

Abstract
Compelling clinical reports reveal that behavioral stress alone is sufficient to cause reversible myocardial dysfunction in selected individuals. We developed a rodent stress cardiomyopathy model by a combination of prenatal and postnatal behavioral stresses (Stress). We previously reported a decrease in percent fractional shortening by echo, both systolic and diastolic dysfunction by catheter-based hemodynamics, as well as attenuated hemodynamic and inotropic responses to the β-adrenergic agonist, isoproterenol (ISO) in Stress rats compared with matched controls (Kan H, Birkle D, Jain AC, Failinger C, Xie S, Finkel MS. J Appl Physiol 98: 77–82, 2005). We now report enhanced catecholamine responses to behavioral stress, as evidenced by increased circulating plasma levels of norepinephrine (P < 0.01) and epinephrine (P < 0.01) in Stress rats vs. controls. Cardiac myocytes isolated from Stress rats also reveal evidence of oxidative stress, as indicated by decreased ATP, increased GSSG, and decreased GSH-to-GSSG ratio in the presence of increased GSH peroxidase and catalase activities (P < 0.01, for each). We also report blunted inotropic and intracellular Ca2+ concentration responses to extracellular Ca2+ (P < 0.05), as well as altered inotropic responses to the intracellular calcium regulator, caffeine (20 mM; P < 0.01). Treatment of cardiac myocytes with N-acetylcysteine (NAC) (10−3 M) normalized calcium handling in response to ISO and extracellular Ca2+ concentration and inotropic response to caffeine (P < 0.01, for each). NAC also attenuated the blunted inotropic response to ISO and Ca2+ (P < 0.01, for each). Surprisingly, NAC did not reverse the changes in GSH, GSSG, or GSH-to-GSSG ratio. These data support a GSH-independent salutary effect of NAC on intracellular calcium signaling in this rodent model of stress-induced cardiomyopathy.
Keywords: prenatal stress, N-acetylcysteine, heart function
compelling clinical reports reveal that behavioral stress alone is sufficient to cause profound, but completely reversible, myocardial dysfunction in selected individuals (1, 5, 45). This stress cardiomyopathy is characterized by the absence of any prior cardiac history, antecedent acute behavioral stress, elevated circulating catecholamine levels, adrenergic desensitization, and complete reversal of myocardial dysfunction within weeks (23). We developed a rodent stress cardiomyopathy model by a combination of prenatal and postnatal behavioral stresses (Stress) (6, 23). Our laboratory previously reported myocardial dysfunction by echo and catheter-based hemodynamics in vivo, isolated cardiac myocyte dysfunction in vitro, as well as attenuated hemodynamic and inotropic responses to the β-adrenergic agonist, isoproterenol (ISO), in vivo and in vitro in Stress compared with matched Con rats (6).
The two prevailing hypotheses for reversible myocardial dysfunction following behavioral stress are enhanced adrenergic stimulation and/or defective nitric oxide-mediated vascular reactivity (5, 36). These two hypotheses are not mutually exclusive and could be related to enhanced oxidative stress. Accordingly, we studied circulating plasma catecholamine concentrations and markers of oxidative stress in our model.
N-acetylcysteine (NAC) has been shown to raise intracellular concentrations of reduced glutathione (GSH) by providing an additional source of cysteine as a precursor (8, 16, 27) to reduce disulfide bonds (17) and scavenge free radicals (26). NAC is currently being studied as an antioxidant in human clinical trials (26). Our laboratory previously reported that NAC reversed myocardial dysfunction in two different genetic cardiomyopathy models (7, 12). We now provide data supporting a GSH-independent salutary effect of NAC on reversing myocyte dysfunction in our rodent stress cardiomyopathy model.
MATERIALS AND METHODS
In Vivo Studies
Stress paradigm.
Virgin 6-wk-old male and female Sprague-Dawley rats were purchased from Hilltop Lab Animals (Scottdale, PA) and housed in the Centers for Disease Control and Prevention/National Institute for Occupational Safety and Health animal quarters. Strict adherence to the protocol dually approved by the West Virginia University Animal Care and Use Committee and National Institute for Occupational Safety and Health was maintained throughout. In brief, during the third and last week (day 14 to day 21), the prenatally stressed (PNS) dam was removed from her cage, received a saline injection (0.9%, 1 ml sc) in the folds of skin on the back of the neck, was placed in a new cage, and was moved to a new location on the cage rack, as our laboratory previously reported (6, 23). Male offspring from control and PNS dams were restrained for 2 h at 6 wk and then again 1 wk later. “Stress” rats are defined as offspring of stressed dams that were subjected to two successive restraint stresses. “Control” (Con) rats are offspring of dams that were undisturbed throughout pregnancy, and that were subjected to the identical restraint stress.
Plasma catecholamine concentrations.
A blood sample of 900 μl was collected in a microfuge tube with 100 μl of 3.8% sodium citrate after rats were anesthetized by pentobarbital (50 mg/kg). The blood sample was centrifuged at 1,000 g for 10 min at 4°C. The plasma was transferred to a new centrifuge tube and stored at −70°C for analysis. Plasma norepinephrine (NE) and epinephrine (Epi) were extracted with ESA plasma catecholamine kit (ESA, Chelmsford, MA), according to the manufacturer's instruction. Separation and quantitation of NE and Epi were accomplished by HPLC with electrochemical detection (Coulochem II, ESA, Chelmsford, MA). Data were analyzed by Waters Millennium 32 software and enabled accurate determination of noradrenaline and adrenaline.
Ex Vivo Experiments
Cardiac tissue.
ATP CONCENTRATION.
Trichloroacetic acid (10%) was added to extract the ATP on ice. Cardiac tissue was homogenized and centrifuged at 2,000 g for 10 min. The supernatant was diluted with 50 mmol/l Tris-acetate buffer containing 2 mmol/l ethylenediaminetetraacetic acid (EDTA) (pH 7.75) to a final concentration of 0.1%. Cardiac ATP content was measured using ENLITEN ATP assay system (Promega, Madison, WI), according to the manufacturer's protocol.
Cardiac myocytes.
ISOLATION OF ADULT RAT VENTRICULAR MYOCYTES.
Cardiac myocytes were isolated from the Con and Stress rats, as previously reported (6, 23). Rats were anesthetized with pentobarbital sodium at 50 mg/kg, and the hearts were removed and perfused with Krebs-Henseleit bicarbonate buffer (KHB) containing (in mM) 118.1 NaCl, 3.0 KCl, 1.8 CaCl2, 1.2 MgSO4, 1.0 KH2PO4, 27.3 NaHCO3, 10.0 glucose, and 2.5 pyruvic acid, pH 7.4, according to the method of Langendorff, at a constant rate of 8 ml/min with a peristaltic pump. Following another 10-min low-Ca2+ KHB (Ca2+ 0.01 mM), hearts were then digested with Liberase (1.25 mg/ml; Roche Diagnostics, Mannheim, Germany). Myocytes were dissociated with vigorous pipetting. The concentration of Ca2+ in KHB was increased in four increments (0.05, 0.4, 0.8, 1.2 mM). The resultant mixture was passed through 225-μm nylon mesh and washed by centrifuging at 50 g for 2 min. The centrifuge procedure was repeated until the preparation was composed of at least 80% viable left ventricular myocytes. Only those myocytes with typical striated and rod-shaped appearance were used for analyses (23).
CARDIAC MYOCYTE CONTRACTILE FUNCTION.
Measurements of the amplitude and velocity of unloaded single cardiac myocyte shortening and relengthening were made on the stage of an inverted phase-contrast microscope (Olympus, IX70-S1F2) using Myocyte Calcium Imaging/Cell Length System, in which the analog motion signal was digitized and analyzed by EDGACQ edge detection software (Ionoptix), as previously reported (6, 23). Electrical field stimulation was applied at 0.5 Hz and ∼8 V to achieve threshold depolarization. Each cell served as its own control by continuous superfusion of buffer and drugs. Cell shortening and relengthening were assessed using the following indexes: percent peak shortening (%PS) and maximal velocities of shortening (+dl/dt) and relengthening (−dl/dt), respectively.
INTRACELLULAR CALCIUM MEASUREMENTS.
To measure calcium concentrations, myocytes were incubated with 1 μM Fura 2-AM for 40 min at room temperature. The cells were alternatively illuminated at 340 and 380 nm at a frequency of 140 Hz. Emission for each excitation wavelength was filtered at 510 nm. The ratio of 340/380 represented the concentration in intracellular calcium concentration.
To detect the response to increasing concentrations of the β-adrenergic agonist ISO, doses of 10−10, 10−9, 10−8, and 10−7 M were used. To measure the response to increasing extracellular calcium concentration, doses of 0.5, 1, 2, 3, and 4 mM were applied. Response to caffeine treatment was as follows. Cardiac myocytes were perifused with 10 mM caffeine for 1 min and then followed by 30-min perfusion of KHB alone or with NAC (10−7, 10−6, 10−5, 10−4, 10−3 M, respectively).
CARDIAC MYOCYTES OXIDATIVE STRESS.
GSH/glutathione disulfide (GSSG) concentration was quantified in isolated cardiac myocytes incubated for 60 min in KHB vehicle alone, or with 10−3 M NAC in KHB. Myocyte lysis buffer (50 mM HEPES, 1 mM EDTA) was added, followed by homogenization, and subsequently centrifuged at 10,000 g for 15 min at 4°C. Supernatant was diluted fivefold, and the ratio of GSH to GSSG (GSH/GSSG) concentration ratio was measured by commercial GSH assay kit (Cayman Chemical), according to the manufacturer's protocol. Glutathione peroxidase (GPX) and catalase were determined according to protocols provided with each specific kit, as our laboratory previously reported (21, 30). Isolated cardiac myocytes were incubated for 60 min in KHB vehicle alone, or with NAC in KHB. Myocytes were washed once with KHB, and then RIPA lysis buffer [20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin; Cellsignal Tech] was added, followed by sonication and centrifuged at 14,000 g for 10 min at 4°C. Supernatants were used to measure GPX activity by commercial GSH assay kit (Cayman Chemical). In brief, oxidation of nicotinamide-adenine dinucleotide phosphate (NADPH) to NADP+ is accompanied by a decrease in absorbance at 340 nm. Under conditions in which the GPX activity is rate limiting, the rate of decrease in the A340 is directly proportional to the GPX activity in the sample. H2O2 concentration was detected in supernatants using Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen). Fifty microliters of the supernatant were incubated with 0.2 U/ml horseradish peroxidase and 100 μM Amplex Red reagent (10-acetyl-3,7-dihydrophenoxazine) at room temperature for 30 min in darkness. The fluorescence was quantified using FLUOStar Optima (BMG Lab Technologies) with excitation at 560 nm and emission at 590 nm. Catalase concentration was assayed in 50-μl fivefold diluted supernatant by Amplex Red Catalase Assay Kit (Invitrogen).
Statistical Methods
Values are expressed as means ± SE. Myocyte physiological data represent the means ± SE of 15 different determinations derived from three individual myocytes from each of five different rats. Biochemical data were derived from 10 different rats. Bonferroni-Holm test was used for paired comparisons. ANOVA was used for multigroup comparisons. Two-way ANOVA with repeated measures was used to compare the values measured in the groups. Statistical significance was accepted at the level of P < 0.05.
RESULTS
In Vivo
The two prevailing hypotheses for reversible myocardial dysfunction following behavioral stress are enhanced adrenergic stimulation and/or defective nitric oxide vascular reactivity (5, 36). These two hypotheses are not mutually exclusive and could be related to enhanced oxidative stress. Accordingly, we compared circulating plasma catecholamine concentrations between Stress and Con rats the day after their second restraint. Both NE and Epi were significantly higher in the Stress compared with Con rats following exposure to the same restraint paradigm (Fig. 1, NE levels, 1.99 ± 0.29 vs. 11.13 ± 0.55 ng/ml, Con vs. Stress, P < 0.01; n = 10; and Epi levels, 7.60 ± 0.80 vs. 21.01 ± 1.02, Con vs. Stress, P < 0.01; n = 10).
Fig. 1.

A: plasma norepinephrine (NE; 1.99 ± 0.29 Con vs. 11.13 ± 0.55 ng/ml Stress). B: epinephrine (Epi; 7.60 ± 0.80 Con vs. 21.01 ± 1.02 ng/ml Stress). Con, control rats; Stress, offspring of stressed dams that were subjected to two successive restraint stresses. Values are means ± SE; n = 10. Con vs. Stress, **P < 0.01.
In Vitro
ATP concentrations, as an indication of mitochondrial function, was significantly lower in Stress compared with Con rats (Fig. 2A, 5.16 ± 0.36 vs. 2.42 ± 0.16 μM/g total protein, Con vs. Stress, P < 0.05; n = 10). Further evidence of enhanced oxidative stress was provided by increased GPX activity (Fig. 2B, 3,125 ± 1,256 vs. 29,355 ± 10,160 mol·min−1·mg protein−1, Con vs. Stress, P < 0.01; n = 10) in the presence of increased antioxidant enzyme activities, such as catalase (Fig. 2C, 229 ± 34 vs. 307 ± 31 fluorescence/mg protein, Con vs. Stress, P < 0.01; n = 10).
Fig. 2.
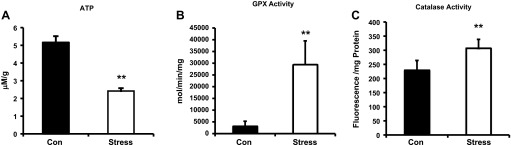
A: ATP levels (5.16 ± 0.36 vs. 2.42 ± 0.16 μM/g total protein, Con vs. Stress, **P < 0.05) measured in extracts from homogenized whole hearts. B: glutathione peroxidase (GPX) activity (3,125 ± 1,256 vs. 29,355 ± 10,160 mol·min−1·mg protein−1, Con vs. Stress, **P < 0.01) from isolated cardiac myocytes. C: catalase activity (229 ± 34 vs. 307 ± 31 fluorescence/mg protein, Con vs. Stress, **P < 0.01) from isolated cardiac myocytes. Values are means ± SE; n = 10.
NAC has been shown to raise intracellular concentrations of reduced GSH by providing an additional source of cysteine as a precursor (8, 16, 27) to reduce disulfide bonds (17) and scavenge free radicals (26). The study of inotropic effects showed that NAC (10−3 M) completely normalized the decrease in %PS in Stress rats without affecting Con (Fig. 3, 11.78 ± 0.34 vs. 7.09 ± 0.38%, Con vs. Stress, P < 0.01; 7.09 ± 0.38 vs. 12.34 ± 0.59%, Stress vs. Stress + NAC, P < 0.01; n = 15).
Fig. 3.

Baseline percent peak shortening (%PS) in Con and Stress myocytes with and without the addition of N-acetylcysteine (NAC; 10−3 M; 11.78 ± 0.34 vs. 7.09 ± 0.38%, Con vs. Stress, **P < 0.01; 7.09 ± 0.38 vs. 12.34 ± 0.59%, Stress vs. Stress + NAC, ##P < 0.01). Values are means ± SE; n = 15.
Blunted/attenuated inotropic response to adrenergic stimulation is one of the hallmark characteristics of virtually all cardiomyopathies (30). Our laboratory has previously shown in both in vitro and in vivo experiments that Stress rats shares this cardiomyopathy feature (6, 25). Accordingly, we sought to determine whether adding NAC would reverse the blunted inotropic responses of Stress rats to the β-adrenergic agonist ISO. NAC completely normalized the concentration-response curves to increasing concentrations of ISO for total %PS (Fig. 4, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15; +dl/dt, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15; and −dl/dt, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15).
Fig. 4.
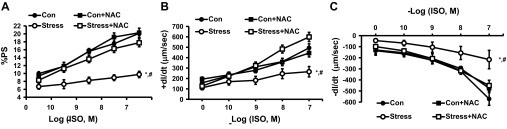
Response of cardiac myocytes to increasing concentrations of the β-adrenergic agonist, isoproterenol (ISO), alone and with incubation with NAC (10−3 M) on %PS (A), positive inotropy (+dl/dt; B), and negative inotropy (−dl/dt; C). Values are means ± SE; n = 15. Con vs. Stress, *P < 0.01; Stress vs. Stress + NAC, #P < 0.01.
ISO mediates its inotropic effects through regulation of intracellular calcium signaling in cardiac myocytes through stimulation of the β-receptor (25). Accordingly, we measured the effect of NAC on intracellular Ca2+ responses to ISO. We confirmed that the NAC-mediated inotropic effects were associated with reversing/normalizing the blunted Ca2+ responses to ISO (Fig. 5, change in Ca2+ concentration, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15; Ca2+ departure velocity, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15; and Ca2+ return velocity, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15).
Fig. 5.
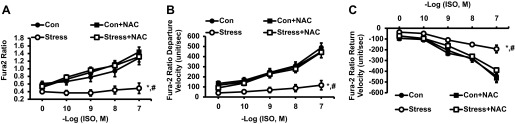
Response of cardiac myocytes to increasing concentrations of the β-adrenergic agonist, ISO, alone and with incubation with NAC (10−3 M) on change in Ca2+ concentration (A), Ca2+ departure velocity (B), and Ca2+ return velocity (C). Values are means ± SE; n = 15. Con vs. Stress, *P < 0.01; Stress vs. Stress + NAC, #P < 0.01.
Our laboratory has recently reported that NAC reversed/normalized the blunted inotropic response of cardiac myocytes to extracellular calcium using a genetic cardiomyopathy model (7). We now report that NAC similarly reversed/normalized the blunted inotropic and intracellular calcium responses to extracellular calcium (Figs. 6 and 7, %PS, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15; +dl/dt, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15, and −dl/dt, Con vs. Stress, P < 0.01, Stress vs. Stress + NAC, P < 0.01, n = 15).
Fig. 6.
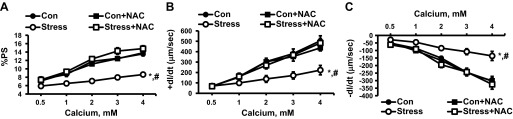
Response of cardiac myocytes to increasing concentrations of extracellular Ca2+ alone and with incubation with NAC (10−3 M) on %PS (A), +dl/dt (B), and −dl/dt (C). Values are means ± SE; n = 15. Con vs. Stress, *P < 0.01; Stress vs. Stress + NAC, #P < 0.01.
Fig. 7.
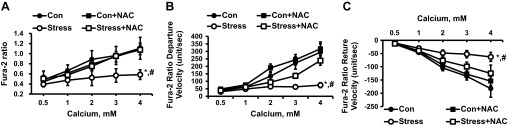
Response of cardiac myocytes to increasing concentrations of extracellular Ca2+ alone and with incubation with NAC (10−3 M) on change in Ca2+ concentration (A), Ca2+ departure velocity (B), and Ca2+ return velocity (C). Values are means ± SE; n = 15. Con vs. Stress, *P < 0.01; Stress vs. Stress + NAC, #P < 0.01.
Our laboratory has previously proposed that NAC mediates inotropic effects through regulation of intracellular calcium (7, 12). Caffeine is well known to cause release of sarcoplasmic reticulum (SR) calcium (14). Accordingly, we explored the inotropic and calcium regulatory effects of caffeine on Stress and Con rats. The addition of caffeine decreased the %PS in cardiac myocytes from both Stress and Con rats (Fig. 8, A1 and A2). However, the decrease was greater in the Stress compared with Con rats (Fig. 8A2, 44.99 ± 5.75 vs. 70.60 ± 5.09%, Con vs. Stress, P < 0.01, n = 15). The same relatively greater effect of caffeine on Stress compared with Con rats was seen in +dl/dt and −dl/dt (Fig. 8, B1 and B2, +dl/dt, 21.59 ± 3.77 vs. 40.96 ± 3.82%, Con vs. Stress, P < 0.01, n = 15; Fig. 8, C1 and C2, −dl/dt, 17.78 ± 2.17 vs. 59.41 ± 3.46%, Con vs. Stress, P < 0.01, n = 15). Caffeine also had a greater effect on the release of intracellular calcium in response to electrical field stimulation in Stress compared with Con rats (Fig. 9, A1 and A2, fura 2 ratio, 37.72 ± 5.68 vs. 60.02 ± 1.04%, Con vs. Stress, P < 0.01, n = 15; Fig. 9, B1 and B2, fura 2 departure velocity, 33.65 ± 6.97 vs. 56.74 ± 5.11%, Con vs. Stress, P < 0.01, n = 15; and Fig. 9, C1 and C2, fura 2 return velocity, 23.21 ± 4.42 vs. 50.88 ± 6.92%, Con vs. Stress, P < 0.01, n = 15). The duration of the inotropic and calcium regulatory effects of caffeine were also longer lasting in the Stress compared with Con myocytes (Figs. 10 and 11; P < 0.01, for each). The addition of NAC following caffeine significantly reversed both the inotropic and calcium regulatory effects of caffeine alone, according to measurements of %PS, +dl/dt, −dl/dt, total calcium, calcium release, and re-uptake velocities (Figs. 12, 13, and 14, P < 0.01, for each).
Fig. 8.
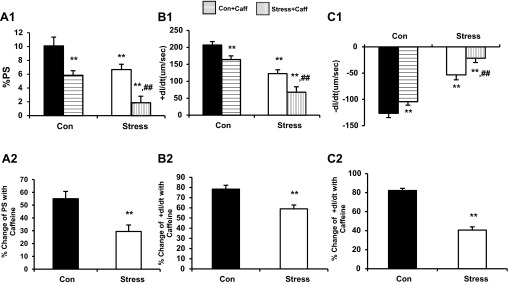
A1: %PS in cardiac myocytes before and after exposure to caffeine (10 mM) (10.10 ± 1.27 vs. 6.68 ± 0.77%, Con vs. Stress, **P < 0.01; 10.10 ± 1.27 vs. 5.8 ± 0.66%, Con vs. Con + Caff, **P < 0.01; 10.10 ± 1.27 vs. 1.88 ± 0.93%, Con vs. Stress + Caff, **P < 0.01; 6.68 ± 0.77 vs. 1.88 ± 0.93%, Stress vs. Stress + Caff, ##P < 0.01). A2: percent change in %PS of Con and Stress cardiac myocytes after exposure to caffeine (10 mM) (44.99 ± 5.75 vs. 70.60 ± 5.09%, Con vs. Stress, **P < 0.01). B1: percent change in +dl/dt of Con and Stress cardiac myocytes after exposure to caffeine (10 mM) (206.98 ± 10.41 vs. 122.74 ± 11.24 μm/s, Con vs. Stress, **P < 0.01; 206.98 ± 10.41 vs. 164.10 ± 11.0824 μm/s, Con vs. Con + Caff, **P < 0.01; 206.98 ± 10.41 vs. 67.98 ± 16.0424 μm/s, Con vs. Stress + Caff, **P < 0.01; 122.74 ± 11.24 vs. 67.98 ± 16.0424 μm/s, Stress vs. Stress + Caff, ##P < 0.01). B2: percent change in +dl/dt of Con and Stress cardiac myocytes after exposure to caffeine (10 mM) (21.59 ± 3.77 vs. 40.96 ± 3.82%, Con vs. Stress, **P < 0.01). C1: percent change in −dl/dt of Con and Stress cardiac myocytes after exposure to caffeine (10 mM) (−126.56 ± 8.06 vs. −53.28 ± 6.84 μm/s, Con vs. Stress, **P < 0.01; −126.56 ± 8.06 vs. −104.23 ± 9.32 μm/s, Con vs. Con + Caff, **P < 0.01; −126.56 ± 8.06 vs. −21.53 ± 8.24 μm/s, Con vs. Stress + Caff, **P < 0.01; −53.28 ± 6.84 vs. −21.53 ± 8.24 μm/s, Stress vs. Stress + Caff, ##P < 0.01). C2: percent change in −dl/dt of Con and Stress cardiac myocytes after exposure to caffeine (10 mM) (17.78 ± 2.17 vs. 59.41 ± 3.46%, Con vs. Stress, **P < 0.01). Values are means ± SE; n = 15.
Fig. 9.
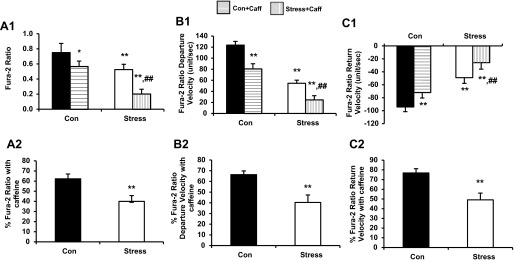
A1: changes in calcium concentration in cardiac myocytes before and after exposure to caffeine (10 mM) (0.75 ± 0.12 vs. 0.53 ± 0.07, Con vs. Stress, **P < 0.01; 0.75 ± 0.12 vs. 0.57 ± 0.07, Con vs. Con + Caff, *P < 0.05; 0.75 ± 0.12 vs. 0.20 ± 0.06, Con vs. Stress + Caff, **P < 0.01; 0.53 ± 0.07 vs. 0.20 ± 0.06, Stress vs. Stress + Caff, ##P < 0.01). A2: percentage change in calcium concentration (37.72 ± 5.68 vs. 60.02 ± 1.04%, Con vs. Stress, **P < 0.01). B1: percentage change in calcium departure velocity (123.60 ± 6.70 vs. 52.90 ± 5.64, Con vs. Stress, **P < 0.01; 123.60 ± 6.70 vs. 80.54 ± 8.91, Con vs. Con + Caff, **P < 0.01; 123.60 ± 6.70 vs. 24.19 ± 7.34, Con vs. Stress + Caff, **P < 0.01; 52.90 ± 5.64 vs. 24.19 ± 7.34, Stress vs. Stress + Caff, ##P < 0.01). B2: percentage change in calcium departure velocity (33.65 ± 6.97 vs. 56.74 ± 5.11%, Con vs. Stress, **P < 0.01). C1: percent change in calcium return velocity (−99.70 ± 6.88 vs. −49.07 ± 8.07, Con vs. Stress, **P < 0.01; −99.70 ± 6.88 vs. −71.94 ± 8.80, Con vs. Con + Caff, **P < 0.01; −99.70 ± 6.88 vs. −25.69 ± 4.52, Con vs. Stress + Caff, **P < 0.01; −49.07 ± 8.07 vs. −25.69 ± 4.52, Stress vs. Stress+Caff, ##P < 0.01). C2: percentage change in calcium return velocity (23.21 ± 4.42 vs. 50.88 ± 6.92%, Con vs. Stress, **P < 0.01). Values are means ± SE; n = 15.
Fig. 10.
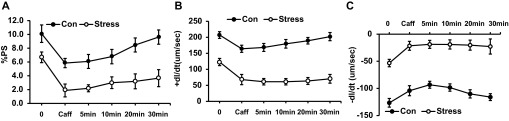
Recovery of cardiac myocytes after exposure to caffeine (10 mM). Results are measured as baseline, caffeine exposure, and time since caffeine exposure. A: %PS. B: +dl/dt. C: −dl/dt. Values are means ± SE; n = 15. Con vs. Stress, P < 0.01.
Fig. 11.
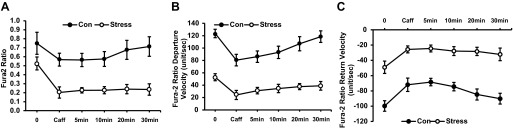
Recovery of cardiac myocytes after exposure to caffeine (10 mM). Results are measured as baseline, caffeine exposure, and time since caffeine exposure. A: change in Ca2+ concentration (Con vs. Stress, P < 0.05). B: Ca2+ departure velocity (Con vs. Stress, P < 0.01). C: Ca2+ return velocity (Con vs. Stress, P < 0.01). Values are means ± SE; n = 15.
Fig. 12.

Tracing of cardiac myocytes contraction (A) and intracellular Ca2+ concentration (B) with 30 min of perifusion of Krebs-Henseleit bicarbonate buffer alone or with NAC (10−3 M) after exposure to caffeine (10 mM).
Fig. 13.
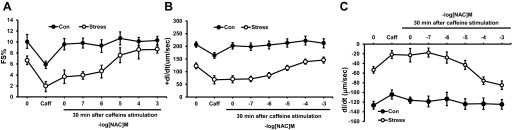
Dose-response curve of NAC and its effect on cardiac myocyte recovery after exposure to caffeine (10 mM). Results are measured as baseline, caffeine exposure, and 30 min after caffeine exposure with perfusion in buffer alone or NAC. Different myocytes were used for each concentration of NAC. A: %PS. B: +dl/dt. C: −dl/dt. Values are means ± SE; n = 15. Con vs. Stress, P < 0.01.
Fig. 14.
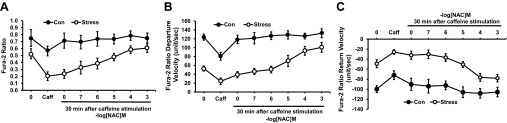
Dose-response curve of NAC and its effect on cardiac myocyte recovery after exposure to caffeine (10 mM). Results are measured as baseline, caffeine exposure, and 30 min after caffeine exposure with perfusion in buffer alone or NAC. Different myocytes were used for each concentration of NAC. A: change in Ca2+ concentration. B: Ca2+ departure velocity. C: Ca2+ return velocity. Values are means ± SE; n = 15. Con vs. Stress, P < 0.01.
GSH is a tripeptide intracellular antioxidant that plays a central role in cellular homeostasis (3). Stress myocytes revealed significantly higher oxidized GSH and lower ratio of GSH/GSSG compared with Con (Fig. 15A, GSH levels, 29.24 ± 3.84 vs. 23.63 ± 2.85 μM/g, Con vs. Stress, P = not applicable, n = 10; Fig. 15B, GSSG levels, 1.23 ± 0.27 vs. 5.46 ± 0.69 μM/g, Con vs. Stress, P < 0.01, n = 10; and Fig. 15C, GSH/GSSG ratio, 23.76 ± 4.20 vs. 5.58 ± 1.42, Con vs. Stress, P < 0.01, n = 10). NAC had no effect on GSH, GSSG, or GSH/GSSG in either Stress or Con rats (Fig. 15).
Fig. 15.
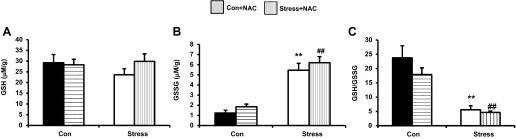
Effects of 10−3 M NAC on reduced glutathione (GSH) concentration (29.24 ± 4.75 vs. 23.63 ± 3.0, Con vs. Stress; 28.29 ± 4.07 vs. 30.05 ± 5.42, Con + NAC vs. Stress + NAC, P > 0.05; A), glutathione disulfide (GSSG) concentration (1.23 ± 0.20 vs. 5.46 ± 0.71, Con vs. Stress, **P < 0.01; 1.87 ± 0.44 vs. 6.20 ± 0.94, Con + NAC vs. Stress + NAC, ##P < 0.01; B), and on the GSH/GSSG ratio (23.76 ± 4.20 vs. 17.93 ± 2.30 μM/mg, Con vs. Con + NAC; 5.58 ± 1.42 vs. 4.66 ± 0.52, Stress vs. Stress + NAC, P = nonsignificant; Con vs. Stress **P < 0.01; Con + NAC vs. Stress + NAC, ##P < 0.01; C). Values are means ± SE; n = 10.
DISCUSSION
A compelling body of basic and clinical literature supports a pathogenic role for oxidative stress in animal models and patients with cardiomyopathies and heart failure (3). Oxidative stress is said to occur when there is an imbalance between the production of reactive oxygen species (ROS) and the antioxidant capacity of the cell. Oxidative stress is increased in failing myocardium, and studies have implicated redox-mediated mechanisms by ROS in several aspects of myocardial failure (40). Prolonged adrenergic stimulation can cause myocardial and systemic oxidative stress, with an increase in ROS without appropriate increase in antioxidants. Evidence of increased adrenergic stimulation was reflected in increased circulating catecholamine levels in Stress rats (Fig. 1).
Mitochondria are the major source of ROS due to electron leakage from the electron transport chain to react with oxygen and form superoxide (40). A delicate balance exists between the production of intracellular oxidants and antioxidants to maintain cellular homeostasis. Enhanced oxidation and/or inadequate reducing capacity result in oxidative stress from uncontrolled free radical activation. Such free radical activation in the heart can cause myocardial contractile dysfunction (2, 15, 48). Stress rats show signs of myocardial dysfunction and oxidative stress with reduced ATP levels (Fig. 2A) and GSH/GSSG (Fig. 15) in the presence of increased GPX (Fig. 2B) and catalase activities (Fig. 2C), respectively.
Both in vivo and in vitro administration of NAC have been shown to raise intracellular concentrations of reduced GSH by providing an additional source of cysteine as a precursor (8, 16, 27). GSH is a tripeptide consisting of glutamate, cysteine, and glycine, with cysteine being the limiting amino acid in the production of GSH. GSH is the main cytosolic redox buffer in cardiomyocytes with the ratio of reduced GSH/GSSG being controlled by the pentose phosphate pathway by providing NADPH, which is needed by GSH reductase to convert GSSG into GSH (40, 48). GSH can directly scavenge free radicals, including ROS, or indirectly through GPX; a high cytosolic GSH/GSSG ratio is critical for antioxidant defenses (48).
NAC can reduce disulfide bonds (17) and scavenge free radicals (26), in addition to serving as a GSH precursor (46). GSH deficiency occurs in many diseases due to a variety of factors, including viral protein-mediated GSH depletion, uncontrolled inflammatory reactions, and increased generation of free radicals. NAC is Food and Drug Administration approved for clinical use in patients as a mucolytic agent, for acetaminophen overdose (5). NAC is also being tested in clinical trials for the treatment of disease states characterized by low cysteine and GSH concentrations like human immunodeficiency virus, heart disease, cancer, and pulmonary diseases (3, 46). The doses of NAC used in humans have typically ranged from 200 to 1,000 mg orally daily (4, 11). These doses have been demonstrated to significantly increase plasma cysteine and GSH, each by ∼100 μM (4, 11). Oral NAC doses of 200–400 mg have also been reported to achieve plasma concentrations of 0.35–4 mg/l (4, 11). Accordingly, we used the concentration range of 10−3 to 10−7 M NAC for in vitro experiments based on a molecular weight of 163 and published pharmacokinetic data in human beings (10, 20).
Chronic heart failure in patients is characterized by myocardial remodeling, left ventricular dysfunction, blunted adrenergic responsiveness, and impaired myocyte calcium handling (10, 20). Contraction and force generation in muscle is regulated by cytosolic concentrations of calcium (Ca2+), which is stored in the SR and is available for immediate release into the cytosol (34). Changes in intracellular Ca2+ handling are associated with myocardial dysfunction presenting with reduced Ca2+ transient amplitude, increased Ca2+ transient duration, prolonged Ca2+ transient decay time, and decreased SR Ca2+ load (18, 19). It is clear that altered myocyte Ca2+ handling is involved in our Stress model of reversible myocardial dysfunction.
Cardiac contraction consists of an electrical excitation phase followed by a contractile phase (E-C coupling) (18, 29). The depolarization of the myocyte allows a small amount of Ca2+ to cross the sarcolemmal L-type calcium channel, where it activates the ryanodine receptor (RyR2) to release a larger amount of Ca2+ into the cytoplasm from the SR. The cytoplasmic Ca2+ then binds to troponin C, enabling actin-myosin binding and sliding of the myofilaments and resulting in sarcomere shortening and myocardial contraction. Calcium is returned to the SR through the sarco-endoplasmic reticulum calcium-ATPase (SERCA2), which resequesters Ca2+ back into the SR at the expense of ATP. Ca2+ also is removed from the cell through the sarcolemmal sodium/calcium exchanger (NCX) and plasmalemmal Ca2+-ATPase to balance the Ca2+ that entered through the L-type calcium channel (18, 29). Therefore, expression and activity of the receptors/transporters of Ca2+ release and uptake are related to the force of myocardial systolic contraction and diastolic relaxation.
Two possibilities may explain the altered Ca2+ handling in our model. First, there could be decreased re-uptake of Ca2+ into the SR due to decreased activity of SERCA2 (due to redox mechanisms altering phosphorylation status of phospholamban) with possible increase in activity of the NCX (increased extrusion of Ca2+ out of the cell helps maintain normal diastolic Ca2+ but decreases the amount available for SR re-uptake). The second possibility is Ca2+ leaking out of the SR during diastole due to dysfunctional activity of RyR2 (31). Altered function of these transporters could explain the reduced %PS, positive inotrophy (+dl/dt), negative inotrophy (−dl/dt), Ca2+ release, departure velocity, and return velocity upon stimulation with ISO (Figs. 4 and 5), extracellular calcium (Figs. 6 and 7), and caffeine (Figs. 8–14) in Stress myocytes.
PNS offspring have prolonged, exaggerated responses when presented with an acute stressor as adults due to impaired negative feedback of their hypothalamic-pituitary-adrenal axis (33). This PNS model has been shown to have increased concentrations of corticotropin-releasing factor (9) and corticosterone (43). We now show that this enhanced stress response leads to an increase in catecholamines in Stress offspring after behavioral stress, as seen by elevated Epi and NE the day after their second restraint (Fig. 1). This is important because prolonged β-adrenergic receptor activity is known to result in cardiac hypertrophy, depressed left ventricular function, and premature mortality (10). Catecholamines activate Gs protein-coupled receptors through βadrenergic receptors, leading to PKA activation (39). Activated PKA will phosphorylate RyR2, activating the Ca2+ release channel partly by increasing the sensitivity of RyR2 to cytosolic Ca2+ (31). Prolonged catecholamine signaling, however, is detrimental, leading to increased activation of PKA and hyperphosphorylation of the RyR2 receptor, Ca2+ leak out of the SR, and contractile dysfunction. This may be due to the dissociation of calstabin 2 (a RyR2 regulatory protein), from RyR2 upon PKA phosphorylation, resulting in increased sensitivity of RyR2 to Ca2+-induced activation. Prolonged increased RyR2 sensitivity to cytosolic Ca2+ leads to inappropriate Ca2+ release during diastole (Ca2+ leak) and reduces SR Ca2+ stores (31). Hyperphosphorylation of RyR2 and the dissociation of calstabin 2 uncouples multiple RyRs and disturbs coupled gating. Coupled gating is important for enabling the opening and closing of RyRs in a given SR T-tubule junction in a coordinated manner. Uncoupled channels will result in some channels not closing during diastole and Ca2+ leak (18, 31). Reduced phosphatase association with RyR2 could play a role in Ca2+ leak with decreased inactivation of RyR2 (31). Stress animals had reduced concentrations of ATP (Fig. 2). This could be due to increased SERCA2 activity in an effort to compensate for Ca2+ leak through RyR2 (18, 48).
Additional evidence that redox mechanisms can affect the activity of several Ca2+ transporters has previously been reported (39, 43). Oxidation via cysteine caused formation of disulfide bonds and opening of the channel and SR Ca2+ release. Conversely, addition of sulfhydryl reducing agents like reduced GSH reduced the disulfide bonds, regenerating the free sulfhydryl groups on SR proteins, closing the channel and activating re-uptake of Ca2+ (39, 43). Prolonged adrenergic stimulation has been suggested to cause myocardial and systemic oxidative stress (40). ROS are capable of reacting with cysteines, causing a conformational change in RyR2 and Ca2+ leak (48). SERCA2 may decrease in activity as a result of oxidation by ROS, while the NCX can be activated by ROS (4, 11, 48). In addition, p38 mitogen-activated protein kinase is redox sensitive and is activated by ROS (37). Stress offspring have evidence of mitochondrial dysfunction and oxidative stress seen by reduced ATP (Fig. 2) and GSH/GSSG (Fig. 15), respectively. Reversible oxidation of components of the E-C coupling machinery is minimal in normal myocytes (42). This concept agrees with our observations that NAC was able to reverse dysfunction in Stress myocytes, but had no effect on Con myocytes (Figs. 3–14).
There is a blunted inotropic response to the β-adrenergic agonist, ISO, in the Stress model (Fig. 4). This may be because RyR2 is already PKA hyperphosphorylated, and further phosphorylation (through β-adrenergic receptors/Gs pathway) cannot occur (31). Phosphorylation of RyR2 increases the probability that it will be in the open state; hyperphosphorylation can result in incomplete channel closure and allows Ca2+ to leak out of the SR. Therefore, upon stimulation, these myocytes have a lower concentration of Ca2+ available for release from the SR compared with normal myocytes (46). Stress myocytes had reduced SR Ca2+ release when stimulated with ISO or calcium (Figs. 5 and 6), NAC (10−3 M) reversed this defect in the Stress myocytes without affecting the Con myocytes (Figs. 5 and 6). NAC (10−3 M) also reverses the positive (+dl/dt) and negative (−dl/dt) inotropic dysfunction in response to ISO and extracellular calcium (Figs. 4 and 6) in the Stress myocytes without affecting the Con myocytes. This may occur through redox mechanisms of NAC on RyR2, closing the channel and stopping the Ca2+ leak.
Exposure of myocytes to caffeine sensitizes RyR2 to activation by Ca2+ and leads to a massive release of Ca2+ from the SR. Caffeine potentiates SR Ca2+ release and myocyte contraction only transiently because it depletes the SR of Ca2+ (46). After initial positive inotropic effects of caffeine exposure, the amplitude of further contractions and SR Ca2+ release will be reduced until the Ca2+ is returned to the SR through SERCA2. This pattern was seen in both Stress and Con myocytes exposed to caffeine (data not shown). Further caffeine exposure resulted in a reduction in amplitude of both contraction and intracellular Ca2+ with return to baseline in Con myocytes within 30 min (Figs. 10 and 11). Stress myocytes were unable to return to baseline within 30 min (Figs. 10 and 11). Perifusion of the myocytes with NAC (10−3) after caffeine exposure allowed the Stress myocytes to return to baseline, while having no effect on the Con myocytes, which returned to baseline without NAC (Figs. 13 and 14). This indicates that the Stress myocytes were not effectively taking Ca2+ back up into the SR, which would point to decreased SERCA2 activity. However, increased Ca2+ leak through RyR2 also could play a role.
In summary, changes in myocyte Ca2+ handling could be attributed partially to redox modifications and/or phosphorylation of Ca2+ transporters and/or their regulatory proteins. Chronically elevated circulating catecholamines can modify these transporters/proteins by phosphorylation through PKA (39) or by redox mechanisms through oxidative stress (42) in the PNS model. NAC, possibly through redox mechanisms, can reverse the myocardial dysfunction seen in this model after behavioral stress. While NAC was able to reverse the dysfunction in Stress myocytes in response to ISO, extracellular calcium, and caffeine, NAC failed to normalize both GSSG and GSH/GSSG levels in Stress myocytes (Figs. 2–7). These data indicate that NAC mediates its inotropic effects independent of increasing GSH and mostly functions through redox mechanisms in the PNS model. Indeed, reversal of both positive and negative inotrophy by NAC is consistent with sulfhydryl regulation of RyR2 or SERCA2. Considerably more detailed physiological and biochemical studies will be necessary before drawing any definitive conclusions regarding which transporters are affected by NAC in this model.
PNS has gained widespread acceptance as an animal model for chronic behavioral stress and mood disorders in humans. PNS rats share important characteristic behavioral, anatomical, and biochemical features with humans suffering from posttraumatic stress disorder and major depressive disorder (13, 41). PNS rats revealed no significant echocardiographic or hemodynamic changes from Con rats at 6 wk of age. However, further behavioral stress alone in PNS rats resulted in both systolic and diastolic myocardial dysfunction and β-adrenergic desensitization in vivo and cardiac myocyte dysfunction in vitro (6, 23). Inhibition of p38 mitogen-activated protein kinase phosphorylation both prevented and reversed the myocardial dysfunction and adrenergic desensitization in vivo and in vitro (6, 24). Our Stress model exhibited myocardial dysfunction following behavioral stress alone, without any of the other known mechanical, chemical, or infectious insults. Reports supporting the clinical relevance of an association between acute behavioral stress alone and myocardial dysfunction appeared in studies of “stress-induced”, “Takotsubo”, or “Broken Heart” cardiomyopathy (1, 45). Characteristics of this pathology include close temporal association with behavioral stress, complete reversibility within weeks, blunted adrenergic responsiveness, and elevated circulating catecholamines. Our Stress model shares these characteristics.
Oxidative stress is one potential biological mechanism relating behavioral stress to adverse cardiovascular outcomes (2, 22, 32). Possible molecular mechanisms linking oxidative stress to myocardial dysfunction include redox-mediated changes in myocardial E-C coupling. We first reported a defect in sulfhydryl gating of the RyR2 in a genetic hamster cardiomyopathy model using the sulfhydryl donor, NAC (12). A defect in a dystrophin-associated protein was subsequently shown by others to be responsible for the genetic hamster cardiomyopathy (38). NAC has also recently been shown to be cardioprotective in the mdx dystrophin mouse model of muscular dystrophy (44). The mechanism of the benefit of NAC was not explained. However, NAC did not reduce free radical generation by NADPH (28, 44). Most recently, our laboratory reported that NAC reversed cardiac myocyte dysfunction in the Tat transgenic cardiomyopathy model through GSH-independent mechanisms (7). The present study provides further support for GSH independent salutary effects of NAC. An alternative possibility is an effect of NAC on sulhydryl regulation of RyR2. This possibility has been suggested by recent reports of the pathogenic role of phosphorylation, nitrosylation, and oxidation of RyR2 in cardiomyopathies and heart failure (29, 35). Future efforts should focus on elucidating the molecular pathways in this animal model that may ultimately serve as pharmacological targets for reversing myocardial dysfunction in cardiomyopathies and heart failure in patients.
GRANTS
This work was supported by a Department of Veteran's Affairs MERIT Award and National Heart, Lung, and Blood Institute Grant RO-1 HL-70565.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.C., C.B., J.M. Hollander, D.B.M., and M.S.F. conception and design of research; F.C., J.M. Hadfield, and C.E.N. performed experiments; F.C. and C.E.N. analyzed data; F.C., J.M. Hollander, and M.S.F. interpreted results of experiments; F.C. and M.S.F. prepared figures; F.C. and J.M. Hadfield drafted manuscript; F.C. and M.S.F. edited and revised manuscript; F.C. and M.S.F. approved final version of manuscript.
REFERENCES
- 1. Akashi YJ, Goldstein DS, Barbaro G, Ueyama T. Takotsubo cardiomyopathy: a new form of acute, reversible heart failure. Circulation 118: 2754–2762, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akki A, Zhang M, Murdoch C, Brewer A, Shah AM. NADPH oxidase signaling and cardiac myocyte function. J Mol Cell Cardiol 47: 15–22, 2009 [DOI] [PubMed] [Google Scholar]
- 3. Atkuri KR, Mantovani JJ, Herzenberg LA. N-acetylcysteine–a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 7: 355–359, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borges-Santos MD, Moreto F, Pereira PC, Ming-Yu Y, Burini RC. Plasma glutathione of HIV(+) patients responded positively and differently to dietary supplementation with cysteine or glutamine. Nutrition 28: 753–756, 2012 [DOI] [PubMed] [Google Scholar]
- 5. Bybee KA, Prasad A. Stress-related cardiomyopathy syndromes. Circulation 118: 397–409, 2008 [DOI] [PubMed] [Google Scholar]
- 6. Chen F, Kan H, Hobbs G, Finkel MS. p38 MAP kinase inhibitor reverses stress-induced myocardial dysfunction in vivo. J Appl Physiol 106: 1132–1141, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen F, Lewis W, Hollander JM, Baseler WA, Finkel MS. N-acetylcysteine reverses cardiac myocyte dysfunction in HIV-Tat proteinopathy. J Appl Physiol 113: 105–113, 2012 [DOI] [PubMed] [Google Scholar]
- 8. Choy KH, Dean O, Berk M, Bush AI, Buuse MV. Effects of N-acetyl-cysteine treatment on glutathione depletion and a short-term spatial memory deficit in 2-cyclohexene-1-one-treated rats. Eur J Pharmacol 649: 224–228, 2010 [DOI] [PubMed] [Google Scholar]
- 9. Cratty MS, Ward HS, Johnson EA, Azzaro AJ, Birkle DL. Prenatal stress increases corticotropin-releasing factor (CRF) content and release in rat amygdala minces. Brain Res 675: 297–302, 1995 [DOI] [PubMed] [Google Scholar]
- 10. Dash R, Schmidt AG, Pathak A, Gerst MJ, Biniakiewicz D, Kadambi VJ, Hoit BD, Abraham WT, Kranias EG. Differential regulation of p38 mitogen-activated protein kinase mediates gender-dependent catecholamine-induced hypertrophy. Cardiovasc Res 57: 704–714, 2003 [DOI] [PubMed] [Google Scholar]
- 11. De Rosa SC, Zaretsky MD, Dubs JG, Roederer M, Anderson M, Green A, Mitra D, Watanabe N, Nakamura H, Tjioe I, Deresinski SC, Moore WA, Ela SW, Parks D, Herzenberg LA. N-acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest 30: 915–929, 2000 [DOI] [PubMed] [Google Scholar]
- 12. Finkel MS, Shen L, Oddis CV, Romeo RC, Salama G. Positive inotropic effect of acetylcysteine in cardiomyopathic Syrian hamsters. J Cardiovasc Pharmacol 21: 29–34, 1993 [DOI] [PubMed] [Google Scholar]
- 13. Fride E, Weinstock M. Prenatal stress increases anxiety related behavior and alters cerebral lateralization of dopamine activity. Life Sci 42: 1059–1065, 1988 [DOI] [PubMed] [Google Scholar]
- 14. Gaburjakova J, Gaburjakova M. Comparison of the effects exerted by luminal Ca2+ on the sensitivity of the cardiac ryanodine receptor to caffeine and cytosolic Ca2+. J Membr Biol 212: 17–28, 2006 [DOI] [PubMed] [Google Scholar]
- 15. Goldhaber JI, Qayyum MS. Oxygen free radicals and excitation-contraction coupling. Antioxid Redox Signal 2: 55–64, 2000 [DOI] [PubMed] [Google Scholar]
- 16. Han YH, Yang YM, Kim SZ, Park WH. Attenuation of MG132-induced HeLa cell death by N-acetyl cysteine via reducing reactive oxygen species and preventing glutathione depletion. Anticancer Res 30: 2107–2112, 2010 [PubMed] [Google Scholar]
- 17. Harada D, Anraku M, Fukuda H, Naito S, Harada H, Suenaga A, Otagiri M. Kinetic studies of covalent binding between N-acetyl-L-cysteine and human serum albumin through a mixed-disulfide using an N-methylpyridinium polymer-based column. Drug Metab Pharmacokinet 19: 297–302, 2004 [DOI] [PubMed] [Google Scholar]
- 18. Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol 34: 951–969, 2002 [DOI] [PubMed] [Google Scholar]
- 19. Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation 103: 1577–1584, 2001 [DOI] [PubMed] [Google Scholar]
- 20. Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clin Pharmacokinet 20: 123–134, 1991 [DOI] [PubMed] [Google Scholar]
- 21. Hollander J, Gore M, Fiebig R, Mazzeo R, Ohishi S, Ohno H, Ji LL. Spaceflight downregulates antioxidant defense systems in rat liver. Free Radic Biol Med 24: 385–390, 1998 [DOI] [PubMed] [Google Scholar]
- 22. Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 295: C849–C868, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kan H, Birkle D, Jain AC, Failinger C, Xie S, Finkel MS. p38 MAP kinase inhibitor reverses stress-induced cardiac myocyte dysfunction. J Appl Physiol 98: 77–82, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Kan H, Failinger CF, Fang Q, Finkel MS. Reversible myocardial dysfunction in sepsis and ischemia. Crit Care Med 33: 2845–2847, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Kan H, Finkel MS. Inflammatory mediators and reversible myocardial dysfunction. J Cell Physiol 195: 1–11, 2003 [DOI] [PubMed] [Google Scholar]
- 26. Kelly GS. Clinical applications of N-acetylcysteine. Altern Med Rev 3: 114–127, 1998 [PubMed] [Google Scholar]
- 27. Kerksick C, Willoughby D. The antioxidant role of glutathione and N-acetyl-cysteine supplements and exercise-induced oxidative stress. J Int Soc Sports Nutr 2: 38–44, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A 107: 15565–15570, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kushnir A, Marks AR. The ryanodine receptor in cardiac physiology and disease. Adv Pharmacol 59: 1–30, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Leeuwenburgh C, Hollander J, Leichtweis S, Griffiths M, Gore M, Ji LL. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am J Physiol Regul Integr Comp Physiol 272: R363–R369, 1997 [DOI] [PubMed] [Google Scholar]
- 31. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101: 365–376, 2000 [DOI] [PubMed] [Google Scholar]
- 32. Masood A, Nadeem A, Mustafa SJ, O'Donnell JM. Reversal of oxidative stress-induced anxiety by inhibition of phosphodiesterase-2 in mice. J Pharmacol Exp Ther 326: 369–379, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mastorci F, Vicentini M, Viltart O, Manghi M, Graiani G, Quaini F, Meerlo P, Nalivaiko E, Maccari S, Sgoifo A. Long-term effects of prenatal stress: changes in adult cardiovascular regulation and sensitivity to stress. Neurosci Biobehav Rev 33: 191–203, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Prabhu SD, Salama G. Reactive disulfide compounds induce Ca2+ release from cardiac sarcoplasmic reticulum. Arch Biochem Biophys 282: 275–283, 1990 [DOI] [PubMed] [Google Scholar]
- 35. Prosser BL, Ward CW, Lederer WJ. Subcellular Ca2+ signaling in the heart: the role of ryanodine receptor sensitivity. J Gen Physiol 136: 135–142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richard C. Stress-related cardiomyopathies. Ann Intensive Care 1: 39, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saitoh S, Kiyooka T, Rocic P, Rogers PA, Zhang C, Swafford A, Dick DM, Viswanathan C, Park Y, Chilian WM. Redox-dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol 293: H3720–H3725, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, Masaki T, Toyo-oka T, Hanaoka F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci U S A 94: 13873–13878, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Salama G, Abramson JJ, Pike GK. Sulphydryl reagents trigger Ca2+ release from the sarcoplasmic reticulum of skinned rabbit psoas fibres. J Physiol 454: 389–420, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol 34: 379–388, 2002 [DOI] [PubMed] [Google Scholar]
- 41. Takahashi LK, Baker Ew, Kalin NH. Ontogeny of behavioral and hormonal responses to stress in prenatally stressed male rat pups. Physiol Behav 47: 357–364, 1990 [DOI] [PubMed] [Google Scholar]
- 42. Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 103: 1466–1472, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ward HE, Johnson EA, Salm AK, Birkle DL. Effects of prenatal stress on defensive withdrawal behavior and corticotropin releasing factor systems in rat brain. Physiol Behav 70: 359–366, 2000 [DOI] [PubMed] [Google Scholar]
- 44. Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol 293: H1969–H1977, 2007 [DOI] [PubMed] [Google Scholar]
- 45. Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, Wu KC, Rade JJ, Bivalacqua TJ, Champion HC. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med 352: 539–548, 2005 [DOI] [PubMed] [Google Scholar]
- 46. Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 60: 6–20, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem 76: 367–385, 2007 [DOI] [PubMed] [Google Scholar]
- 48. Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 71: 310–321, 2006 [DOI] [PubMed] [Google Scholar]