

Abstract
Inhibition of GABAA receptors by Cu2+ has been appreciated for some time, but differences between synaptic and extrasynaptic GABAA receptors have not been explored. We show that Cu2+ potently blocks steady-state GABA currents mediated by extrasynaptic δ subunit-containing GABAA receptors (δ-GABAARs) with an IC50 of 65 nm. This compares with an IC50 of 85 μm for synaptic γ subunit-containing GABAARs (γ-GABAARs). To test the significance of this subunit selectivity, we examined the blocking action of Cu2+ on neurons of the mouse cerebellum and striatum, brain regions that are known to express both types of receptor. Cu2+ was shown to significantly reduce tonic inhibition mediated by extrasynaptic δ-GABAARs with little action on phasic inhibition mediated by conventional synaptic γ-GABAARs. We speculate on the implications of these observations for conditions, such as Wilson's disease, that can involve raised Cu2+ levels in the brain.
Introduction
Conventional γ subunit-containing GABAA receptors (γ-GABAARs) are blocked by Cu2+ in the micromolar range (Narahashi et al., 1994; Fisher and Macdonald, 1998; Kim and Macdonald, 2003), but the Cu2+ sensitivity of extrasynaptic δ subunit-containing GABAA receptors (δ-GABAARs) has not been established. Low extracellular GABA concentrations (Attwell et al., 1993; Richerson and Wu, 2003) persistently activate extrasynaptic δ-GABAARs (Nusser et al., 1995, 1998) because of the high GABA affinity of these receptors (Bright et al., 2011). The resulting tonic inhibition modifies the excitability of cerebellar granule cells both in vitro (Brickley et al., 2001) and in vivo (Chadderton et al., 2004). High-affinity δ-GABAARs are also found in brain regions, such as the hippocampus (Stell et al., 2003), thalamus (Porcello et al., 2003), and striatum (Santhakumar et al., 2010), and disturbances in tonic inhibition are now associated with a number of neurological disorders, including depression, epilepsy, and Parkinson's disease (Brickley and Mody, 2012).
During vesicular release, Cu2+ is coreleased into the synaptic cleft at concentrations of a few 100 μm (Kardos et al., 1989). Free Cu2+ is likely to be much lower (Hopt et al., 2003) given the prevalence of Cu2+ binding proteins in the vicinity of the synapse. It is known that a diversity of ion channel types (Mathie et al., 2006), including voltage-gated (Ma et al., 2008) and voltage-independent potassium channels (Gruss et al., 2004), are sensitive to <10 μm free Cu2+ with a particularly high-affinity block being reported for GABAARs (Sharonova et al., 1998). Raised Cu2+ levels are associated with Wilson's disease resulting from a rare mutation in the copper transporter gene (Das and Ray, 2006). If untreated, raised Cu2+ can lead to neurological symptoms, including bradykinesia, tremor, ataxia, dystonia, and long-term cognitive impairment. The neuronal damage associated with Wilson's disease is often restricted to the striatum and cerebellum (Trocello et al., 2011). We have explored the possibility that extracellular Cu2+ selectively blocks δ-GABAARs in cerebellum and striatum, and the neurological deficits associated with altered Cu2+ homeostasis may, in part, reflect disturbances in tonic inhibition.
Materials and Methods
Recombinant expression.
HEK-293 cells were grown in DMEM (supplemented with 1% nonessential amino acids, 10% heat-inactivated FBS, 1% penicillin (10,000 U/ml)/streptomycin (10 mg/ml), and 200 mm l-glutamine in humidified 5% CO2 at 37°C before transfection. Plasmids contained the ampicillin resistance gene, a cytomegalovirus promoter, and the SV40 early promoter. Mouse cDNA was subcloned into either the pRK5 or the pcDNA3.1 vector, and cells were transfected using a calcium phosphate technique (Houston et al., 2012). Transfected cells were plated onto poly-d-lysine-coated coverslips and kept in a humidified incubator overnight before electrophysiological recording.
Acute slice preparations.
After cervical dislocation of male C57BL/6J mice (35 ± 6 d postnatal), acute 250 μm sagittal cerebellar slices were cut with a vibratome tissue slicer (Campden Instruments) at ∼4°C in slicing aCSF containing the following in mm: KCl 2.5, CaCl2 1, MgCl 5, NaH2PO4 1.25, NaHCO3 26, glucose 11, glycerol 250, pH 7.4, when bubbled with 95%O2/5%CO2. Slices were stored at room temperature while the glycerol-based aCSF was exchanged for control aCSF (in mm as follows): NaCl 125, KCl 2.5, CaCl2 2, MgCl 2, NaH2PO4 1.25, NaHCO3 26, glucose 11. Coronal striatal slices were sliced in control aCSF with 5 mm MgCl and 1 mm kynurenic acid.
Electrophysiology.
Glass electrodes were pulled to a final resistance of 9 MΩ (cerebellar granule cells) and 4MΩ (HEK-293 cells, medium spiny neurons) from borosilicate glass capillaries, and whole-cell recordings were made using an Axopatch 200B amplifier (Molecular Devices) at room temperature for HEK-293/granule cells (22–23°C) and 37°C for medium spiny neurons. Pipette solution contained (in mm) the following: CsCl 140, NaCl 4, CaCl2 0.5, HEPES 10, EGTA 5, Mg-ATP 2; the pH was adjusted to 7.3 with CsOH and QX-314; 100 μm was added to block voltage-gated sodium channels, and 1.5 mg/ml of biocytin (Sigma) was also included to allow confocal imaging of filled neurons. Cells were visualized using a fixed-stage upright microscope (BX51W1, Olympus or Scientifica) fitted with high numerical aperture water-immersion objectives. GABA (Sigma), gabazine (Tocris Bioscience), and copper (ii) chloride hydrate (Sigma) were prepared in control aCSF. Drug application was performed with a gravity-fed system via an eight unit solenoid driver that flowed solutions into a small diameter manifold tip placed close to the cell. All hardware was controlled via a National Instruments digitization board (PCI-6052E; National Instruments) linked to a dedicated PC running either WinEDR or WinWCP software (John Dempster, University of Strathclyde, Glasgow, United Kingdom). Data were passed through a 2 kHz, −3 dB, 8-pole Bessel filter before digitization at a rate of 20 kHz.
Imaging.
After electrophysiological recording, slices were fixed in PBS containing 4% paraformaldehdye for 24 h. Biocytin-filled neurons were conjugated to a streptavidin-linked Alexa-555 dye (Invitrogen) at 1:200 dilution in 1% serum and 0.2% Triton-X at room temperature for 3–4 h at room temperature. Slices were washed in PBS and mounted in Vectashield medium and imaged using a Zeiss LSM 510 CLSM microscope, with a He-Ne 543 nm laser with filter settings optimized for an Alexa-555 emission peak at 565 nm.
Statistical analysis.
Statistical analysis was performed using Origin 8.5 (Microcal) using unconstrained least-squared fitting procedures, and statistical tests were performed at a 95% confidence level.
Results
Peak and steady-state GABA responses in cells expressing α6β3δ GABAARs were clearly reduced by preapplication of 1 μm Cu2+ (Fig. 1A). At 30 μm Cu2+, the response to GABA exhibited little desensitization because of the action of Cu2+ on the peak response. On average, 1 μm Cu2+ reduced the peak response to 1 μm GABA by 50% (1486 ± 253 pS/pF vs 735.3 ± 91.1 pS/pF, n = 18) compared with 37% (378 ± 37 pS/pF vs 240.2 ± 31.1 pS/pF, n = 18) for the steady-state response. Figure 1B illustrates how Cu2+ rapidly blocks the steady-steady response to 1 μm GABA. On average, the speed of Cu2+ block was described by a single exponential with an average time constant of 4.7 ± 0.5 s (n = 13). The slower recovery from Cu2+ block was described with an average time constant of 37.0 ± 6.1 s (n = 13). However, as shown in Figure 1B (inset), the speed of block and unblock was independent of Cu2+ concentration (linear regression analysis, R2 = 0.06 for block, R2 = −0.06 for unblock, n = 12).
Figure 1.
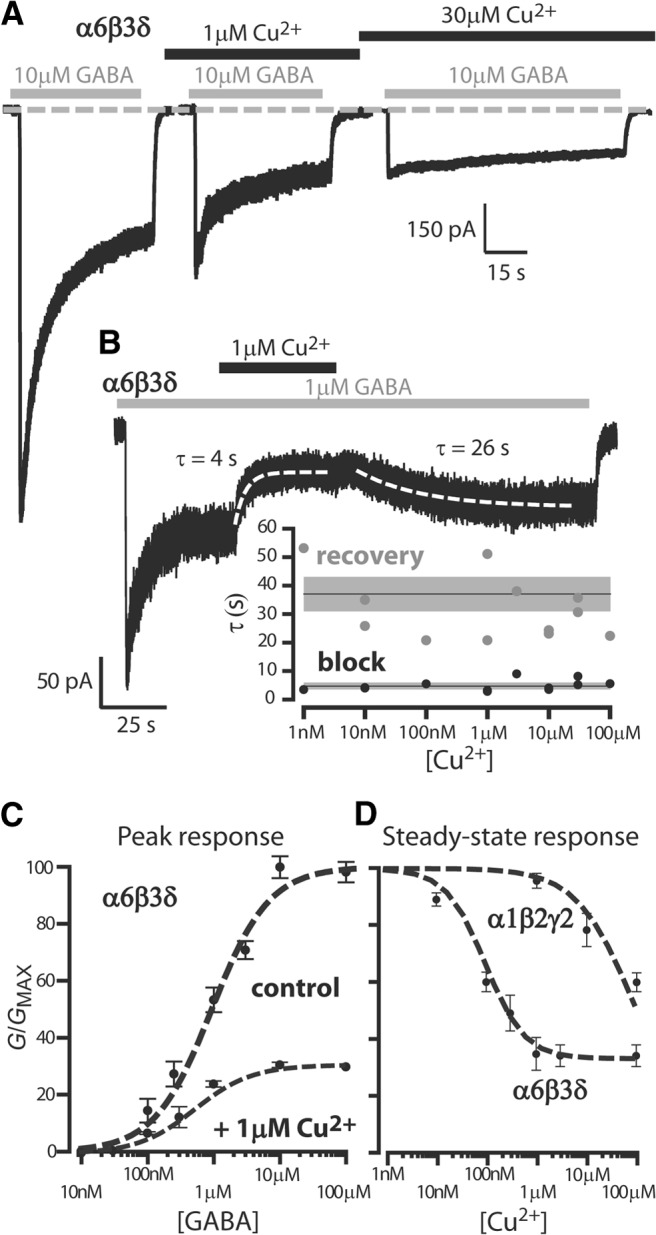
High-affinity Cu2+ block of GABAARs. A, Whole-cell voltage-clamp traces during 10 μm GABA applications to HEK-293 cells expressing α6β3δ subunits. In control conditions (left trace), GABA resulted in a clear peak response that desensitized to a steady-state level. Prior application of 1 μm Cu2+ (middle trace) resulted in an attenuated peak and steady-state response. Higher concentrations of Cu2+ (right trace) resulted in a GABA response that exhibited little desensitization because of the large attenuation of the peak response. B, A whole-cell response to 1 μm GABA (gray bar) and coapplication of 1 μm copper (black bar). The Cu2+ block was rapid and well described by a single exponential function with a time constant of 4 s. Recovery from block was slower and described by a single exponential function with a time constant of 26 s. The speed of block and recovery was estimated at varying Cu2+ concentrations. The data plotted in the inset to B demonstrate that the Cu2+ concentration does not influence the speed of block or recovery. The solid lines indicate the average kinetics, and the shaded areas represent the SEM. C, GABA dose–response curves constructed from the peak response (G/GMAX) recorded in the presence and absence of 1 μm Cu2+. The mean peak conductance ± SEM are plotted and data fitted with a Hill equation (dashed line). D, Plot of the average steady-state conductance (G/GMAX) ± SEM obtained in response to a 1 μm GABA application at a range of Cu2+ concentrations. The dashed lines are the results of a fit using the Hill equation to compare the potency of Cu2+ block for two GABAA receptor types. The IC50 estimate for α6β3δ subunit-containing GABAARs was 65 nm compared with 85 μm for α1β2γ2s subunit-containing GABAARs.
The peak GABA dose–response curve in Figure 1C demonstrates that the apparent affinity of α6β3δ GABAARs for GABA was not altered by Cu2+. Fitting control data with a Hill equation gave an EC50 of 873 ± 156 nm. There was no rightward shift in the position of the GABA dose–response curve in 1 μm Cu2+ (EC50 = 359 ± 40 nm), but there was a 67% reduction in the maximum conductance. A similar profile was apparent for steady-state GABA dose–response curves with an EC50 of 120 ± 45 nm in control compared with 124 ± 16 nm in 1 μm Cu2+ with a 65% reduction in the maximum steady-state response. The potency of this noncompetitive block was determined at different Cu2+ concentrations. As shown in Figure 1D, a significant reduction in the steady-state response to 1 μm GABA was evident at Cu2+ concentrations as low as 10 nm (10.5 ± 1.5% inhibition, n = 12). The average reduction in the steady-state current observed at 100 μm Cu2+ was 65 ± 6%. Fitting the copper inhibition data with a Hill equation resulted in an IC50 of 65 ± 24 nm (R2 = 0.96) for α6β3δ GABAARs with a maximum inhibition of 68%.
Previous studies examining the Cu2+ block of recombinant γ subunit-containing GABAARs reported IC50 values in the μm range (Fisher and Macdonald, 1998). Therefore, we also examined Cu2+ block of the α1β2γ2s subunit combination. In contrast to the α6β3δ GABAARs, sub μm Cu2+ concentrations had no discernible blocking action on GABA-evoked responses from α1β2γ2s GABAARs. For comparison, the average steady-state conductance in response to 1 μm GABA alone was 106 ± 46 pS/pF (n = 6) for these α1β2γ2s GABAARs. A small suppression of these GABA-evoked responses was first evident at 1 μm Cu2+ (5 ± 3%, n = 8). As shown in Figure 1D, fitting the copper inhibition data obtained from α1β2γ2s GABAARs with a Hill equation that was constrained to the maximum inhibition (88 ± 3% at 1 mm Cu2+, n = 4) gave an IC50 of 85 μm.
These recombinant expression data raise the possibility that, in native neurons, the tonic inhibition mediated by δ-GABAARs will be more sensitive to Cu2+ block than the phasic inhibition mediated by γ-GABAARs. To test this prediction, we recorded from cerebellar granule cells in acute slice preparations of the adult cerebellum and compared the Cu2+ block of tonic versus phasic inhibition. As shown in Figure 2A, cerebellar granule cells exhibit spontaneous GABAA receptor-mediated synaptic responses or sIPSCs (Fig. 2A) and a clearly discernible tonic conductance that has been shown to be generated by δ-GABAARs (Brickley et al., 2001). As expected, the phasic and tonic conductance was blocked by the GABAAR antagonist gabazine. However, application of 10 μm Cu2+ to the extracellular solution had little action on the properties of sIPSCs but at the same time produced an obvious reduction in the tonic conductance. Figure 2B illustrates the average holding current measured during each 1 s epoch of recording, demonstrating a clear reduction in the holding current after Cu2+ application. At the same time, Figure 2C demonstrates little change in either the peak amplitude or the frequency of sIPSCs. On average, the tonic conductance was reduced from 96.7 ± 26 pS/pF in control conditions to 44.4 ± 17 pS/pF in 10 μm Cu2+ (p = 0.003, paired sample t test; n = 8). However, there was no significant change in the frequency (0.8 ± 0.1 Hz vs 0.5 ± 0.1 Hz; p = 0.07) or the peak amplitude (22.5 ± 5 pA vs 21.4 ± 4 pA; p = 0.6) of sIPSCs. We also examined the 10–90% rise-time and the weighted decay time of sIPSCs, but neither of these parameters was significantly altered by 10 μm Cu2+; the average decay time was 7.3 ± 0.8 ms in control versus 7.0 ± 0.9 ms in 10 μm Cu2+ (p = 0.3), and the average 10–90% rise-time was 1.7 ± 0.1 ms in control versus 1.5 ± 0.2 ms in Cu2+ (p = 0.1).
Figure 2.
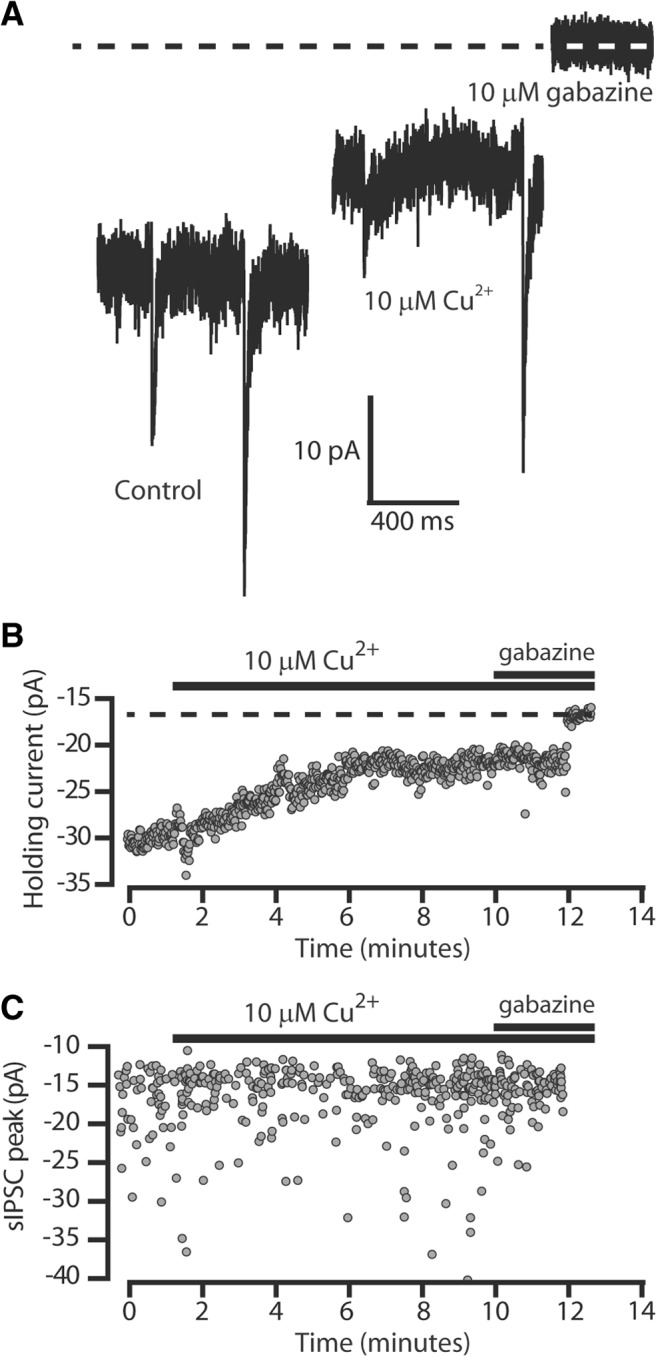
Cu2+ reduces the tonic conductance recorded from cerebellar granule cells. A, Example traces recorded in control conditions, in the presence of 10 μm Cu2+ and in the presence of 10 μm gabazine. B, Time course data from a different adult cerebellar granule cell. The average holding current was calculated during each 1 s epoch at a command voltage of −60 mV. After a period of stable control recording, 10 μm Cu2+ was included in the aCSF, and the holding current was monitored before coapplying 10 μm gabazine to block all GABAARs. C, The peak amplitude of IPSCs is plotted during the course of the same experiment as in A. The peak amplitude and frequency of IPSCs were not dramatically altered by 10 μm Cu2+, but application of 10 μm gabazine blocked all IPSCs.
These data suggest that high Cu2+ levels will reduce tonic inhibition in neurons that express high-affinity extrasynaptic δ-GABAARs. It is possible that the low free Cu2+ levels in the extracellular space would be sufficient to block extrasynaptic δ-GABAARs. To test this possibility, we chelated extracellular Cu2+ with histidine to determine whether the tonic conductance was increased. We established that 100 μm histidine did not directly alter the steady-state current generated by recombinant α6β3δ GABAARs. The steady-state conductance recorded from α6β3δ GABAARs after 10 μm GABA application was 299 ± 101 pS/pF compared with 303 ± 95 pS/pF when 100 μm histidine was coapplied (n = 5). On average, the tonic conductance recorded from cerebellar granule cells was increased from 45.6 ± 16 pS/pF to 66.6 ± 16 pS/pF (n = 7, p = 0.008, paired sample t test) in the presence of 100 μm histidine. Histidine application did not influence IPSC parameters, and it would appear that conventional synaptic γ-GABAARs are not influenced by the resting Cu2+ levels in the acute brain slice preparation. Average IPSC amplitude was 17.9 ± 1.7 pA in control versus 20.6 ± 2 pA in histidine (p = 0.1). Average IPSC frequency was 0.6 ± 0.1 Hz in control versus 0.6 ± 0.2 Hz in histidine (p = 0.9). Average IPSC decay was 6.7 ± 0.4 ms in control versus 7.2 ± 0.4 in histidine (p = 0.2). Average 10–90% rise-time was 1.4 ± 0.1 ms in control versus 1.6 ± 0.1 ms in histidine (p = 0.2, paired sample t test). The 30% block of the tonic conductance indicates a resting Cu2+ concentration of at least 80 nm (Fig. 1D).
The motor deficits associated with Cu2+ accumulation in the brain are very similar to those observed during Parkinson's disease, and an obvious common feature of these disorders is pathology within the caudate/putamen and globus pallidus. Extrasynaptic δ-GABAARs are known to be expressed by medium spiny neurons of the striatum, prompting us to examine Cu2+ block of tonic inhibition within these neurons (Ade et al., 2008; Janssen et al., 2009; Santhakumar et al., 2010). Medium spiny neurons were identified from their characteristic morphological (Fig. 3A) and electrophysiological parameters. As shown in Figure 3B, medium spiny neurons exhibited IPSCs and an obvious tonic conductance. Both of these conductance types could be blocked by the broad-spectrum GABAA receptor antagonist gabazine, and the rate of vesicular GABA release was reduced by 66% in the presence of 500 nm TTX (data not shown). The tonic conductance recorded from medium spiny neurons was significantly reduced from an average conductance of 20.4 ± 4 pS/pF in control conditions to 12.7 ± 3 pS/pF in 10 μm Cu2+ (n = 7, p = 0.003), whereas the average IPSC properties were unaltered. IPSC amplitude was 38.4 ± 5 pA in control versus 38.5 ± 6 pA in Cu2+ (p = 0.9). IPSC frequency was 1.8 ± 0.5 Hz in control versus 1.6 ± 0.4 Hz in Cu2+ (p = 0.6). IPSC decay was 7.2 ± 0.7 ms in control versus 7.9 ± 0.5 ms in Cu2+ (p = 0.06). IPSC 10–90% rise-time was 2.1 ± 0.3 ms in control versus 2.2 ± 0.3 ms in Cu2+ (p = 0.9). The scatter plot in Figure 3C demonstrates how the reduction in tonic conductance consistently occurred in the absence of any change in the frequency of GABA release.
Figure 3.
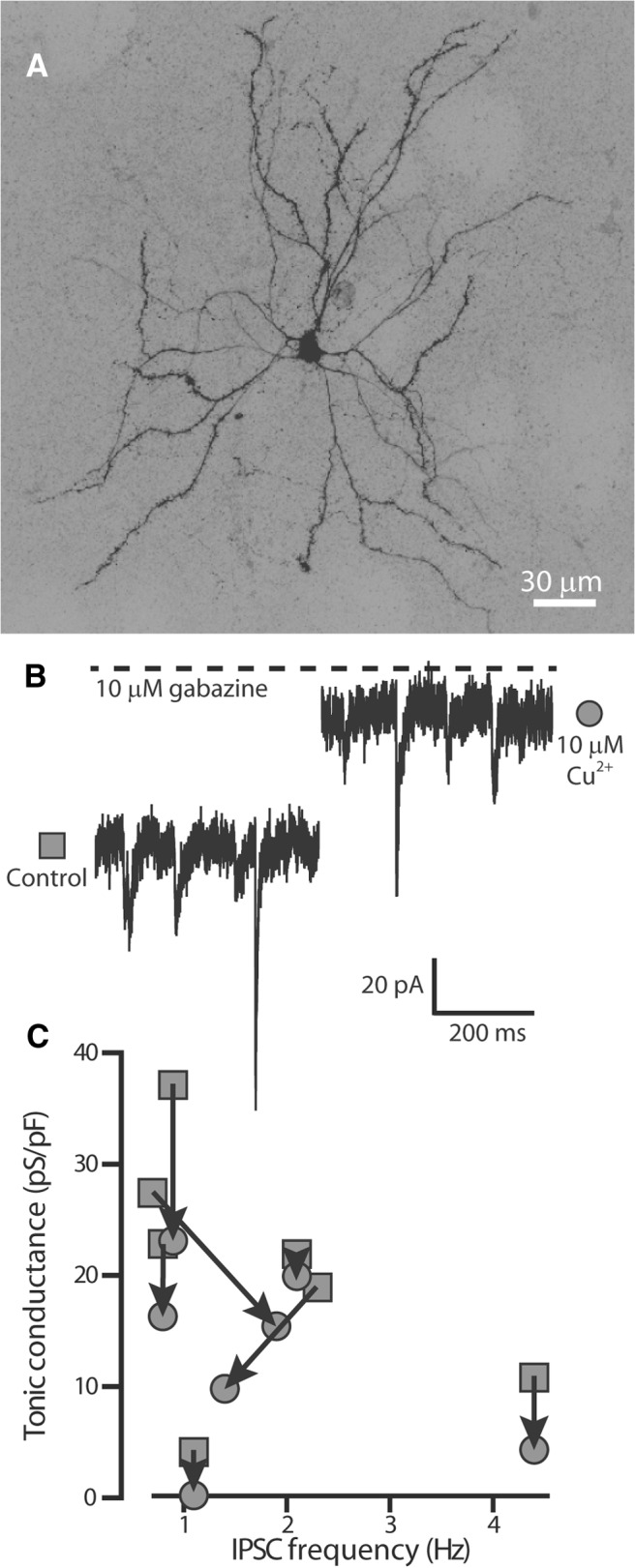
Cu2+ reduces the tonic conductance recorded from striatal medium spiny neurons. A, Projection from confocal stacks obtained after whole-cell voltage recording from a putative medium spiny neuron. The characteristic morphology of medium spiny neurons was confirmed from this image. B, Current traces illustrating the reduction in holding current that occurs after application of 10 μm Cu2+ in the absence of any change in the properties of IPSCs. The dashed line indicates the average holding current recorded in the presence of 10 μm gabazine. C, Plot of the IPSC frequency against tonic conductance calculated for individual medium spiny neurons in the presence (filled gray circles) and absence (filled gray squares) of 10 μm Cu2+. The arrows link the paired data.
Discussion
We have demonstrated, for the first time, that extrasynaptic δ-GABAARs are particularly sensitive to Cu2+ block. The noncompetitive nature of this subunit-selective block is particularly relevant given that extrasynaptic δ-GABAARs are likely to experience varying ambient GABA concentrations depending upon the contribution of vesicular (Bright and Brickley, 2008) and nonvesicular GABA release mechanisms (Lee et al., 2010) and the equilibrium state of the GABA transporters (Richerson and Wu, 2003). Indeed, the tonic conductance recorded from both cerebellar granule cells and medium spiny neurons of the striatum was selectively reduced by extracellular Cu2+ with no action on synaptic GABAARs. We also find that chelating Cu2+ enhanced the tonic conductance recorded from cerebellar granule cells; and by comparing these data with the recombinant expression information presented in Figure 1D, we predict that extracellular Cu2+ will rest >80 nm. The fact that we observed Cu2+ block in native neurons further indicates that the resting Cu2+ concentration is <1 μm in these recording conditions. This is consistent with previous estimates (Hopt et al., 2003) and suggests that free Cu2+ levels should be capable of reducing the tonic inhibition mediated by δ-GABAARs. This could in part explain the surprisingly small size of the tonic conductance that is typically recorded from native neurons. For example, we have previously estimated that cerebellar granule cells express in the region of 2000 δ-GABAARs on their somatic and dendritic membrane, but the tonic conductance recorded from these cells represents the superimposed opening of just 3 or 4 channels (Houston et al., 2012). Low receptor occupancy coupled with profound desensitization (Bright et al., 2011) will result in low open probability, but it is also plausible that endogenous Cu2+ block reduces the tonic conductance further. Indeed, from Figure 1D, we would predict that 65% of δ-GABAARs will be blocked in situations where free Cu2+ reaches just 1 μm. It is also worth noting that Cu2+ block of δ-GABAARs will also reduce the apparent desensitization of these receptors because of a greater fractional reduction in the peak response to GABA.
These novel observations may also have implications for conditions, such as Wilson's disease where the steady-state Cu2+ concentration can more than double. It has not been appreciated until now that these changes could influence tonic inhibition, and it is widely assumed that the pathology associated with Wilson's disease is primarily the result of oxidative damage (Ferenci, 2005). Cu2+ is a cofactor for a variety of enzymes, and its inherent reactivity leads to the formation of reactive oxygen species. However, there is considerable variability in the appearance of neurological symptoms in Wilson's disease, even among siblings, and the impact of raised Cu2+ levels for brain pathology is often restricted to regions, such as the striatum and cerebellum. Wilson's disease is also unusual, compared with other neurodegenerative disorders, in that it has a simple treatment (reducing free Cu2+ in the brain). The fact that this rare disease can be effectively treated by normalizing free Cu2+ levels may have implications when considering the treatment of other neurodegenerative disorders, such as Parkinson's and Alzheimer's, that may also involve changes in Cu2+ homeostasis.
Footnotes
This work was supported by a Medical Research Council program grant and a Wellcome Trust project grant. T.P.M. received a Biotechnology and Biological Sciences Research Council PhD studentship.
The authors declare no competing financial interests.
References
- Ade KK, Janssen MJ, Ortinski PI, Vicini S. Differential tonic GABA conductances in striatal medium spiny neurons. J Neurosci. 2008;28:1185–1197. doi: 10.1523/JNEUROSCI.3908-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–407. doi: 10.1016/0896-6273(93)90145-H. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Mody I. Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron. 2012;73:23–34. doi: 10.1016/j.neuron.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Bright DP, Brickley SG. Acting locally but sensing globally: impact of GABAergic synaptic plasticity on phasic and tonic inhibition in the thalamus. J Physiol. 2008;586:5091–5099. doi: 10.1113/jphysiol.2008.158576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Renzi M, Bartram J, McGee TP, MacKenzie G, Hosie AM, Farrant M, Brickley SG. Profound desensitization by ambient GABA limits activation of δ-containing GABAA receptors during spillover. J Neurosci. 2011;31:753–763. doi: 10.1523/JNEUROSCI.2996-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Das SK, Ray K. Wilson's disease: an update. Nat Clin Pract Neurol. 2006;2:482–493. doi: 10.1038/ncpneuro0291. [DOI] [PubMed] [Google Scholar]
- Ferenci P. Wilson's disease. Clin Gastroenterol Hepatol. 2005;3:726–733. doi: 10.1016/S1542-3565(05)00484-2. [DOI] [PubMed] [Google Scholar]
- Fisher JL, Macdonald RL. The role of an α subtype M2–M3 His in regulating inhibition of GABAA receptor current by zinc and other divalent cations. J Neurosci. 1998;18:2944–2953. doi: 10.1523/JNEUROSCI.18-08-02944.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruss M, Mathie A, Lieb WR, Franks NP. The two-pore-domain K(+) channels TREK-1 and TASK-3 are differentially modulated by copper and zinc. Mol Pharmacol. 2004;66:530–537. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- Hopt A, Korte S, Fink H, Panne U, Niessner R, Jahn R, Kretzschmar H, Herms J. Methods for studying synaptosomal copper release. J Neurosci Methods. 2003;128:159–172. doi: 10.1016/S0165-0270(03)00173-0. [DOI] [PubMed] [Google Scholar]
- Houston CM, McGee TP, Mackenzie G, Troyano-Cuturi K, Rodriguez PM, Kutsarova E, Diamanti E, Hosie AM, Franks NP, Brickley SG. Are extrasynaptic GABAA receptors important targets for sedative/hypnotic drugs? J Neurosci. 2012;32:3887–3897. doi: 10.1523/JNEUROSCI.5406-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen MJ, Ade KK, Fu Z, Vicini S. Dopamine modulation of GABA tonic conductance in striatal output neurons. J Neurosci. 2009;29:5116–5126. doi: 10.1523/JNEUROSCI.4737-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J, Kovács I, Hajós F, Kálmán M, Simonyi M. Nerve endings from rat brain tissue release copper upon depolarization: a possible role in regulating neuronal excitability. Neurosci Lett. 1989;103:139–144. doi: 10.1016/0304-3940(89)90565-X. [DOI] [PubMed] [Google Scholar]
- Kim H, Macdonald RL. An N-terminal histidine is the primary determinant of α subunit-dependent Cu2+ sensitivity of αβ3γ2L GABA(A) receptors. Mol Pharmacol. 2003;64:1145–1152. doi: 10.1124/mol.64.5.1145. [DOI] [PubMed] [Google Scholar]
- Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- Ma Z, Wong KY, Horrigan FT. An extracellular Cu2+ binding site in the voltage sensor of BK and Shaker potassium channels. J Gen Physiol. 2008;131:483–502. doi: 10.1085/jgp.200809980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Sutton GL, Clarke CE, Veale EL. Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther. 2006;111:567–583. doi: 10.1016/j.pharmthera.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Narahashi T, Ma JY, Arakawa O, Reuveny E, Nakahiro M. GABA receptor-channel complex as a target site of mercury, copper, zinc, and lanthanides. Cell Mol Neurobiol. 1994;14:599–621. doi: 10.1007/BF02088671. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Roberts JD, Baude A, Richards JG, Somogyi P. Relative densities of synaptic and extrasynaptic GABAA receptors on cerebellar granule cells as determined by a quantitative immunogold method. J Neurosci. 1995;15:2948–2960. doi: 10.1523/JNEUROSCI.15-04-02948.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcello DM, Huntsman MM, Mihalek RM, Homanics GE, Huguenard JR. Intact synaptic GABAergic inhibition and altered neurosteroid modulation of thalamic relay neurons in mice lacking δ subunit. J Neurophysiol. 2003;89:1378–1386. doi: 10.1152/jn.00899.2002. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wu Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol. 2003;90:1363–1374. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Jones RT, Mody I. Developmental regulation and neuroprotective effects of striatal tonic GABAA currents. Neuroscience. 2010;167:644–655. doi: 10.1016/j.neuroscience.2010.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharonova IN, Vorobjev VS, Haas HL. High-affinity copper block of GABA(A) receptor-mediated currents in acutely isolated cerebellar Purkinje cells of the rat. Eur J Neurosci. 1998;10:522–528. doi: 10.1046/j.1460-9568.1998.00057.x. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by δsubunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trocello JM, Guichard JP, Leyendecker A, Pernon M, Chaine P, El Balkhi S, Poupon J, Chappuis P, Woimant F. Corpus callosum abnormalities in Wilson's disease. J Neurol Neurosurg Psychiatry. 2011;82:1119–1121. doi: 10.1136/jnnp.2009.204651. [DOI] [PubMed] [Google Scholar]