable puncta (Fig. 6c,d). These data suggest that blockade of GABA transmission may suppress the elimination of weak and transient contact, resulting in increased Syn-GFP puncta stability and density.
Figure 6.

Blockade of GABA release reduces elimination of Syn-GFP puncta in developing basket cell axons. a, Dynamics of Syn-GFP puncta in a Ctrl cell over a 3 h period at EP24. Note that punctum d is lost at t120. Scale bar, 1 μm. b, Dynamics of a Syn-puncta in vGAT−/− cell at EP24. Note that three new puncta appeared over the 3 h period. Puncta 10 and 11 appeared and disappeared (the puncta 10 and 11 intensity dropped below threshold,) and then appeared again. c, d, Plot of puncta intensity fluctuation over time for puncta annotated in the Ctrl (c) and vGAT−/− (d) example. Note that newly formed puncta (j and k, for example) have much lower intensity.
Release blockade increases filopodia extension
In the target innervation area, synapse formation is an integral component of axon arbor growth. Synapse formation not only anchors and stabilizes a newly extended axon branch but also serves as new site of branch extension. To explore the influence of release blockade on basket cell axon growth, we imaged the dynamics of filopodia, which often extend from presynaptic boutons, sample local targets, and form transient contacts during axon development (Niell et al., 2004; Wierenga et al., 2008), and a subset becomes stabilized and mature into axon the branch (Hua et al., 2005). To assess the effect of blocking GABA release on axon growth, we labeled basket cell axons with PG67-GFP or PG67-GFP/ PG67-Cre and examined filopodia dynamics at EP19 (Fig. 7a–c). Time-lapse Z-stack images were taken every 10 min for 3 h. We defined filopodia as processes of 0.3–1.5 μm in length that protrude from presynaptic boutons. These boutons and filopodia were frequently located and dynamic around pyramidal cell soma, the site of perisomatic synapses (data not shown). In GABAblock cells, filopodium density was significantly higher compared with control (Fig. 7d; 0.09 ± 0.01 and 0.08 ± 0.01/μm vs 0.05 ± 0.01/μm; mean ± SD; one-way ANOVA, post hoc Dunn's test, p < 0.05). This result suggests that without transmission, basket cell axons extend more filopodia to search for postsynaptic targets, which might, in part, be the result of the increased number and stability of boutons labeled by Syn-GFP puncta (Fig. 4i). The likelihood of a filopodium to protrude from a bouton (defined by [number of filopodium]/[number of bouton]) was higher in GABAblock cells than in controls (Fig. 7e; p < 0.05; n = 5 cells for each genotype). In addition, more boutons were added in the GABAblock cells than in the control cells (Fig. 7f; one-way ANOVA, post hoc Dunn's test, p < 0.05; n = 5 cells for each genotype) over the 3 h imaging session, which may contribute to the increased bouton density in the GABAblock axons (Fig. 1).
Figure 7.
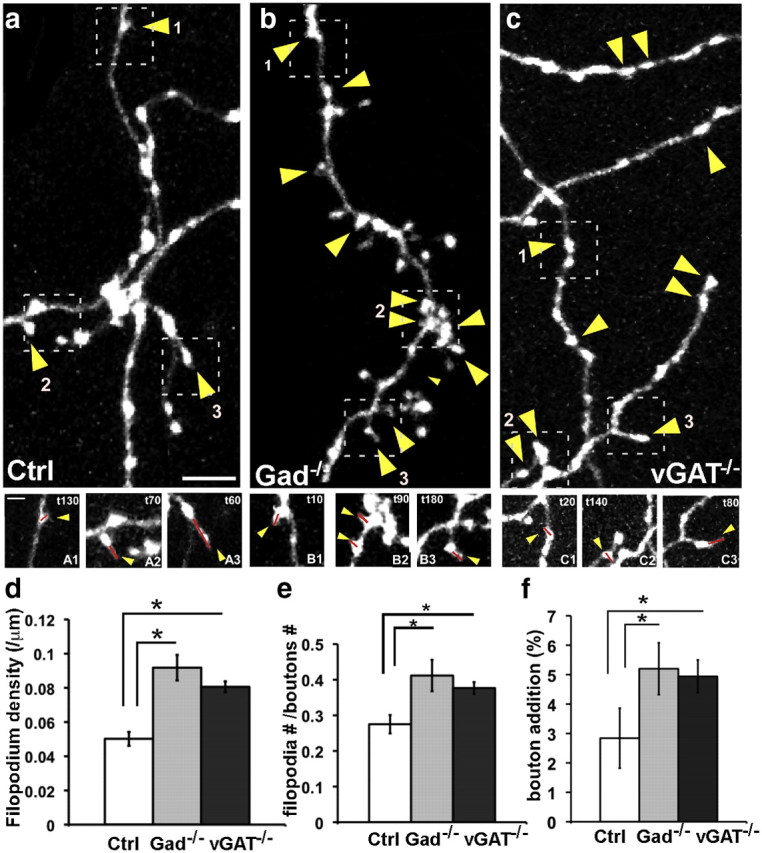
Blockade of GABA release results in more filopodia extension in developing basket cell axons. a–c, Filopodia dynamics in control (a), Gad−/− (b), and vGAT−/− (c) basket cells at EP19. Yellow arrowheads indicate the sites of filopodium extension over a 3 h period. Top, Axon arbor at t0. Scale bar, 5 μm. Bottom (A1–C3), Filopodia extensions at different time points during the imaging sessions. Scale bar, 2 μm. Red lines are drawn to annotate the filopodia. d, Filopodium density is significantly increased in both Gad−/− and vGAT−/− compared wit controls (one-way ANOVA, post hoc Dunn's test, p < 0.05; n = 5 cells for each group). Error bar represents SEM. e, The ratio of boutons bearing filopodia ([number of filopodium]/[number of bouton]) is significantly increased in both Gad−/− and vGAT−/− compared with controls (one-way ANOVA, post hoc Dunn's test, p < 0.05; n = 5 cells for each group). f, The percentage of newly acquired boutons is significantly increased in both Gad−/− and vGAT−/− compared with controls (one-way ANOVA, post hoc Dunn's test, p < 0.05; n = 5 cells for each group). Asterisks indicate p < 0.05.
Blockade of transmission promotes axon extension and suppresses branch retraction
To examine how chronic blockade of GABA transmission results in axon overgrowth, we performed time-lapse imaging of GFP-labeled axons over a 3 d period in organotypic cultures. We tracked the axon arbor structure and followed the fate of each dynamic branch. In total, we characterized 299 branches from 9 control cells and 443 branches from 11 vGAT−/− cells (see examples in Fig. 8a–d). The distribution of the change in branch length appears skewed toward larger changes in the vGAT−/− cells than in the control cells (Fig. 8e). Because the distributions of branch changes do not fit any standard distribution (e.g., normal distribution, gamma distribution, exponential distribution, etc.), to determine whether the distributions of the Ctrl and vGAT−/− cells is statistically significant, we performed a 1000 times bootstrap resampling and calculated the mean, median, skewness, and kurtosis on each resampling. The mean is 0.04 μm in the Ctrl cells versus 6.78 μm in vGAT−/− cells. The median is 0 μm in the Ctrl cells versus 2 μm in vGAT−/− cells. The distributions of skewness and kurtosis obtained by this bootstrap analysis are significantly larger in vGAT−/− cells than in the control cells (Fig. 8e; Student's t test, p ∼ 0; skewness are 0.040 in Ctrl versus 1.318 in vGAT−/−; kurtosis is 5.050 in Ctrl versus 5.965 in vGAT−/−; p = 3.37e−223). To estimate the measurement error, we measured the distance between 20 pairs of fiducial points, defined as markers of unchanged structures, such as two easily identifiable and stable points on the primary dendrite of a basket cell, over 3 d. By plotting the histogram of residual values, we determined that our measurement error is ±2.27 μm (data not shown). We therefore categorized branch changes ≤2.5 μm as “stable.” Branches that changed >2.5 μm were classified into four categories (Sin et al., 2002): (1) “extend,” present on all 3 d and grew longer; (2) “added,” did not exist on day 1; (3) “retracted,” present on all 3 d but became shorter at day 3; (4) “lost,” present on day 1 and then disappeared.
Figure 8.
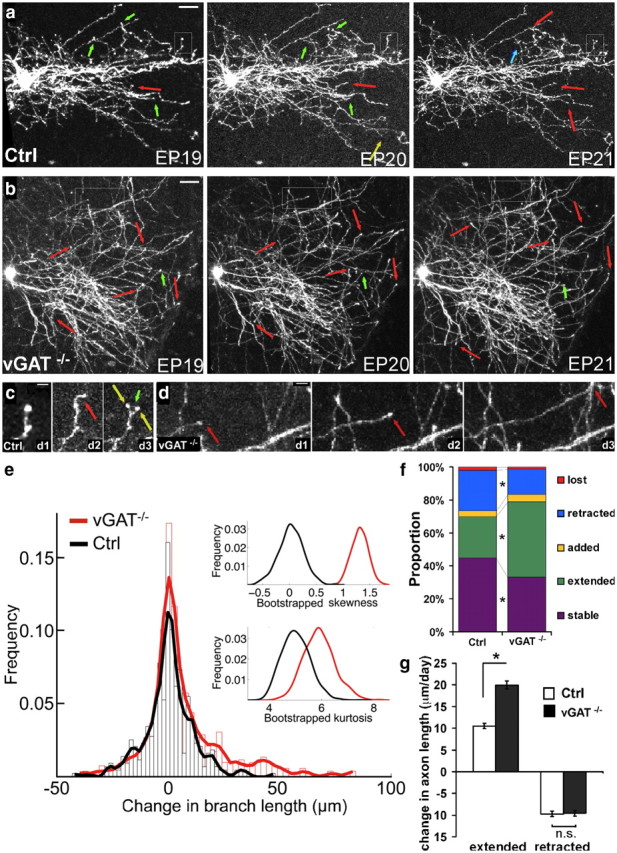
Blockade of GABA release results in reduced branch pruning and increased branch extension. a, From EP19 to EP21, the growth of a basket cell axon arbor involves branch addition (yellow arrows)/extension (red arrows) and loss (green arrows)/retraction (blue arrows). b, vGAT−/− axon arbors show much more extension events (red arrows) and much less loss/retraction (green arrows) events. Scale bar. 50 μm. c, d, Higher-magnification view of the boxed axon terminals in the control (c) and vGAT−/− (d) cells. c, Note that in the control cell, a branch extended (red arrow) in day 2 and then retracted in day 3 (green arrow); another branch was added (yellow arrow) in day3. d, In the vGAT−/− cell, a branch extended (red arrow) in day 2 and then further extended in day 3 (red arrow). Scale bar, 10 μm. e, Histogram showing branch dynamics of Ctrl and vGAT−/− cells with a lowest smoothed line drawn over the bin heights. Each bin contains 50 data points. Insets, Distributions of skewness and kurtosis of the 1000 bootstrapped values. f, The distribution of each of five branch behaviors in control and vGAT−/− cell axons are significantly different (χ2 test, p < 0.001), with twice as many extended branches (green) and half as many retracted (blue) branches in vGAT−/− cells. g, In vGAT−/− cells, the increase in axon length in extended branch group is significantly greater than that in control cells (Student's t test, p < 0.001). However, there is no significance between the reduction in axon length in the retracted branch group compared with that in control cells. Error bars represent SEM. Asterisks indicate p < 0.05.
The proportions of different mode branch dynamics were, overall, rather balanced between growth and retraction in control cells, with 3.5% added, 25.0% extended, 2.0% lost, and 24.6% retracted versus 44.9% stable. vGAT−/− cells, on the other hand, exhibited branch behaviors that significantly favored axon growth: 4.6% added, 45.4% extended, 1.2% lost, and 15.4% retracted versus 33.4% stable (Fig. 8f; χ2 test, p < 0.001). Notably, there was almost a doubling of the proportion of the extended branches and a near-50% reduction in the proportion of the retracted branches in vGAT−/− cells compared with controls. In addition, the average growth rate of the “extended branch” group in vGAT−/− cells was twofold higher than in controls (+20.0 and +10.5 μm/d, respectively), whereas the retraction rate in the “retracted branch” group did not differ between the two cell groups (−9.7 vs −9.6 μm) (Fig. 8g). These data suggest that developing basket cell axons are highly dynamic during EP19–EP21. The overall arbor growth is superimposed after an approximate equilibrium between progressive and regressive events with a subtle bias toward branch addition/extension over branch loss/retraction. Branch addition and extension co-occur with synapse formation (Fig. 9). It has been suggested that branch addition and extension are stabilized by synapse formation in Xenopus motor neuron axon growth (Javaherian and Cline, 2005). Branch loss and retraction likely reflect the exploration of potential synaptic targets and may, in part, result from the elimination of transient or weak contacts with unfavorable targets. Blockade of transmission resulted in more frequent branch extension with a higher growth rate and less frequent branch retraction, which together lead to a higher net growth of axon arbors. These results suggest that basket axon growth is continuously verified through a mechanism that involves GABA transmission, likely at developing synaptic contact sites. In particular, GABA transmission may prune branches bearing inappropriate contacts by promoting the elimination of inappropriate transient synapses.
Figure 9.

Basket cell axon branch growth is accompanied by bouton formation. Two-photon images of the same basket axon terminal branches on 2 consecutive days. From P18 to P19, branch extension (long arrow), branch retraction (short arrow), and bouton formation (arrowheads) are observed. a/b and c/d are from two different basket cells. Note the appearance of numerous distinct boutons along axon branches from c to d. Scale bar, 10 μm.
Discussion
As an integral step of neural circuit assembly, synapse formation is guided by genetic programs and neural activity (Lichtman and Colman, 2000; Zhang and Poo, 2001; Hua and Smith, 2004). As a direct mediator of neural activity, synaptic transmission not only reflects the functional status of synapse maturation but also provides the appropriate spatial and temporal precision to validate or eliminate connections. At the vertebrate neuromuscular junction, although acetylcholine transmission is not necessary for the differentiation of motor axon terminals, it regulates multiple aspects of synapse development including the distribution of synapses in the target field, the number of synaptic sites per target cell, and the number of axons per synaptic site (Misgeld et al., 2002). In the vertebrate CNS, glutamate transmission regulates numerous aspects of excitatory synapse development and plasticity (Zheng et al., 1994; Wong and Wong, 2001; Tashiro et al., 2003; Hua and Smith, 2004; Hua et al., 2005). In the past few years, accumulating evidence indicates that, similar to glutamate at developing excitatory synapses, GABA may coordinate presynaptic and postsynaptic maturation at inhibitory synapses (Huang, 2009). However, the precise role of GABA, and neurotransmitter in general, in distinct steps of CNS synapse formation, such as initial contact, validation, and maturation, remains not well understood.
Although several studies have examined the role of transmitter release in CNS synapse development (Verhage et al., 2000; Varoqueaux et al., 2002), their interpretation has been complicated by the broad effects caused by a germline deletion of components of transmitter release machinery. These manipulations block transmission and eliminate neural activity in the entire CNS and throughout development, leading to neonatal lethality and grossly altering the synapse formation process. As a result, much of the synapse development that proceeds during postnatal period could not be examined. Here we blocked GABA release in single interneurons and used live imaging to examine the effects on synapse and axon development from minutes to several days. These studies allowed us to tease apart the specific role of GABA at different steps of synapse development with spatial and temporal resolution that has not previously been achieved.
Opposite effects of GABA reduction vs blockade on inhibitory synapse formation
We have shown previously that reduction of GABA synthesis by GAD67 knockdown in basket cells resulted in reduced synapse density, size, and axon arbor, indicating that GABA signaling promotes the maturation of inhibitory synapses (Chattopadhyaya et al., 2007). Here we show that a complete blockade of GABA release, either by removing GAD67 and GAD65 or by removing vGAT in single basket cells, yields an opposite effect, overproliferation of small synapse and overgrowth of axons. Our current data not only demonstrate that GABA is not necessary for synapse formation but also suggest that GABA regulates synapse elimination. Indeed, this latter novel facet of GABA function probably can only be uncovered by a complete blockade of GABA release at single-cell level. These results do not contradict our previous findings because reduction and blockade of GABA release are qualitatively different manipulations, and together they reveal two different and likely interrelated aspects of GABA function in inhibitory synapse development. The key to reconcile and integrate the two seemingly paradoxical effects of GABA is to consider the dynamic process of inhibitory synapse formation shown by our live imaging experiments, which revealed frequent formation of transient contacts and the elimination as well as maturation of these nascent synapses.
Role of transient contacts and their activity-dependent elimination in synapse development
CNS synapse assembly and disassembly proceed in the time course of minutes to hours (Ziv and Garner, 2004; McAllister, 2007). Our study is the first to observe developing inhibitory synapses in the minute-range time resolution and has yielded insights into the dynamic process of inhibitory synapse formation. Along developing basket cell axons, we found tight correlation between Syn-GFP puncta, which label vesicular release sites, and morphological boutons, consistent with our previous results (Chattopadhyaya et al., 2007). This indicates that in our system, as in other developing circuits validated by EM (Ruthazer et al., 2006), Syn-GFP puncta are an excellent proxy of synaptic contact sites, although we cannot rule out the possibility that some of the smallest particles could represent transport packets (Ahmari et al., 2000; Ahmari and Smith, 2002; Sabo et al., 2006). We found that Syn-GFP puncta frequently appear, disappear, and translocate along the axon during the period of inhibitory synapse formation. These results provide evidence that developing basket axons form a large number of transient contacts, many of which are quickly eliminated. It is likely that in a complex cellular environment such as the developing cortex, even short-range diffuse cues are insufficient to guide axon–target recognition. Therefore, a general strategy for the developing basket cell axon might be to form contacts, more or less, indiscriminately throughout its length as a necessary step to engage cell-surface cues and neurotransmission to evaluate the targets. This could be followed by selective pruning of contacts via neurotransmission. Importantly, GABA transmission seems to have a dominant role in the validation, and especially the elimination, of nascent contacts. Acute blockade of transmission results in increased stability of release sites and nascent contacts and reduced elimination. Chronic blockade results in a bias of axon growth dynamics toward extension over retraction, and eventually an over exuberant axon arbor studded with dense and small “synaptic contacts.”
GABA transmission may promote the elimination of certain transient contacts while stimulating the maturation of other nascent synapses
We propose a model that integrates current and previous findings (Fig. 10). A developing cortical basket cell axon explores a complex cellular milieu consisting of intermixed and closely juxtaposed potential targets by forming a large number of transient contacts throughout its length. Many of these nascent contacts are eliminated either because of the mismatch of cell type and subcellular compartments or weaker strength compared with neighboring contact sites. Importantly, these transient contacts contain release machinery and mediate GABA transmission. The strength of transmission might provide a measure of relative strength among nearby synaptic contact sites and contribute to competition among these sites. For example, GABA transmission at one contact site may emit a “punishing signal” (Lichtman and Colman, 2000) to weaken the neighboring sites, unless the latter are protected by their own transmission activities. Nearby contacts may also compete for limited presynaptic resource, such as components of vesicle release machinery. Therefore, GABA transmission may mediate activity-dependent redistribution of synaptic resource, such as removal from weak sites to promote their elimination and supplement to stronger sites to promote further maturation. Reduction of GABA transmission (e.g., in GAD67 knockdown or deletion) preserves such a competition mechanism but reduces the capacity to support synapse maturation, therefore resulting in reduced synapse formation and density (Chattopadhyaya et al., 2007). On the other hand, a complete blockade of transmission disables this key mechanism for measuring the strength of nascent synaptic contacts. As a result, most nascent contacts cannot be eliminated, evenly share synaptic resources (e.g., synaptic adhesion molecules, vesicle release machinery, etc.), and manifest as small and similar-sized “synapses.” Therefore, GABA transmission may act as a “double-edged sword” to eliminate weak or inappropriate contacts as well as to promote the maturation of appropriate contacts, thereby regulating activity-dependent GABAergic synapse formation. Because synapse formation stabilizes the axon branch and promotes branch growth, and synapse elimination may precede and result in branch retraction, we suggest that the effect of GABA transmission on axon arbor development is likely rooted in its function in synapse elimination and validation. All together, our study provides deeper insight into the regulatory role of GABA in activity-dependent development of inhibitory synapses and axons.
Figure 10.

A model on the role of GABA transmission in inhibitory synapse and axon development. A developing GABAergic axon (light blue) explores potential synaptic targets (pink rectangles) by making transient synaptic contacts. Dark pink rectangles represent appropriate targets; light pink rectangles represent inappropriate targets. These transient contacts contain release machinery (e.g., synaptic vesicles represented as green circles) and mediate GABA transmission; the strength of transmission might provide a measure of the relative strength among nearby contact sites. GABA transmission may result in an activity-dependent redistribution of synaptic resource, such as removal from weak sites to promote their elimination and supplement to stronger sites to promote their further maturation, thereby promoting competition among these sites. Through GABA transmission (arrow), transient contacts at the inappropriate targets are eliminated, whereas those at the appropriate targets are validated and strengthened. Synapse maturation may result in branch stabilization, new branch extension and growth (WT). At inappropriate contacts, the elimination of transient synapses may lead to the pruning of branches. Reduction of GABA transmission (GABA+/−) preserves such competition mechanism for synaptic elimination but may limit the capacity to support synapse maturation, therefore resulting in reduced synapse density and strength, and a simple axon arbor. Without GABA transmission (GABAblock), GABAergic axons cannot discriminate between appropriate and inappropriate targets and strong and weak contacts. Thus, appropriate contacts cannot be validated and mature, and inappropriate contacts cannot be eliminated, resulting in increased bouton density and small and more homogenously sized boutons. Such increases in bouton density and stability result in more branch extension and less pruning, leading to massive growth of axon arbor.
Our findings raise a number of questions. First, the mechanism of synapse elimination downstream to GABA transmission is unknown. Whether there is a punishing signal or competition for a presynaptic resource, such mechanisms likely are sensitive to the relative strength of transmission among nearby sites along the axon. Second, the substantial increase in synapse density throughout GABAblock axons raises the question of whether some of these aberrant synapses are made onto ectopic targets, such as incorrect cell types or subcellular compartments. In the rodent retina, blocking neurotransmission from presynaptic ON bipolar cell alters synapse number but not the connectivity to ON retinal ganglion cells indicating that transmission is not necessary for synapse specificity (Kerschensteiner et al., 2009). In the cortex, although basket interneurons normally form perisomatic synapses, only ∼30% are on the pyramidal soma and the rest are on proximal dendrites (Tamas et al., 1997; Wang et al., 2002). Thus, only a rigorous quantitative analysis with simultaneous presynaptic and postsynaptic labeling could reveal whether the cellular and subcellular specificity of synapse formation is compromised by transmission blockade in basket cell axons. This possibility warrants investigation because it would provide evidence that although synapse specificity might be guided by molecular cues, the execution of molecular cues in developing vertebrate circuits may be intertwined with and dependent on synaptic transmission, such as shown for the neurexin–neuroligin system (Fu and Huang, 2010).
Footnotes
This work was supported by a grant from the Harold and Leila Mathers Foundation and by the Robertson Neuroscience Fund at Cold Harbor Spring Laboratory. We thank the members of the Huang laboratory for helpful comments on this manuscript. We thank Drs. Hollis Cline, Gary Matthews, Mirjana Maletic-Savatic, and Karel Svoboda for guidance on this project.
References
- Ahmari SE, Smith SJ. Knowing a nascent synapse when you see it. Neuron. 2002;34:333–336. doi: 10.1016/s0896-6273(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Cardona A, Saalfeld S, Preibisch S, Schmid B, Cheng A, Pulokas J, Tomancak P, Hartenstein V. An integrated micro- and macroarchitectural analysis of the Drosophila brain by computer-assisted serial section electron microscopy. PLoS Biol. 2010;8:e1000502. doi: 10.1371/journal.pbio.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgau S, Schierbeck H, Aanstoot HJ, Aagaard L, Begley K, Kofod H, Hejnaes K, Baekkeskov S. Pancreatic beta cells express two autoantigenic forms of glutamic acid decarboxylase, a 65-kDa hydrophilic form and a 64-kDa amphiphilic form which can be both membrane-bound and soluble. J Biol Chem. 1991;266:21257–21264. [PubMed] [Google Scholar]
- Doischer D, Hosp JA, Yanagawa Y, Obata K, Jonas P, Vida I, Bartos M. Postnatal differentiation of basket cells from slow to fast signaling devices. J Neurosci. 2008;28:12956–12968. doi: 10.1523/JNEUROSCI.2890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Huang ZJ. Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at developing GABAergic synapses. Proc Natl Acad Sci U S A. 2010;107:22699–22704. doi: 10.1073/pnas.1011233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat Neurosci. 2004;7:327–332. doi: 10.1038/nn1218. [DOI] [PubMed] [Google Scholar]
- Hua JY, Smear MC, Baier H, Smith SJ. Regulation of axon growth in vivo by activity-based competition. Nature. 2005;434:1022–1026. doi: 10.1038/nature03409. [DOI] [PubMed] [Google Scholar]
- Huang ZJ. Activity-dependent development of inhibitory synapses and innervation pattern: role of GABA signalling and beyond. J Physiol. 2009;587:1881–1888. doi: 10.1113/jphysiol.2008.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Di Cristo G, Ango F. Development of GABA innervation in the cerebral and cerebellar cortices. Nat Rev Neurosci. 2007;8:673–686. doi: 10.1038/nrn2188. [DOI] [PubMed] [Google Scholar]
- Javaherian A, Cline HT. Coordinated motor neuron axon growth and neuromuscular synaptogenesis are promoted by CPG15 in vivo. Neuron. 2005;45:505–512. doi: 10.1016/j.neuron.2004.12.051. [DOI] [PubMed] [Google Scholar]
- Ji F, Kanbara N, Obata K. GABA and histogenesis in fetal and neonatal mouse brain lacking both the isoforms of glutamic acid decarboxylase. Neurosci Res. 1999;33:187–194. doi: 10.1016/s0168-0102(99)00011-5. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner D, Morgan JL, Parker ED, Lewis RM, Wong RO. Neurotransmission selectively regulates synapse formation in parallel circuits in vivo. Nature. 2009;460:1016–1020. doi: 10.1038/nature08236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. 2000;25:269–278. doi: 10.1016/s0896-6273(00)80893-4. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR, Greenberg ME. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/s0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- Nakata T, Terada S, Hirokawa N. Visualization of the dynamics of synaptic vesicle and plasma membrane proteins in living axons. J Cell Biol. 1998;140:659–674. doi: 10.1083/jcb.140.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, Meyer MP, Smith SJ. In vivo imaging of synapse formation on a growing dendritic arbor. Nat Neurosci. 2004;7:254–260. doi: 10.1038/nn1191. [DOI] [PubMed] [Google Scholar]
- Patrizi A, Scelfo B, Viltono L, Briatore F, Fukaya M, Watanabe M, Strata P, Varoqueaux F, Brose N, Fritschy JM, Sassoe-Pognetto M. Synapse formation and clustering of neuroligin-2 in the absence of GABAA receptors. Proc Natl Acad Sci U S A. 2008;105:13151–13156. doi: 10.1073/pnas.0802390105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinal CS, Tobin AJ. Uniqueness and redundancy in GABA production. Perspect Dev Neurobiol. 1998;5:109–118. [PubMed] [Google Scholar]
- Rasmussen H, Rasmussen T, Triller A, Vannier C. Strychnine-blocked glycine receptor is removed from synapses by a shift in insertion/degradation equilibrium. Mol Cell Neurosci. 2002;19:201–215. doi: 10.1006/mcne.2001.1074. [DOI] [PubMed] [Google Scholar]
- Ruthazer ES, Li J, Cline HT. Stabilization of axon branch dynamics by synaptic maturation. J Neurosci. 2006;26:3594–3603. doi: 10.1523/JNEUROSCI.0069-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Gomes RA, McAllister AK. Formation of presynaptic terminals at predefined sites along axons. J Neurosci. 2006;26:10813–10825. doi: 10.1523/JNEUROSCI.2052-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin WC, Haas K, Ruthazer ES, Cline HT. Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature. 2002;419:475–480. doi: 10.1038/nature00987. [DOI] [PubMed] [Google Scholar]
- Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamas G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Lottspeich F, Greengard P, Mehl E, Jahn R. A synaptic vesicle protein with a novel cytoplasmic domain and four transmembrane regions. Science. 1987;238:1142–1144. doi: 10.1126/science.3120313. [DOI] [PubMed] [Google Scholar]
- Tamas G, Buhl EH, Somogyi P. Fast IPSPs elicited via multiple synaptic release sites by different types of GABAergic neurone in the cat visual cortex. J Physiol. 1997;500:715–738. doi: 10.1113/jphysiol.1997.sp022054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Dunaevsky A, Blazeski R, Mason CA, Yuste R. Bidirectional regulation of hippocampal mossy fiber filopodial motility by kainate receptors: a two-step model of synaptogenesis. Neuron. 2003;38:773–784. doi: 10.1016/s0896-6273(03)00299-x. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gupta A, Toledo-Rodriguez M, Wu CZ, Markram H. Anatomical, physiological, molecular and circuit properties of nest basket cells in the developing somatosensory cortex. Cereb Cortex. 2002;12:395–410. doi: 10.1093/cercor/12.4.395. [DOI] [PubMed] [Google Scholar]
- Wiedenmann B, Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. 1985;41:1017–1028. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Becker N, Bonhoeffer T. GABAergic synapses are formed without the involvement of dendritic protrusions. Nat Neurosci. 2008;11:1044–1052. doi: 10.1038/nn.2180. [DOI] [PubMed] [Google Scholar]
- Wong WT, Wong RO. Changing specificity of neurotransmitter regulation of rapid dendritic remodeling during synaptogenesis. Nat Neurosci. 2001;4:351–352. doi: 10.1038/85987. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM. Electrical activity and development of neural circuits. Nat Neurosci. 2001;4:1207–1214. doi: 10.1038/nn753. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Felder M, Connor JA, Poo MM. Turning of nerve growth cones induced by neurotransmitters. Nature. 1994;368:140–144. doi: 10.1038/368140a0. [DOI] [PubMed] [Google Scholar]
- Ziv NE, Garner CC. Cellular and molecular mechanisms of presynaptic assembly. Nat Rev Neurosci. 2004;5:385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]