

Abstract
Microglia, the resident immune cells of the central nervous system (CNS), play an important role in CNS homeostasis during development, adulthood and ageing. Their phenotype and function have been widely studied, but most studies have focused on their local interactions in the CNS. Microglia are derived from a particular developmental niche, are long-lived, locally replaced and form a significant part of the communication route between the peripheral immune system and the CNS; all these components of microglia biology contribute to maintaining homeostasis. Microglia function is tightly regulated by the CNS microenvironment, and increasing evidence suggests that disturbances, such as neurodegeneration and ageing, can have profound consequences for microglial phenotype and function. We describe the possible biological mechanisms underlying the altered threshold for microglial activation, also known as ‘microglial priming’, seen in CNS disease and ageing and consider how priming may contribute to turning immune-to-brain communication from a homeostatic pathway into a maladaptive response that contributes to symptoms and progression of diseases of the CNS.
Keywords: Microglia, Neurodegeneration, Priming, Systemic inflammation
The concept that the central nervous system (CNS) is an immunologically privileged organ has had a profound impact on the study of immune-to-brain interactions. Even though it is now well recognized that this immune privilege is only partial [1], for many neuroscientists, the nervous system is still perceived to be isolated from the immune system, with its components playing little or no role in the neural networks and communicating pathways. It is also the case that for many immunologists, the CNS is a foreign land. It is clear that the interaction between the nervous and immune systems is a two-way communication, each having a profound influence on the other. These influences extend from homeostasis to pathology, with the immune system acting as a major transducer of both internal and external environmental challenges to the host. Involvement of the immune system can arise from CNS resident innate immune cells or from cellular and soluble factors from the periphery that act on the CNS via neural or humoural routes. The heterogeneity of the macrophage populations in the CNS is reflected in the differential responses in different tissue compartments each playing distinct roles in homeostasis and pathology.
The macrophage populations of the CNS include the microglia, perivascular macrophages (PVM), meningeal macrophages (MM), macrophages of the circumventricular organs and the macrophages of the choroid plexus. The most abundant and best studied of these are the microglia (Fig. 1a, c). We focus here on recent findings that highlight the role of ‘priming’ of microglia during neurodegenerative disease and ageing and suggest how this may contribute to neurological disease.
Fig. 1.
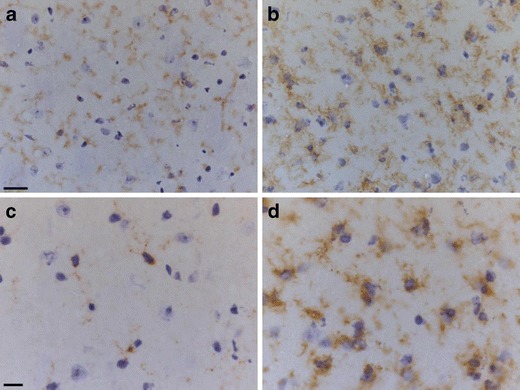
Microglia in the naïve mouse brain express CD11b (a) and CD68 (c). In an animal with a chronic neurodegenerative disease, prion disease, the microglia increase in number and become activated, altering their morphology with upregulation of CD11b (b) and CD68 (d). Scale bar in a, b = 20 μm, c, d = 10 μm
Microglia in the developing brain
The notion that the microglia are the resident macrophages of the CNS is well accepted, but precisely from where they originate and when they populate the tissue has been a matter of much debate [2]. Microglia are long-lived cells, and studies in mouse have suggested that they are predominantly maintained via self-renewal under steady-state conditions [3]. The recent study of Ginhoux and colleagues provides good evidence in mouse that a significant proportion of these cells arise from the yolk sac and populate the neuroepithelium in early (before E10.5) embryogenesis [4]. However, what is less clear is whether, and to what extent, there is a contribution from a bone marrow myeloid progenitor at later stages of embryonic development or in the postnatal period. Pioneering work by Rio Hortega in the early 1900s and subsequent studies showed that the microglia appear to invade the developing brain from the vasculature at a time when bone marrow hematopoiesis is ongoing [5, 6]. Ginhoux et al. [4] report that only a proportion of the cells were labelled by the genetic manipulation that defined them as of yolk sac origin, which was attributed to the inefficiency of Cre activation by the Runx promotor. While these studies have provided essential insight into the developmental origins of microglia, the potential contribution of non-yolk sac origin macrophages throughout the entire life course and after repeated immune challenges remains to be resolved.
The yolk-sac-derived macrophages that invade the primitive nervous system, the neuroepithelium, from the early vasculature have a typical macrophage morphology and phenotype, characterized by high expression of F4/80 and CD11b. As the neural tissue matures, there is widespread apoptotic cell death of both neurons and glia, and the embryonic and postnatal microglia are involved in the phagocytosis of these apoptotic bodies, implying that embryonic microglia are involved in determining the circuitry of the developing CNS [6]. It has been proposed that microglia may actively kill some neurons during the period of cell death, but to date, this has only been convincingly demonstrated in vitro [7]. The role of microglia in sculpting nervous system circuitry during this period of development, when there is exuberant axonal and synaptic connectivity, is a subject of much interest. In mice lacking components of the complement cascade, there is an excess convergence of connections from the retinal ganglion cells of the eye onto neurons in the dorsal lateral geniculate nucleus (dLGN) of the thalamus that is maintained until later in development than normal [8, 9]. It is proposed that complement component C1q decorates synapses that are destined to degenerate and then microglia recognize and remove these opsonized synapses [8, 9]. The notion that microglia are actively involved in the removal of synapses from their postsynaptic targets has a long history, and it was first invoked as a response by microglia in the removal of synapses from damaged or injured neurons, so-called synaptic stripping. As reviewed elsewhere [10], the evidence that microglia are active participants in synaptic stripping is unclear, and direct evidence that microglia recognize healthy synapses and select those which are about to lose the competition for a postsynaptic connection remains unproven. In contrast to the visual system, studies on the hippocampus of CX3C-receptor (CX3CR)-null mice describe the microglia as phagocytosing material derived from the postsynaptic density of the dendritic spine, the protein PSD-95 [11], rather than the pre-synaptic elements as described in the visual system. Although it was suggested that this form of synaptic pruning by microglia was necessary for normal development, the number of spines was only transiently different from normal at 15 days postnatal and by 40 days postnatal had achieved normal levels.
If the microglia are exclusively derived from the yolk sac during development, the interesting question is raised as to how the population is subsequently maintained during life: what are the relative contributions of local proliferation and recruitment from the blood, and are these pathways similar under healthy and diseased conditions? Lawson et al. [3], using tritiated thymidine with immunocytochemistry, demonstrated that microglia, under physiological conditions, proliferate more slowly than other tissue macrophage populations and are maintained in large part by local proliferation. To study the degree of recruitment from the blood, a number of laboratories have used bone marrow chimeras (BM-chimeras) with protein polymorphisms, genetic labels, Y-chromosome or more recently GFP to identify the donor cells [12, 13]. However, these studies have been shown to suffer from a serious artefact as a consequence of the irradiation strategy [14]. Evidence from parabiotic animals [15] or following the generation of BM-chimeras with a head-shield to prevent conditioning of the CNS by the radiation [13, 16] showed very little evidence of invasion of the brain parenchyma from the bone marrow. It appears, as previously described [3], that local division regulates the microglia population at least in mice in pathogen-free laboratory conditions. Using BrdU labelling of dividing cells in animals in which the microglia are labelled with GFP driven from the colony-stimulating factor receptor-1 (CSFR1) promotor [17], there is indeed significant proliferation of the resident cells with some variation from one region of the brain to another [18]. The turnover of the microglia is influenced by systemic events: microglia in the neurohypophysis proliferate during osmotic stress [19] and more generally following systemic challenge with lipopolysaccharide (LPS) [20]. The increase in macrophages and microglia in the autoimmune disease experimental allergic encephalomyelitis (EAE) comes from both the proliferation of the microglia and invasion of monocytes [21].
One important message from the generation of BM-chimeras is that microglia morphology is not unique to yolk-sac-derived cells since some donor BM-derived cells can invade the parenchyma and develop morphology typical of microglia. This simple observation indicates that microglia morphology is a property largely imposed by the local microenvironment rather than their developmental origin. Deletion of microglia by another paradigm, using local cortical injection of ganciclovir to locally kill CD11b + microglia expressing HSV-thymidine kinase, led to re-population of the cortex by blood-derived monocytes within 2 weeks. While these cells developed morphological features of microglia, they were subtly distinct from the resident cells, and they had higher CD45 expression, even many months after invasion [22].
An important point to highlight is the marked species difference in the response to generation of BM-chimeras. The invasion of the irradiated CNS by grafted bone-marrow-derived cells may be a particular property of mice since the same experimental approach in rats, including irradiation of the brain, results in population of the perivascular space by BM-derived cells but not the brain parenchyma [23]. Few studies have been carried out in larger animals, but in macaque monkey, the evidence suggests that transplanted adult CD34-positive stem cell progeny does not invade the parenchyma but only the perivascular space [24], and similarly, in humans with bone marrow transplants, BM-derived cells appear only to enter the perivascular space [25].
Perivascular and meningeal macrophages
In contrast to the microglia, the other populations of CNS macrophages have received much less attention with regard to their developmental origin and turnover. These macrophages have a distinct morphology and phenotype when compared to the microglia, illustrating the profound effect of the local microenvironment. The BM-chimera studies, be they in mouse with a head shield [16], rats or primates, show that perivascular macrophages are regularly replaced by cells recruited from the blood [23, 24]. The precise time period for replacement is not well described but usually reported as periods of months rather than days. The turnover of the macrophages in the circumventricular organs (CVOs) and choroid plexus has not been systematically studied. Recent studies have described the role of pericytes in the regulation of local blood flow and blood-brain barrier permeability, but their role in neuroinflammation and interactions with PVMs and microglia remains to be defined [26].
Cellular and solute drainage from the brain
An important question with regard to immune–brain communication is whether immune cells in the CNS can migrate from the CNS to lymphoid organs. The brain lacks a typical lymphatic drainage system, although interestingly, it is not known what components of the CNS microenvironment prevent the formation of lymphatic vessels. Despite the absence of lymphatics, there are well-described drainage routes for soluble molecules from the brain parenchyma along the perivascular space of the arteries into the cerebrospinal fluid and hence to the cervical lymph nodes in the neck [27]. It is a contentious issue as to whether these routes might also be a lymphatic-like drainage route for PVMs and MMs to carry CNS or pathogen-derived antigens to cervical lymph nodes. In the normal healthy brain, it has been argued that the absence of dendritic cells in the brain parenchyma or perivascular space and the lack of migratory capacity of both microglia and PVMs account for the immune privilege of the CNS parenchyma [1] and permit bacterial and viral particles to persist in the parenchyma undetected by the immune system [28]. By contrast, delivery of bacterial or viral particles to the ventricles or surface of the brain leads to a prompt immune response [29]. This distinct compartmentalization of macrophage phenotypes may also be related to the very different responses of the parenchyma and meninges to challenge with agents such as lipopolysaccharide or cytokines that evoke stereotypic acute inflammatory responses in the meninges but not in the brain parenchyma [30].
Regulation of microglia phenotype
Under steady-state conditions, microglia display a striking down-regulated phenotype when compared with other CNS macrophages with low expression of CD45, Fc receptors and MHC class II. A recent study assessing gene expression in tissue macrophages from various mouse organs showed that all tissue macrophages express mRNA encoding diverse receptors such as the Mer tyrosine kinase receptor (MerTK), involved in phagocytosis of apoptotic cells, and mRNA encoding the toll-like receptors TLR4, TLR7, TLR8 and TLR13 involved in the recognition of microbial pathogen-derived molecules [31]. Microglia in the normal healthy CNS display particularly low-level expression of hundreds of transcripts that are expressed in other macrophage populations, but enrichment of genes related to oxidative metabolism. This study not only extends the notion of the down-regulated phenotype of microglia under steady-state conditions, but also points to the role of microglia in homeostasis and first line of defence: they are well equipped to remove debris and apoptotic cells, respond to infectious and non-infectious danger signals and regulate oxidative processes.
The local microenvironment and, in particular, neurons and astrocytes play an integral role in regulating microglia phenotype via interaction with both soluble and membrane-bound mediators. Neurotropic factors released by neurons contribute to the down-regulated state of microglia and dampen inflammation in the CNS. For example, nerve growth factor inhibits MHC class II expression on microglia [32], and brain-derived neurotrophic factor down-regulates expression of co-stimulatory molecules B7 and CD40 [33]. Microglia express diverse receptors for neurotransmitters [34], and neurotransmitters such as norepinephrine (NE) and γ-aminobutyric acid (GABA) have immune modulatory effects by suppressing pro-inflammatory cytokine production. For example, microglia and astrocytes, which are in intimate communication with microglia, express GABA(A) and GABA(B) receptors; activation of these receptors suppresses LPS or interferon-γ-mediated inflammation by inhibition of NFκB and P38 MAP kinase [35].
Apart from soluble factors, cell–cell-mediated modulation of microglia also contributes to a down-regulated phenotype. Membrane-bound proteins on neurons include CD200 and the chemokine fractalkine (CX3CL1) [36, 37], and each binds their receptor on microglia CD200R and CX3CR1, respectively. Both CD200R and CX3CR1 are transmembrane proteins on microglia that carry ITIM motifs (immunoreceptor tyrosine-based inhibitory motif) in their intracellular domain. Upon crosslinking, these ITIM-bearing receptors recruit SHP-1 and SHP-2 phosphatases, which inhibit down-stream immune signalling [38]. Thus, these neuron-microglia ligand-receptor interactions play an important role in the down-regulation of the microglia phenotype. This is in contrast to transmembrane receptors that signal via ITAM motifs (immunoreceptor tyrosine-based activation based motif), which, following crosslinking, recruit Syk which results in phosphorylation of down-stream signalling pathways and subsequent cell activation [39]. There is a growing list of ligand-receptor interactions regulating the microglia phenotype which includes the receptors TREM-2 and Siglecs expressed on microglia, for example, Siglec 11 (human) or Siglec F (rodent) contribute to immune regulation by binding sialic acid residues on neurons (Fig. 2) [40]. TREM-2 may contribute to immune regulation via recruitment of the ITAM-containing adapter protein DAP-12, which results in inhibitory cell signalling following tonic or monovalent engagement with intact neurons [41].
Fig. 2.
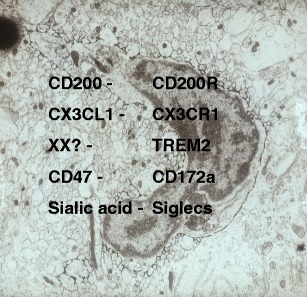
Electron micrograph of a microglia cell in the mouse brain labelled with F4/80. Note the sparse cytoplasm and limited organelles typical of the down-regulated phenotype of microglia. Ligands expressed by neurons (left column) bind receptors on microglia (right column) that inhibit their activation
An important consequence of these interactions between neurons and microglia is that in conditions where neurons degenerate, either acutely or in chronic neurodegenerative disease, the microglia will be released from the tonic inhibition provided by neurons. There is also evidence that in animal models mimicking aspects of human neurodegenerative disease, the neuropathology may be enhanced by blockade of CD200–CD200R interactions [42] or deletion of CX3CR1 [43]. Mutations in TREM-2 and CD33 have been associated with increased risk of Alzheimer’s disease, highlighting the importance of immune suppression, mediated by this class of immune receptors in maintenance of CNS homeostasis [44–46].
Microglia in the adult brain
Microglia are ubiquitously distributed in the adult CNS, but their distribution is not uniform. In the rodent brain, their density varies from regions with a high density, for example, the substantia nigra, to a low density, such as the molecular layer of the cerebellum; the fibre tracts also have a low density [47]. The microglia each occupy their own territory, and in vivo imaging studies reveal that they continually palpate the neurons and glia within their territory such that they survey their ‘sphere of influence’ about every hour [48]. The significance of the differential density distribution and the differences in morphology, the length and density of processes and local territory covered is largely a matter of conjecture, but it is not simply related to the extent of developmental cell death, the density of the vasculature or routes of entry into the CNS [47]. The microglia express receptors for diverse neurotransmitters [34], but the distribution of microglia has not been readily related to any particular transmitter.
The regional differences in microglia density and morphology have led to investigations into possible functional differences in the adult naïve state, but expression of immune molecules such as CD80/CD86, CD11b, F4/80 and CXCR3 has only revealed subtle differences in young rodents [49]; the similarities are more striking than the differences. Recent evidence suggests that there is heterogeneity in the microglia response to a local inflammatory challenge, but whether this represents the response of distinct subgroups of microglia [50] or the stochastic nature of the transcriptional response [51] remains to be established. In the ageing CNS, the microglia show readily detectable regional differences, as discussed below.
CNS macrophages and microglia: the immune–brain interface
Although the phenotype and function of macrophages and microglia have predominantly focused on their role within the CNS and their very local interactions, it is known that these cells play an important role in communication between the systemic immune system and the brain. Systemic infection and inflammation lead to metabolic and behavioural changes that play an important role in protecting an organism and promoting recovery. The typical signs of a systemic infection lead to behaviours commonly referred to as “sickness behaviours” that include fever, malaise, lethargy, anorexia and depression [52]. The routes by which systemic inflammation communicates with the brain include both neural and humoural pathways. Receptors for cytokines and other inflammatory mediators on afferent vagus nerve fibres respond to these molecules and signal to the nucleus of the solitary tract and hence to other regions of the CNS [53]. Circulating cytokines and inflammatory mediators may communicate with macrophages in the circumventricular organs: regions of the CNS that lack a typical tight blood-brain barrier. Signals generated in the CVOs then communicate with neural signalling pathways in the brain [54]. A further route of communication involves signalling from mediators in blood to the cerebral endothelial cells, which signal to the perivascular macrophages which, in turn, signal to the microglia. The signalling from the cerebral endothelium involves activation of COX-1 and COX-2 and the generation of prostaglandins and likely other mediators [55]. The synthesis of cytokines such as interleukin-1β (IL-1) in the PVMs, microglia and cerebral endothelial cells has been demonstrated following systemic LPS challenge [56]. Indeed, microinjection of IL-1 and other cytokines into the cerebral ventricles or local regions of the CNS can evoke sickness behaviours [57].
The signalling of systemic inflammation to the CNS, arising as a consequence of injury or disease, is part of normal homeostasis; it takes place across an intact blood-brain barrier and does not lead to damage of the neurons of the CNS. Although most of what we know about signalling from the peripheral immune system to the brain has been studied in rodents, experimental studies in humans provide evidence for similar effects of systemic inflammation. LPS challenge in humans leads to fever and neuropsychological symptoms. For example, low-dose endotoxin in healthy volunteers reduced declarative memory performance that was inversely correlated with cytokine increases [58] and increased symptoms of depression [59]. Other clinical symptoms include fatigue and decrease in social interest. Similar findings have been described following influenza H1N1 virus infection; individuals reported symptoms of anxiety and depression up to 1 year after the infection [60]. The use of biologicals as therapeutic agents has also provided valuable insight, and it is well known that peripheral injection of β-interferon leads to flu-like symptoms in patients with multiple sclerosis, that α-interferon therapy in patients with hepatitis C may lead to serious symptoms of depression in some individuals and that anti-tumour necrosis factor-α (anti-TNF) therapy in patients with psoriasis can lead to improvements in mood [61]. There are likely multiple systemic inflammatory mediators that impact on innate immune cells in the CNS, leading to changes in behaviour.
The regulation of the microglia phenotype by the microenvironment of the CNS appears to be an important part of CNS immune physiology. The fact that these cells are derived from a particular developmental niche, are long lived, locally replaced and play a key role in signalling from the peripheral immune system to the brain, raises interesting questions on how disturbances of the CNS microenvironment such as neurodegeneration and ageing affect immune system to brain signalling. There is now a growing body of evidence to show that during neurodegeneration and normal ageing, the loss of the down-regulated phenotype of the microglia can have profound consequences for signalling from the immune system to brain: what was once an important part of homeostasis can become a maladaptive response.
Microglia priming in chronic neurodegeneration
The microglia are highly responsive to almost any form of injury or disease of the CNS and become activated. This activation is characterized by alterations in their morphology, such as hypertrophy of the cell soma, increased branching, upregulation or de novo synthesis of cell surface or intracellular molecules, and proliferation (Fig. 1b, d). Some of the most important diseases afflicting society today are the chronic neurodegenerative diseases of the CNS such as Alzheimer’s disease (AD), Parkinson’s disease, frontotemporal dementia and amyotrophic lateral sclerosis. Age is the single most important risk factor for the sporadic forms of these diseases, and with the demographic shift to longer lifespan, it is clear that the prevalence of these diseases will increase [62]. The role of inflammation in disease progression is of much interest as manipulations of inflammation may help to ameliorate disease progression. There are many useful reviews on the possible roles of microglia in chronic neurodegeneration [63, 64], but we focus here on the concept of microglia priming and the impact of systemic inflammation.
Although there are numerous animal models that mimic aspects of AD, the deposition of amyloid and the accumulation of phosphorylated tau, these models are not models of chronic neurodegeneration. These models do not replicate the widespread and devastating loss of neurons in the AD brain: the loss of neurons is modest or absent. A tractable laboratory model of chronic neurodegeneration that involves the accumulation of a misfolded protein and is a fatal progressive neurodegenerative disease is murine prion disease. In this disease, microglia take on an activated morphology at discrete sites in the brain early in disease, and as the pathology spreads in a systematic fashion through the brain, the microglia take on an activated morphology [65]. The stimulus that leads to this activation is not known but may include the loss of inhibitory contact with the neuronal ligands such as CD200, C3XCL1 described above, the accumulation of the misfolded protein, phagocytosis of apoptotic bodies and neuronal debris and other yet unidentified routes. Despite their apparent morphological activation, the microglia appear to have a predominantly anti-inflammatory phenotype.
Most tissue macrophages, following the appropriate stimulus, can polarize into M1 and M2 or M2-like phenotypes [66]. Classical activation and M1 polarization require interferon-γ (IFN-γ) combined with TLR4 signalling and are characterized by increased expression of pro-inflammatory mediators and effectors enabling phagocytosis and killing of pathogens. Alternative activation and M2 polarization occur in response to IL-4 and are distinguished by increased expression of transforming growth factor-β (TGFβ), IL-10, scavenger receptor CD206, MS4A4a/6A and fibrinogenic and coagulation factors, enabling regulation of wound healing, tissue repair, collagen formation and recruitment of Th2 cells. ‘Regulatory’ macrophages with an M2-like phenotype share many characteristics with M1 macrophages, including expression of pro-inflammatory cytokines, but have an immunosuppressive function due to secretion of large amounts of IL‑10. These regulatory macrophages, or M2b macrophages, appear following Fc receptor ligation, have both pro- and anti-inflammatory function and recruit regulatory T cells. Finally, in response to IL-10, TGFβ, glucocorticoid or after ingestion of apoptotic cells, microglia polarize into deactivated or M2c cells, characterized by increased expression of scavenger receptor CD163 and CD206 and down-regulation of pro-inflammatory mediators. Microglia can exhibit different phenotypes that have some of the characteristics of M1- and M2-phenotypes, but they are also highly plastic cells and may transition between different states depending on both local and systemic influences.
The microglia in prion diseased brain, even early in the disease, are associated with high levels of TGFβ, prostaglandin-E2 (PGE2), colony-stimulating factor-1 (CSF1) and IL-34 [18, 67]. The microglia appear to have a relatively anti-inflammatory phenotype, which we have previously referred to as ‘benign’ with regard to driving disease progression [65, 68]. The microglia do not appear to be involved in the removal of the misfolded protein [69] nor in the removal of the degenerating synapses [70], although it is clear that they have phagocytic potential. Furthermore, a microarray analysis revealed upregulation of various genes which encode receptors of immune regulation (i.e. CD33, TREM-2, FcRs, Dectin-1) and down-stream ITIM signalling pathways [71]. It has only recently became clear that this phenotype is not ‘benign’ since preventing microglia proliferation in the prion diseased animals using an inhibitor of the CSF1R kinase activity led to a delay in the onset of behavioural symptoms and prolonged their survival [18]. The CSF1R kinase inhibitor not only reduced the numbers of microglia but also led to a further shift towards an M2-like phenotype with further upregulation of arginase-1 (ARG1) and YM1 mRNA [18].
The apparent anti-inflammatory or M2-like profile of the microglia in murine prion disease is in accord with that seen in some murine models of AD [72], where mice express human genes encoding for the AD-related proteins, but at odds with reports of pro-inflammatory cytokines in postmortem tissue from AD patients. To investigate how systemic inflammation, a common component of end-stage disease in AD patients, might impact on the microglia phenotype, we injected LPS into the peritoneum of mice with prion disease. These experiments showed that systemic inflammation can lead to a dramatic change in the phenotype of the microglia, which switch towards a pro-inflammatory phenotype with the expression of cytokines such as IL-1, IL-6 and TNF [73, 74]. We have also shown that systemic LPS challenge results in increased expression of a wide range of immune regulatory receptors that mediate their function via activating ITAM signalling, including IgG Fc receptors [71]. The upregulation of FcRs was further investigated using prion-infected Fcγ-deficient mice, which failed to increase IL-1 expression in the CNS following systemic LPS challenge, despite a normal peripheral immune response. The systemic inflammation led to increased expression of serum-derived IgG in the brain parenchyma, suggesting a role for IgG-FcR interaction in switching primed microglia to an aggressive pro-inflammatory phenotype [71]. A similar switch in microglia phenotype is seen with systemic challenge with the TLR3 agonists poly I:C to mimic a viral infection [75] and following intranasally administered live PR3 influenza (Teeling et al., unpublished observations). These changes in microglia phenotype are associated with an exaggeration of sickness behaviours, such as fever, loss of activity, loss of burrowing behaviour and an acceleration of disease progression with earlier onset of cognitive impairments and motor deficits [74, 76]. We proposed that the microglia are ‘primed’ by the ongoing neurodegeneration and that the systemic mediators signalling across the blood-brain barrier lead to a switch in microglia phenotype.
The concept of macrophage priming is well known in the context of in vitro systems with priming of macrophages by IFN-γ prior to a challenge with a TLR agonist. Cross regulation of innate immune receptors such as TLR and IFN-γ receptors that activate PI3K and/or NFkB may explain this enhanced response to the TLR agonist [77]. Although in the prion diseased brain there are small numbers of T-cells recruited to sites of pathology [78], these cells do not synthesize detectable amounts of IFN-γ, and indeed, they appear to be anergic (Teeling et al., unpublished observations). Further insight into macrophage priming has come from in vitro cultures of macrophages with high and low concentrations of LPS. Low-dose LPS (5–50 pg/ml) failed to induce PI3K and NFkB and instead utilized IRAK-1 to induce mitochondrial ROS, which, in turn, allows the transcription factor ATF2 to bind DNA, resulting in low-grade but prolonged expression of pro-inflammatory cytokines [79, 80]. Other possible candidates for priming the microglia in the absence of IFN-γ are the cytokines CSF1 and IL-34, which both signal through CSFR1. In vitro and in vivo mouse studies show that pre-exposure to CSF1 can lead to increased synthesis of cytokines IL-6 and TNF to a subsequent LPS challenge [81].
The data thus suggest that the normal homeostatic signalling from the peripheral immune system to brain is distorted in the diseased brain and leads to the generation of a response in the brain that can increase disease symptoms, increase neuronal loss and accelerate disease progression. The primed microglia now produce a maladaptive response during systemic inflammation.
Evidence for this microglia priming phenomenon has now been described in a number of different animal models of neurodegeneration with a similar switch in the cytokine profile following systemic challenges. APP transgenic mice challenged with LPS synthesized increased pro-inflammatory cytokines in the CNS [82]. Degeneration of cells in the substantia nigra (SN) can be induced with a number of different protocols to generate a model of Parkinson’s disease, and subsequent systemic challenge of animals with IL-1 leads to enhanced degeneration of SN neurons and pro-inflammatory cytokine production [83]. It has long been known that systemic infection is associated with relapses in multiple sclerosis, and there is evidence to suggest that relapses precipitated by an infection may have long-term consequences on clinical disease [84]. In the animal model of multiple sclerosis, EAE, systemic challenge with LPS or other bacterial toxins can exacerbate neurological symptoms [85]. Even in the absence of clinical symptoms following a systemic challenge, there is increased axon injury associated with inducible nitric oxide synthase (iNOS) induction in microglia [86]. There was, however, significant heterogeneity in the appearance of the lesions after LPS challenge, with pronounced induction of axon injury in some and minimal axon injury in others. Laser dissection of individual lesions and further characterization showed that lesions with ongoing axon injury were associated with pro-inflammatory cytokine mRNA profile and iNOS synthesis, while the lesions with no or limited axon injury were dominated by the cytokine IL-10 [86]. These data show that the local regulation by the CNS microenvironment plays an important role in response to the systemic challenge.
Microglia priming and ageing
It is well known that ageing is characterized by a decline in the function of the adaptive immune system, resulting in increased antigenic stress, increased oxidative stress and increased incidence of autoimmunity. The innate immune system remains intact and functional, and the reduced activity of the adaptive immune system results in a low-grade chronic pro-inflammatory state, so called inflamm-ageing [87]. This increased pro-inflammatory peripheral tissue environment may impact on the brain by activation of cerebral endothelial cells as well as on innate immune cells in the CNS. We, and others, have shown that microglia in the rodent brain take on an activated phenotype during ageing, characterized by increased expression of CD11b, CD68, F4/80, FcRI and CD11c [88]. Although microglia adopt this activated phenotype, pro-inflammatory cytokines are generally not increased above baseline levels. The phenotype and morphological changes in microglia are region specific, with caudal regions such as the cerebellum more affected than rostral forebrain regions. Microglia in white matter showed significant increased expression of CD11c and FcRI relative to microglia in grey matter.
The change in cell surface antigen repertoire seen in the aged brain is also associated with a primed microglia phenotype: challenge of aged rodents with systemic LPS leads to exaggerated sickness behaviours and enhanced cytokine synthesis of IL-1 and IL-10, and increased expression of TLR2, MHCII and indoleamine 2,3-dioxygenase (IDO) mRNA, and the latter is associated with depressive-like behaviours [89, 90]. The primed phenotype of microglia may be a consequence of the loss of molecules, both soluble and cell-bound mediators, involved in maintaining the down-regulated phenotype and may also reflect changes in the responsiveness of the microglia to inhibitory molecules such as IL-10 [89].
The priming of microglia in the ageing brain is of particular importance as one of the components by which systemic inflammation, associated with infection, trauma or drug insult, in elderly individuals with no apparent brain disease, may lead to delirium. Delirium is a condition in which a patient shows a neuropsychiatric episode typified by fluctuating degree of disorientation, confusion, memory loss and disturbances of attention. The pathology underlying delirium is not well understood, but the exaggerated sickness behaviours precipitated by systemic inflammation signalling across an intact blood-brain barrier to primed microglia suggest cellular and molecular pathways that may underpin delirium [91, 92]. Even in the absence of delirium, low-level systemic inflammation and infections are associated with more rapid cognitive decline in AD patients [93].
Peripheral routes to microglia priming
Local changes in soluble and cell mediators expressed by neurons and astrocytes are one potential route by which microglia priming arises and the cells escape from the inhibitory CNS microenvironment. Since transient systemic inflammation, such as that induced by a peripheral LPS or poly I:C challenge, can affect microglia, it is of interest to consider whether the environment in which animals are raised also plays a role. Rats raised and maintained in a conventional animal house showed a robust immune response in the brain parenchyma following the microinjection of an adenoviral vector engineered to express beta-galactosidase. In contrast, animals raised in a specific pathogen-free unit show a much reduced inflammatory response to the virus and prolonged expression of the beta-galactosidase gene [94]. More recently, we have shown that peripheral challenge with Salmonella typhimurium leads to activation of the cerebral endothelium and microglia [95]. To investigate whether the microglia in the brain parenchyma were primed by the systemic inflammation, we microinjected a very small amount of LPS into the brain parenchyma, which, in normal animals, evoked little or no inflammatory response. In animals previously challenged with S. typhimurium, there was a more robust inflammatory response at the site of the LPS injection. The priming of the innate immune response in the CNS is associated with ageing, neurodegeneration and systemic inflammation.
Conclusion
Experimental and clinical studies have each provided evidence for a detrimental role of microglia in age-related CNS disorders, but the biological factors that initiate innate immune activation in the CNS are still elusive. Normal ageing is associated with loss of synapses, changes in the integrity of the fibre tracts and increases in oxidative stress, and it is likely that all these changes affect microglial function, due to altered neuroglial interactions. A heightened innate immune response has been referred to as inflamm-aging, an adaptive response to aid re-establishment of homeostasis. This process is under tight control, and when it fails, chronic inflammation occurs, which may contribute to tissue damage and disease progression.
We propose that age-related changes in the CNS microenvironment and age-related neurodegenerative diseases prime microglia (Fig. 3). Primed microglia respond more vigorously to inflammatory challenges and, in response to further innate immune activation, switch from an anti-inflammatory, potentially protective phenotype to an aggressive inflammatory phenotype, with tissue damage and cell loss. In healthy individuals, systemic inflammation associated with infection and tissue injury induces transient activation of microglia, which is part of the strategy to promote recovery, repair and a return to homeostasis. Microglia are long-lived cells derived from the yolk sac, and repeated or chronic systemic infections during a lifetime may alter the phenotype and function of these long-lived cells, such that they become primed for subsequent immune challenges. Infections are not exclusively responsible for microglial activation, as evidence is emerging that smoking, obesity and atherosclerosis are associated with increased onset and progression of cognitive decline. These lifestyle choices induce low-grade inflammation which may explain the increased incidence of chronic degenerative diseases of the CNS such as AD and PD, and possibly age-related macular degeneration. Further understanding of the biological pathways underlying microglial priming in experimental models and humans will not only provide insight into the pathogenesis of CNS disorders, but may also generate novel targets for drug discovery.
Fig. 3.
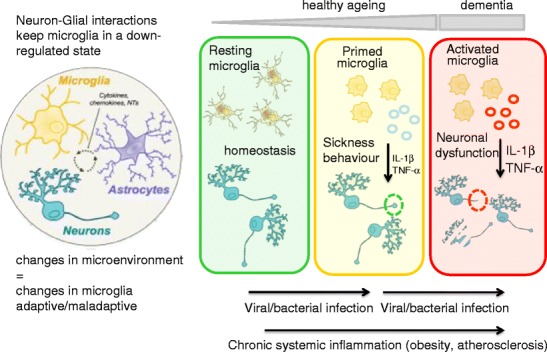
Communication between the CNS and the immune system contributes to homeostasis during systemic inflammation, resulting in transient, reversible microglial activation and adaptive behavioural and metabolic changes. Normal ageing and low-grade systemic inflammation result in prolonged ‘priming’, microglial activation. We hypothesize that multiple or chronic systemic inflammation not only accelerates the normal ageing process, resulting in irreversible pathological changes in the CNS, but may also contribute to neuronal dysfunction, disease symptoms and disease progression in the diseased CNS
Acknowledgments
Work in the authors’ laboratory is supported by funding from the MRC (UK), the Wellcome Trust, Fight for Sight, ARUK and the EU.
Footnotes
This article is a contribution to the special issue on Macrophage Heterogeneity, Subsets and Human Disease - Guest Editor: Siamon Gordon
References
- 1.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28(1):12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Greter M, Merad M. Regulation of microglia development and homeostasis. Glia. 2012;61:121–127. doi: 10.1002/glia.22408. [DOI] [PubMed] [Google Scholar]
- 3.Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48(2):405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- 4.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hume DA, Perry VH, Gordon S. Immunohistochemical localization of a macrophage-specific antigen in developing mouse retina: phagocytosis of dying neurons and differentiation of microglial cells to form a regular array in the plexiform layers. J Cell Biol. 1983;97(1):253–257. doi: 10.1083/jcb.97.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15(2):313–326. doi: 10.1016/0306-4522(85)90215-5. [DOI] [PubMed] [Google Scholar]
- 7.Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, et al. Microglia promote the death of developing Purkinje cells. Neuron. 2004;41(4):535–547. doi: 10.1016/S0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- 8.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 9.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry VH, O’Connor V. The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro. 2010;2(5):e00047. doi: 10.1042/AN20100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka R, Komine-Kobayashi M, Mochizuki H, Yamada M, Furuya T, et al. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117(3):531–539. doi: 10.1016/S0306-4522(02)00954-5. [DOI] [PubMed] [Google Scholar]
- 13.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10(12):1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 14.Ransohoff RM. Microgliosis: the questions shape the answers. Nat Neurosci. 2007;10(12):1507–1509. doi: 10.1038/nn1207-1507. [DOI] [PubMed] [Google Scholar]
- 15.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- 16.Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci. 2011;31(31):11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, et al. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 18.Gómez-Nicola D, Fransen NL, Suzzi S, Perry VH. Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci. 2013;33(6):2481–2493. doi: 10.1523/JNEUROSCI.4440-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawson LJ, Perry VH, Gordon S. Microglial responses to physiological change: osmotic stress elevates DNA synthesis of neurohypophyseal microglia. Neuroscience. 1993;56(4):929–938. doi: 10.1016/0306-4522(93)90139-7. [DOI] [PubMed] [Google Scholar]
- 20.Shankaran M, Marino ME, Busch R, Keim C, King C, et al. Measurement of brain microglial proliferation rates in vivo in response to neuroinflammatory stimuli: application to drug discovery. J Neurosci Res. 2007;85(11):2374–2384. doi: 10.1002/jnr.21389. [DOI] [PubMed] [Google Scholar]
- 21.Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14(9):1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 22.Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, et al. Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A. 2012;109:18150–18155. doi: 10.1073/pnas.1210150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambertsen KL, Deierborg T, Gregersen R, Clausen BH, Wirenfeldt M, et al. Differences in origin of reactive microglia in bone marrow chimeric mouse and rat after transient global ischemia. J Neuropathol Exp Neurol. 2011;70(6):481–494. doi: 10.1097/NEN.0b013e31821db3aa. [DOI] [PubMed] [Google Scholar]
- 24.Soulas C, Donahue RE, Dunbar CE, Persons DA, Alvarez X, et al. Genetically modified CD34+ hematopoietic stem cells contribute to turnover of brain perivascular macrophages in long-term repopulated primates. Am J Pathol. 2009;174(5):1808–1817. doi: 10.2353/ajpath.2009.081010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unger ER, Sung JH, Manivel JC, Chenggis ML, Blazar BR, et al. Male donor-derived cells in the brains of female sex-mismatched bone marrow transplant recipients: a Y-chromosome specific in situ hybridization study. J Neuropathol Exp Neurol. 1993;52(5):460–470. doi: 10.1097/00005072-199309000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 28.Matyszak MK, Perry VH. Bacillus Calmette-Guérin sequestered in the brain parenchyma escapes immune recognition. J Neuroimmunol. 1998;82(1):73–80. doi: 10.1016/S0165-5728(97)00190-2. [DOI] [PubMed] [Google Scholar]
- 29.Matyszak MK, Perry VH. A comparison of leucocyte responses to heat-killed bacillus Calmette-Guérin in different CNS compartments. Neuropathol Appl Neurobiol. 1996;22(1):44–53. doi: 10.1111/j.1365-2990.1996.tb00845.x. [DOI] [PubMed] [Google Scholar]
- 30.Schnell L, Fearn S, Schwab ME, Perry VH, Anthony DC. Cytokine-induced acute inflammation in the brain and spinal cord. J Neuropathol Exp Neurol. 1999;58(3):245–254. doi: 10.1097/00005072-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumann H, Misgeld T, Matsumuro K, Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc Natl Acad Sci U S A. 1998;95(10):5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei R, Jonakait GM. Neurotrophins and the anti-inflammatory agents interleukin-4 (IL-4), IL-10, IL-11 and transforming growth factor-beta1 (TGF-beta1) down-regulate T cell costimulatory molecules B7 and CD40 on cultured rat microglia. J Neuroimmunol. 1999;95(1–2):8–18. doi: 10.1016/S0165-5728(98)00248-3. [DOI] [PubMed] [Google Scholar]
- 34.Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30(10):527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59:152–165. doi: 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- 36.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 37.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 38.Billadeau DD, Leibson PJ. ITAMs versus ITIMs: striking a balance during cell regulation. J Clin Invest. 2002;109:161–168. doi: 10.1172/JCI14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daeron M. Fc receptor biology. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 40.Angata T, Kerr SC, Greaves DR, Varki NM, Crocker PR, et al. Cloning and characterization of human Siglec-11. A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J Biol Chem. 2002;277(27):24466–24474. doi: 10.1074/jbc.M202833200. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201(4):647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S, Wang XJ, Tian LP, Pan J, Lu GQ, et al. CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J Neuroinflammation. 2011;6(8):154. doi: 10.1186/1742-2094-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, et al. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;6(68(1)):19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karch CM, Jeng AT, Nowotny P, Cady J, Cruchaga C, et al. Expression of novel Alzheimer’s disease risk genes in control and Alzheimer’s disease brains. PLoS One. 2012;7(11):e50976. doi: 10.1371/journal.pone.0050976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-W. [DOI] [PubMed] [Google Scholar]
- 48.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 49.de Haas AH, Boddeke HW, Biber K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia. 2008;56(8):888–894. doi: 10.1002/glia.20663. [DOI] [PubMed] [Google Scholar]
- 50.Scheffel J, Regen T, Van Rossum D, Seifert S, Ribes S, et al. Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia. 2012;60(12):1930–1943. doi: 10.1002/glia.22409. [DOI] [PubMed] [Google Scholar]
- 51.Ravasi T, Wells C, Forest A, Underhill DM, Wainwright BJ, et al. Generation of diversity in the innate immune system: macrophage heterogeneity arises from gene-autonomous transcriptional probability of individual inducible genes. J Immunol. 2002;168(1):44–50. doi: 10.4049/jimmunol.168.1.44. [DOI] [PubMed] [Google Scholar]
- 52.Dantzer R, Bluthe RM, Laye S, Bret-Dibat JL, Parnet P, et al. Cytokines and sickness behavior. Ann N Y Acad Sci. 1998;840:586–590. doi: 10.1111/j.1749-6632.1998.tb09597.x. [DOI] [PubMed] [Google Scholar]
- 53.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 54.Lacroix S, Feinstein D, Rivest S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol. 1998;8:625–640. doi: 10.1111/j.1750-3639.1998.tb00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teeling JL, Cunningham C, Newman TA, Perry VH. The effect of non-steroidal anti-inflammatory agents on behavioural changes and cytokine production following systemic inflammation: implications for a role of COX-1. Brain Behav Immun. 2010;24:409–419. doi: 10.1016/j.bbi.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Konsman JP, Tridon V, Dantzer R. Diffusion and action of intracerebroventricularly injected interleukin-1 in the CNS. Neuroscience. 2000;101:957–967. doi: 10.1016/S0306-4522(00)00403-6. [DOI] [PubMed] [Google Scholar]
- 58.Krabbe KS, Reichenberg A, Yirmiya R, Smed A, Pedersen BK, et al. Low-dose endotoxemia and human neuropsychological functions. Brain Behav Immun. 2005;19:453–460. doi: 10.1016/j.bbi.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 59.Dellagioia N, Devine L, Pittman B, Hannestad J (2012) Bupropion pre-treatment of endotoxin-induced depressive symptoms. Brain Behav Immun. doi:10.1016/j.bbi.2012.10.008. [DOI] [PubMed]
- 60.Luyt CE, Combes A, Becquemin MH, Beigelman-Aubry C, Hatem S, et al. Long-term outcomes of pandemic 2009 influenza A(H1N1)-associated severe ARDS. Chest. 2012;142:583–592. doi: 10.1378/chest.11-2196. [DOI] [PubMed] [Google Scholar]
- 61.Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry. 2004;56:819–824. doi: 10.1016/j.biopsych.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 62.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, et al. Alzheimer’s Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366(9503):2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129(2):154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- 65.Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010;120(3):277–286. doi: 10.1007/s00401-010-0722-x. [DOI] [PubMed] [Google Scholar]
- 66.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perry VH, Cunningham C, Boche D. Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol. 2002;15(3):349–354. doi: 10.1097/00019052-200206000-00020. [DOI] [PubMed] [Google Scholar]
- 68.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7(2):161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 69.Hughes MM, Field RH, Perry VH, Murray CL, Cunningham C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia. 2010;58(16):2017–2030. doi: 10.1002/glia.21070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sisková Z, Page A, O’Connor V, Perry VH. Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping. Am J Pathol. 2009;175(4):1610–1621. doi: 10.2353/ajpath.2009.090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lunnon K, Teeling JL, Tutt AL, Cragg MS, Glennie MJ, et al. Systemic inflammation modulates Fc receptor expression on microglia during chronic neurodegeneration. J Immunol. 2011;186:7215–7224. doi: 10.4049/jimmunol.0903833. [DOI] [PubMed] [Google Scholar]
- 72.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Combrinck MI, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience. 2002;112(1):7–11. doi: 10.1016/S0306-4522(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 74.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25(40):9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24(6):996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, et al. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65(4):304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schroder K, Sweet MJ, Hume DA. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology. 2006;211(6–8):511–524. doi: 10.1016/j.imbio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 78.Betmouni S, Perry VH, Gordon JL. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience. 1996;74(1):1–5. doi: 10.1016/0306-4522(96)00212-6. [DOI] [PubMed] [Google Scholar]
- 79.Maitra U, Deng H, Glaros T, Baker B, Capelluto DG, et al. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J Immunol. 2012;189:1014–1023. doi: 10.4049/jimmunol.1200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deng H, Maitra U, Morris M, Li L. Molecular mechanism responsible for the priming of macrophage activation. J Biol Chem. 2013;288(6):3897–3906. doi: 10.1074/jbc.M112.424390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chapoval AI, Kamdar SJ, Kremlev SG, Evans R. CSF-1 (M-CSF) differentially sensitizes mononuclear phagocyte subpopulations to endotoxin in vivo: a potential pathway that regulates the severity of gram-negative infections. J Leukoc Biol. 1998;63(2):245–252. doi: 10.1002/jlb.63.2.245. [DOI] [PubMed] [Google Scholar]
- 82.Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, et al. Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res Bull. 2001;56(6):581–588. doi: 10.1016/S0361-9230(01)00730-4. [DOI] [PubMed] [Google Scholar]
- 83.Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain. 2008;131:1880–1894. doi: 10.1093/brain/awn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buljevac D, Flach HZ, Hop WC, et al. Prospective study on the relationship between infections and multiple sclerosis exacerbations. Brain. 2002;125:952–960. doi: 10.1093/brain/awf098. [DOI] [PubMed] [Google Scholar]
- 85.Schiffenbauer J, Johnson H, Butfiloski E, Wegrzyn L, Soos J. Staphylococcal enterotoxins can reactivate experimental allergic encephalomyelitis. Proc Natl Acad Sci USA. 1993;90:8543–8546. doi: 10.1073/pnas.90.18.8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moreno B, Jukes JP, Vergara-Irigaray N, Errea O, Villoslada P, et al. Systemic inflammation induces axon injury during brain inflammation. Ann Neurol. 2011;70(6):932–942. doi: 10.1002/ana.22550. [DOI] [PubMed] [Google Scholar]
- 87.Cevenini E, Monti D, Franceschi C. Inflamm-ageing. Curr Opin Clin Nutr Metab Care. 2013;16(1):14–20. doi: 10.1097/MCO.0b013e32835ada13. [DOI] [PubMed] [Google Scholar]
- 88.Hart AD, Wyttenbach A, Perry VH, Teeling JL. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun. 2012;26:754–765. doi: 10.1016/j.bbi.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wynne AM, Henry CJ, Godbout JP. Immune and behavioral consequences of microglial reactivity in the aged brain. Integr Comp Biol. 2009;49:254–266. doi: 10.1093/icb/icp009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perry VH, Newman TA, Cunningham C. The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci. 2003;4(2):103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- 92.Cunningham C, Maclullich AM. At the extreme end of the psychoneuroimmunological spectrum: delirium as a maladaptive sickness behavior response. Brain Behav Immun. 2013;28:1–13. doi: 10.1016/j.bbi.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73(10):768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ohmoto Y, Wood MJ, Charlton HM, Kajiwara K, Perry VH, et al. Variation in the immune response to adenoviral vectors in the brain: influence of mouse strain, environmental conditions and priming. Gene Ther. 1999;6(4):471–481. doi: 10.1038/sj.gt.3300851. [DOI] [PubMed] [Google Scholar]
- 95.Püntener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]