

Abstract
Disruption of ionic homeostasis and neuronal hyperexcitability contribute to early brain injury after subarachnoid hemorrhage (SAH). The hyperpolarization-activated/cyclic nucleotide (HCN)-gated channels play critical role in the regulation of neuronal excitability in hippocampus CA1 region and neocortex, in which the abnormal neuronal activities are more readily provoked. This study was to investigate the interactions between HCN channels and hyperneuronal activity after experimental SAH. The present results from whole-cell recordings in rat brain slices indicated that (1) perfusion of hemoglobin (Hb)-containing artificial CSF produced neuronal hyperexcitability and inhibited HCN currents in CA1 pyramidal neurons, (2) nitric oxide/Spermine (NO/Sp), a controlled releaser of nitric oxide, attenuated neuronal excitability and enhanced HCN currents in CA1 pyramidal neurons, while l-nitroarginine (l-NNA), an inhibitor of nitric oxide synthase, reduced the HCN currents; and (3) the inhibitory action of Hb on HCN currents was reversed by application of NO/Sp, which also reduced neuronal hyperexcitability; conversely, l-NNA enhanced inhibitory action of Hb on HCN currents. Additionally, Hb perfusion scavenged the production of nitric oxide and decreased the expression of HCN1 subunits in CA1 region. In the rat SAH model, the expression of HCN1, both at mRNA and protein level, decreased in hippocampus CA1 region at 24 h and more pronounced at 72 h after SAH. These observations demonstrated a reduction of HCN channels expression after SAH and Hb reduced HCN currents in hippocampus CA1 pyramidal neurons. Inhibition of HCN channels by Hb may be a novel pathway for inducing the hyperneuronal excitability after SAH.
Introduction
It has been estimated that 1–6% of the world population may harbor an intracranial aneurysm and that each year ∼10/100,000 people suffer from an aneurysmal subarachnoid hemorrhage (SAH) (McCormick and Nofzinger, 1965; Schievink et al., 2004). Despite major advances in surgical techniques, radiology, and anesthesiology, the mortality and morbidity rates after spontaneous SAH have shown a moderate reduction in recent years (Schievink et al., 2004; Nieuwkamp et al., 2009; Lovelock et al., 2010). Early brain injury including electrophysiological disorders, rather than cerebral vasospasm (Vajkoczy et al., 2005), may contribute to the high mortality and morbidity rate of SAH (Cahill et al., 2006; Pluta et al., 2009).
Electrophysiological disorders such as “cortical spreading depolarization,” which was originally described by Leão (1944), first appeared at the early stage after SAH (Dreier et al., 2006). This kind of neuronal hyperexcitability originates from a temporary disruption of local ionic homeostasis and ultimately contributes to the neuronal swelling and death (Dreier, 2011). In SAH patients, intense neural activity leads to increases in extracellular K+ and excitatory neurotransmitters, which, in turn, induce additional depolarization and neuronal excitability (Somjen, 2001; Iadecola, 2009). In addition, it has been reported that cortical spreading depolarization is associated with a sustained reduction in cerebral blood flow and profound brain hypoxia (Dreier et al., 2009; Bosche et al., 2010), which may lead to delayed ischemic neurological deficit in patients with SAH (Dreier et al., 2006). However, the mechanism of neuronal hyperexcitability after SAH remains unclear.
The hyperpolarization-activated/cyclic nucleotide (HCN)-gated channels are a mixed-cation conductance, encoded by 4 genes (HCN1–4), and widely distributed in peripheral and central neurons (Robinson and Siegelbaum, 2003; Biel et al., 2009). An abundance of evidence has demonstrated that HCN1 channels are critical for the regulation of neuronal excitability in hippocampus CA1 region and neocortex (Day et al., 2005; Shin and Chetkovich, 2007; Wang et al., 2007; George et al., 2009; Li et al., 2010). In these brain areas, HCN1 subunits are densely distributed and expressed on the dendritic spines of pyramidal neurons (Magee, 1998; Lörincz et al., 2002; Notomi and Shigemoto, 2004; Nusser, 2009), and are involved in the integration of excitatory synaptic input and thereby influences the excitability of neural network (Magee, 1999; Williams and Stuart, 2003; Oviedo and Reyes, 2005; Tsay et al., 2007). Interestingly, cortical spreading depolarization is more readily provoked in hippocampus CA1 sector and neocortex (Somjen, 2001), and its generation as well as propagation is also greatly dependent on the apical dendrites of pyramidal neurons (Canals et al., 2005; Henning et al., 2005; Takano et al., 2007). Therefore, we speculate that HCN1 channels are potential regulative targets which contribute to the formation of neuronal hyperexcitability after SAH.
First, in the present study, the changes of HCN1 currents in hippocampus CA1 pyramidal neurons were investigated under a hemoglobin (Hb)-containing environment. Then in the rat SAH model, we further observed the changes of HCN1 expression in hippocampus CA1 area. The results revealed a functional reduction of HCN channel in the special environment of SAH, which participated in the emerging of neural hyperexcitability in hippocampus CA1 region.
Materials and Methods
Whole-cell clamp recordings.
First, for slice preparation, the Sprague Dawley rat (P21–P24) was rapidly decapitated, and then the brain was quickly removed and submerged in a 0°C sucrose solution containing the following (in mm): 220 sucrose, 2.5 KCl, 1.25 Na2HPO4, 26 NaHCO3, 6 MgCl2, 1 CaCl2, 10 glucose, and was saturated with 95%O2-5%CO2. Horizontal slices (400 μm), including the hippocampal structures (Fig. 1A), were prepared using an oscillating tissue slicer (Leica, VT1000) and transferred to a holding chamber containing artificial CSF (aCSF) for a minimum of 90 min at room temperature (22−24°C) before recording. aCSF containing the following (in mm): 124 NaCl, 3 KCl, 26 NaHCO3, 2 MgCl2, 2 CaCl2, 10 glucose. Slices were then transferred to a submersion-type recording chamber and perfused at a rate of 1–2 ml/min with aCSF. All aCSF solutions were constantly aerated with a mixture of 95% O2-5% CO2 to maintain pH ∼7.4. Whole-cell current-clamp recordings in brain slice neurons were made as described previously (Li et al., 2010). Pyramidal neurons in CA1 region were targeted for recording using an upright microscope equipped with Leica infrared-differential interference contrast (IR-DIC) optics, a 340 water-immersion objective, and a video imaging camera. Patch pipette (3–7 MΩ) filled with internal solution containing the following (in mm): 125 potassium gluconate, 20 KCl, 10 HEPES, 1 EGTA, 2 MgCl2, 4 ATP, pH 7.2–7.4. The pipette resistance, as measured in the bath, was typically 4 ± 0.5 MΩ. After gigaohm seal formation and patch rupture, neurons were given at least 5 min to stabilize before data were collected. Following patch rupture, series resistance was compensated 50–70% and continually monitored throughout the experiment. Cells were discarded if series resistance increased by >15%. The signal was amplified using an EPC10 amplifier (HEKA Elektronik) and stored for off-line analysis with Pulse/Pulsefit v.8.74 (HEKA Elektronik) and Igor Pro v.4.03 (WaveMetrics). Recordings were performed at room temperature unless stated otherwise. Additionally, some cells were intracellularly labeled with biocytin (0.5%) to confirm identification.
Figure 1.
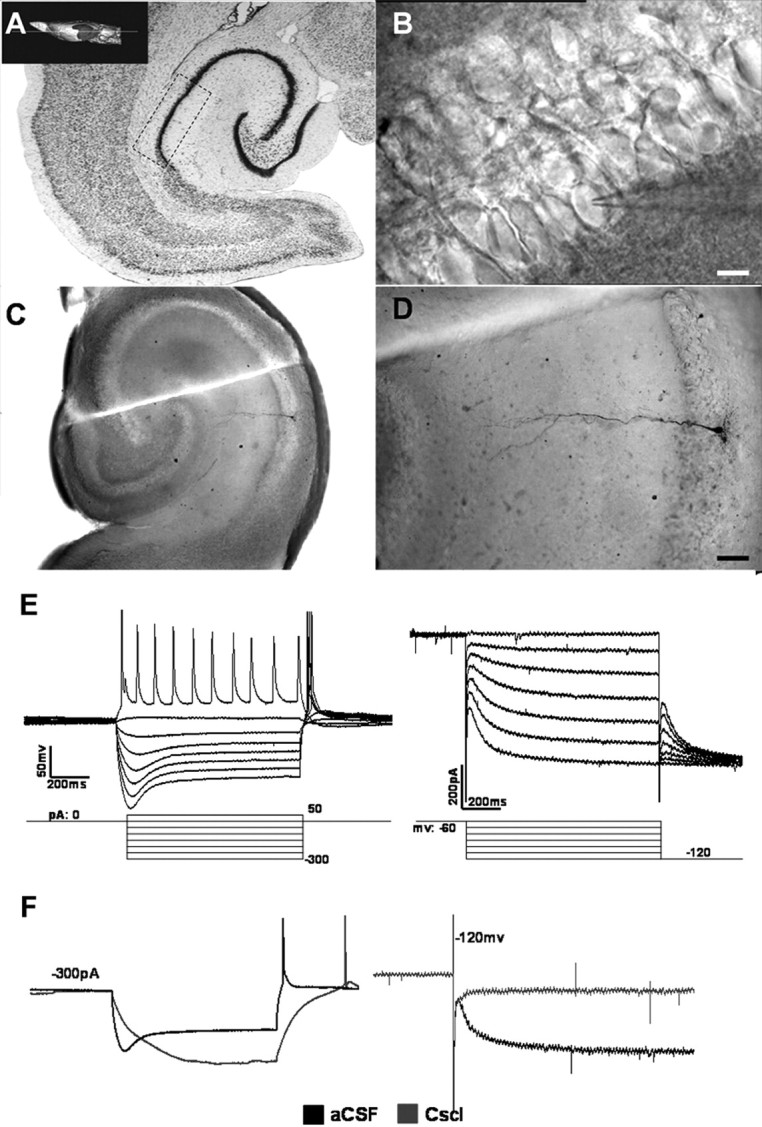
CA1 pyramidal neurons display electrophysiological feature of HCN currents. A, Schematic representation of the place of CA1 region of rat hippocampus. B, IR-DIC image of CA1 stratum pyramidale and the position of recording electrode on pyramidal neruon in hippocampus slice. Scale bar, 10 μm. C, D, Biocytin labeling revealed recording cells as pyramidal cells located in stratum pyramidale with a long apical dendrite passing through the stratum radiatum of CA1. E, Left, Control current-clamp recording from CA1 pyramid neuron at rest with current steps from −300 to 50 pA (step = 50 pA), the voltage sag was induced by negative current steps. Right, HCN currents activated by 1000 ms hyperpolarizing voltage steps from −60 to −120 mV in 10 mV increments, then all sweeps were subjected to a voltage jump to −120 mV to obtain full activation of HCN currents. F, HCN currents were blocked after application of CsCl (1 mm). Left, the voltage sag vanished when stimulated by negative current (−300 pA). Right, the amplitude of HCN currents nearly reduced to 0 pA at −120 mV.
Measurement of NO levels.
At 1, 2, 3 and 4 h after perfusion of hippocampus slices with Hb (10 μm), the circulative artificial CSF were collected, and the NO levels were assayed according to the instructions in the NO detection kit (Nanjing Jiancheng Technology). Briefly, the collective aCSF was collected and assayed in triplicate, and a standard curve using NaNO2 was generated for each experiment for quantification. Briefly, 50 μl of the supernatants or standard NaNO2 was mixed with 50 μl of Griess reagent I and 50 μl of Griess reagent II in a 96-well plate at room temperature for 10 min, and the absorbance was measured at 540 nm using a microplate reader.
Experimental model of SAH.
All experiments were performed in accordance with China Animal Welfare Legislation and were approved by the Third Military Medical University Committee on Ethics in the Care and Use of Laboratory Animals. The endovascular perforation model of SAH was produced as reported earlier (Kusaka et al., 2004; Luo et al., 2010; Suzuki et al., 2010). Briefly, Male Sprague Dawley rats (240–270 g) were anesthetized with sodium pentobarbital (40 mg/kg, intraperitoneal). A sharpened 4–0 nylon suture was introduced into the right external carotid artery and advanced through the intracranial internal carotid artery until resistance was felt and then pushed 3 mm further to perforate the internal carotid artery wall. Then, the suture was withdrawn into the external carotid artery, and the internal carotid artery was reperfused. In the sham surgery, the filaments were advanced without arterial perforation. The severity of SAH was blindly assessed in all animals after they were killed, as previously described (Sugawara et al., 2008). The subarachnoid cistern was divided into 6 segments, and each segment was allotted a grade from 0 to 3 as follows; grade 0 = no subarachnoid blood; grade 1 = minimal subarachnoid blood; grade 2 = moderate blood clot with recognizable arteries; and grade 3 = blood clot obliterating all arteries within the segment. Each animal received a total score ranging from 0 to 18 after the individual scores were summed.
H&E staining and immunohistochemistry.
At designated time points (24 and 72 h post-SAH), animals were anesthetized and perfused through the ascending aorta with 0.9% saline, followed by 400 ml of 4% paraformaldehyde (PFA) in phosphate buffer (PB; 0.1 m sodium phosphate, pH 7.4). Brains were removed, postfixed for 24 h in phosphate-buffered 4% PFA, embedded with paraffin, and cut into 6 μm sections on a vibratome. The protocol of H&E staining was not shown. For immunohistochemistry, sections were first treated with 0.3% Triton X-100 and 3%H2O2, and then incubated for 24 h at 4°C with a rabbit monoclonal antibody to rat HbA2 (Epitomics) diluted 1: 100 in a solution of 2% goat serum. After three 10 min PB washes, the sections were treated according to the instruction of Streptavidin-Peroxidase kit (SP9003, Zhongshan Goldenbridge Biotechnology). Finally, sections were incubated with a solution of 0.6 mg/ml diaminobenzidine (DAB) and 0.05% H2O2 for 2 min. This incubation was terminated by three 10 min PB washes. Sections were mounted onto gelatin-coated slides and dried overnight before placing a coverslip on them. Representative sections from each animal were then photographed.
Western blotting analyses.
At designated time points (24 and 72 h post-SAH) animals were killed, brains rapidly removed, and plunged into sucrose solution as described above. Transverse slices (600 μm) of brain tissue through the hippocampus were cut and subdissected into CA1, CA3, dentate gyrus (DG) and entorhinal cortex (EC) regions. Figure 9A (see below) shows a schematic illustration of the dissecting parts in hippocampus. CA1 tissue samples were immediately stored at −80°C until required. For in vitro study, CA1 tissue samples were collected at 1, 2, 3 and 4 h after Hb perfusion. For Western blotting, CA1 tissues were homogenized, and aliquots of each fraction were used to determine the protein concentration of each sample using a detergent compatible assay (Bio-Rad). Equal amounts of protein (50 μg) were fractionated by SDS-PAGE and subsequently transferred to a nitrocellulose membrane (GE Healthcare Pharmacia Biotech). The membrane was incubated in fresh blocking buffer (containing Tris-buffered saline, 0.1% Tween 20 in Tris-buffered saline, pH 7.4) at room temperature for 1 h and then incubated with rabbit polyclonal anti-HCN1 (1:500; Abcam) antibody, overnight at 4°C. The blots were washed three times in TBS-T for 5 min and then incubated with appropriate secondary antibodies (HRP-goat anti rabbit, 1:1000, Zhongshan Goldenbridge Biotechnology). The bound antibodies were visualized using an enhanced chemiluminescent detection system and then exposed to x-ray films (Kodak). The images were scanned with a GS800 Densitometer Scanner (Bio-Rad), and OD data were analyzed using Quantity One software (Bio-Rad). In these analyses, β-actin was used as an internal reference.
Figure 9.

Changes of HCN1 protein and mRNA expression in hippocampus CA1 region after SAH. A, Schematic illustration of sectioning of hippocampus technique for regional tissue acquisition and analysis. B, Western blot for HCN1 in the right hippocampus CA1 region at 24 and 72 h after SAH. The expression levels are expressed as a ratio of β-actin levels for normalization. n = 5 rats per group. C, Top, Reverse transcription-PCR assay of HCN1 mRNA expression in the right hippocampus CA1 region at 24 and 72 h after SAH. Bottom, Real-time quantitative PCR measurements of HCN1 mRNA expression in the right hippocampus CA1 region at 24 and 72 h after SAH. mRNA expressed as a percentage change from control. n = 6 rats per group.
Reverse transcription-PCR and quantitative PCR.
Total RNA (5–20 μg) was isolated using TRIzol (Invitrogen) following the manufacturer's protocol. A subsequent DNase I treatment was performed to remove any residual DNA contamination (QIAGEN). Isolated RNA (1 μg) was reverse transcribed and amplified using a One-Step RT-PCR (reverse transcription PCR) kit (Invitrogen). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene for the internal standard. The primers corresponding to the rat HCN1 and to the reference gene (GAPDH) are listed as follows: GAPDH, 5′TGGGGTGATGCTGGTGCTGAGT3′ (forward sense), 5′AGGTTTCTCCAGGCGGCATGTC3′ (reverse sense); HCN1, 5′CCCCAACGTCGCGCATGAG3′ (forward sense,), 5′GCTGAGGCTGCTGCATGAGTG3′ (reverse sense). For quantitative PCR, total RNA was isolated and used to prepare cDNA as described above. A master mix was aliquoted to a 384-well plates (Applied Biosystems) with each well containing: 2.85 μl of water, 1 μl of 10× PCR buffer, 0.2 μl of primer mix (or 0.1 μl of forward, 0.1 μl of reverse), 1.2 μl of 25 mm MgCl2, 0.2 μl of deoxynucleotide triphosphate mix (10 mm each), 0.025 μl of Platinum Taq polymerase (Invitrogen), 0.3 μl of SYBR green stock (Invitrogen). The real-time PCR protocol used was as follows: heat activation of polymerase at 95°C for 3 min, followed by 40 cycles of: 95°C for 10 s, 65°C for 15 s and 72°C for 20 s. Readings were performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems) and compared against a standard curve created from genomic DNA.
Drugs.
All reagents were obtained from (Sigma) with the exception of l-nitroarginine (l-NNA, Tocris Bioscience). Pure hemoglobin (oxyhemoglobin) was generally prepared by the method of Martin et al. (1985). First, a tenfold molar excess of the reducing agent, sodium dithionite (Na2S2O4) was added to a 1 mm solution of commercial hemoglobin in distilled water. Then, sodium dithionite was then removed by dialysis against 100 volumes of distilled water for 2 h at 4°C. The concentrations of the solutions hemoglobin was determined spectrophotometrically, and the solution was frozen in aliquots at −20°C and stored for up to 14 d. During patch-clamp recording, all drugs were applied by switching perfusion from artificial CSF to a solution containing the desired drug. Ba2+ was bathed in at a concentration (200 μm) that blocks Kir2 channels to keep HCN currents intact. Additionally, CsCl (1 mm) was used to block HCN channel in preference to the organic blockers, such as ZD7288, which require a long time to exert their effect. All drugs were prepared as concentrated stock solutions and frozen at −20°C until use.
Statistical analysis.
All quantitative values are given as the mean ± SEM. Statistical comparisons were made by using ANOVA or the unpaired or paired Student's t test with SPSS (version 13) software, as appropriate. A p value of <0.05 was considered statistically significant.
Results
Whole-cell recordings of pyramidal neurons in hippocampus CA1 region
Whole-cell recordings were obtained from a total of 114 pyramidal cells located within the rat hippocampus CA1 area. These neurons were identified using IR-DIC video microscopy by their compacted collocation and large pyramidal shaped cell body with a long apical dendrite passing through the stratum radiatum of CA1 (Fig. 1B). A subset of these cells was injected with biocytin for further morphological identification. All the labeled cells had morphological features of pyramidal neurons as described previously: a pyramidal soma and a prominent apical dendrite (Fig. 1C,D). The mean resting membrane potential of neurons recorded in current clamp was 64 ± 5.8 mV.
HCN currents recording in CA1 pyramidal neurons
Injection of negative current steps (−300 to 50 pA in 50 pA steps) in CA1 pyramidal neurons produced a rapid hyperpolarization, followed by a depolarizing sag (Fig. 1E, left, n = 28). This response pattern is typical of neurons expressing HCN channel currents. Meanwhile, the whole-cell voltage-clamp recordings showed that HCN-like inward currents were activated by a series of hyperpolarized voltages (−60 to −120 mV, −10 mV step), which are typical for HCN channels. The amplitude of the HCN-like current at −120 mV was 205.7 ± 38.5 pA (Fig. 1E, right, n = 28), with the leak currents controlled at ±10.0 pA. The relative small somatic HCN-like currents recorded in these neurons may reflect current generation at sites distant from the soma (as in dendritic spines) and subsequent signal attenuation resulting from a large dendritic electrotonic length (Destexhe et al., 1996). Barium was bathed in artificial CSF at a concentration of 200 μm that blocked Kir2 channels to leave the HCN currents intact. To further examine the HCN currents in CA1 pyramidal neurons, the protocol was repeated in the presence of the HCN channels blocker CsCl. CsCl (1 mm) was applied to the bath for 10 min before recording. At current-clamp recordings of −300 pA, the voltage sag vanished when applying CsCl (Fig. 1F, left, n = 8). Accordingly, the amplitude of HCN currents was nearly close to zero at −120 mV (Fig. 1F, right, n = 8).
Hemoglobin inhibited HCN currents in CA1 pyramidal neurons
Hb (10 μm) was bathed in circulatory artificial CSF ∼20 min before recording. Whole-cell current-clamp recordings from CA1 pyramidal neurons show that Hb induced a moderate fluctuation of membrane potential, accompanied with a rapid firing of action potentials (APs). After washout of Hb, the membrane potential and AP frequency returned to control levels (Fig. 2A,D, n = 12 cells per group). Interestingly, Hb synchronously produced a significant decrease in the amplitude of HCN currents evoked by −60 to −120 mV voltage-ramp protocol (Fig. 2B). The suppression of Hb on HCN currents at −120 mV was amplified in Figure 2, C and E (n = 12 cells per group). After washout of Hb for at least 20 min, the amplitude of HCN currents also recovered to the control level, indicating that the inhibitory effect of Hb on HCN currents was reversible.
Figure 2.

HCN channels were inhibited during perfusion of aCSF containing Hb in CA1 pyramidal neurons. A, Perfusion of aCSF containing Hb (10 μm) induced fast spike firing of CA1 pyramidal neurons at resting potential. Summary of the comparison of AP number per minute between control and in the presence of Hb is shown in D. B, HCN currents were obviously inhibited during the perfusion of aCSF containing Hb, then the amplitude of HCN currents recovered to control level after washout of Hb. The inhibition of Hb on HCN currents at −120 mV was amplified in C. E, Bar graph summarizes the statistical data of comparison of normalized HCN amplitude between control, perfusion and washout of Hb. The amplitude of HCN currents at −120 mV in baseline was integrated as 100%. n = 12 cells per group. *p < 0.05 and **p < 0.01 in this and the following figures.
To further evaluate the correlation between the effect of Hb on neural excitability and HCN channel in CA1 region, CsCl (1 mm), a HCN blocker, was pretreated before Hb administration. The results indicated that there was no significant increase of spike firing between application of CsCl alone and CsCl/Hb combined (Fig. 3A). Accordingly at voltage clamp, there was also no HCN currents recorded during application of both CsCl alone and CsCl/Hb combined (Fig. 3B). But, it should be pointed out that the recording cells had already discharged quickly and the membrane potential fluctuated vigorously during CsCl application, and this phenomenon is consistent with the results of previous reports (Wang et al., 2007; George et al., 2009; Li et al., 2010).
Figure 3.

Perfusion of Hb did not enhance the spike firing of CA1 pyramidal neurons when HCN channels were blocked. A, Changes of AP firing and membrane potential during the application of Cs+ (1 mm) and Cs+ + Hb (10 μm) in aCSF. B, HCN currents were inhibited totally during the application of Cs+ (1 mm) and Cs+ + Hb (10 μm) in aCSF.
Changes of NO level and HCN1 expression in CA1 slices during Hb perfusion
At 1, 2, 3, and 4 h after perfusion of hippocampus slices with Hb (10 μm), the circulative artificial CSF were collected, respectively, for NO analysis and CA1 tissues were prepared for the detection of HCN1 expression. As indicated in Figure 4, the level of NO released from hippocampus slices had a dramatic drop at 1 h after Hb perfusion, and then slowly decreased at 2, 3, and 4 h after Hb perfusion. Simultaneously, the protein expression of HCN1 in CA1 region decreased moderately at 1 (decreased by 31.3%) and 2 (decreased by 37.8%) h after Hb perfusion, and then had a significant decrease at 3 (decreased by 75.3%) and 4 (decreased by 73.8%) h after Hb perfusion.
Figure 4.

Changes of NO level in hippocampus slices and HCN1 expression in CA1 region during Hb perfusion. A, Concentration of NO released from CA1 slices at 1, 2, 3, and 4 h after Hb perfusion. n = 5 rats per group. B, Western blot for HCN1 in CA1 slices at 1, 2, 3, and 4 h after Hb perfusion. The expression levels are expressed as a ratio of β-actin levels for normalization. n = 5 rats per group.
Role of NO signaling on HCN channels and neuronal excitability
The membrane potential was initially adjusted to −56 to ∼−53 mV using intracellular injection of direct current and the recording neurons fired spontaneously under this condition. Bath application of nitric oxide/Spermine (NO/Sp, 100 μm), a controlled releaser of nitric oxide, decreased the spike firing of recording cells (from 19.6 ± 3.6 to 4.2 ± 1.7 APs per minute, p < 0.01, n = 8; Fig. 5A,C). The decreased firing activity recovered close to the control (predrug) level after 10 min washout. Simultaneously, the effect of NO/Sp on HCN currents was examined during the above-mentioned process. As indicated in Figure 5B, the amplitude of HCN currents was elevated during application of NO/Sp (increased by 32.6 ± 14.6% at −120 mV, p < 0.05, n = 8; Fig. 5D). After washout, the increased HCN currents recovered to the control level.
Figure 5.

The effect of NO/Spermine on neuronal excitability and HCN currents in CA1 pyramidal neurons. A, Changes of spike firing during application of NO/Spermine (100 μm) when the membrane potential was holding at −53 to ∼−56 mV. B, Changes of HCN currents during application of NO/Spermine (100 μm). C, Summary of the comparison of AP number per minute between control, NO/Sp and washout. D, Summary of the comparison of normalized HCN amplitude between control, NO/Sp, and washout at −120 mV. n = 8 cells per group.
Furthermore, we tested whether endogenous NO could affect the function of HCN channels in CA1 pyramidal neurons. l-NNA, an inhibitor of nitric oxide synthase (NOS), was applied under the same protocol in the bath. As shown in Figure 6, application of l-NNA produced a moderate decrease in the amplitude of HCN currents (decreased by 23.2 ± 6.5% at −120 mV, p < 0.05, n = 8), accompanied by spike firing of recording cells increased (from 23.5 ± 4.3 to 67.8 ± 9.6 APs per minute, p < 0.01, n = 8).
Figure 6.

The effect of l-NNA on neuronal excitability and HCN currents in CA1 pyramidal neurons. A, Changes of spike firing during application of l-NNA (100 μm) when the membrane potential was holding at −53 to ∼−56 mV. B, Changes of HCN currents during application of l-NNA (100 μm). C, Summary of the comparison of AP number per minute between control, l-NNA and washout. D, Summary of the comparison of normalized HCN amplitude between control, l-NNA and washout at −120 mV. n = 8 cells per group.
Interaction of Hb and NO on HCN channels and neuronal excitability in CA1 pyramidal neurons
The above-mentioned results indicated that Hb inhibited HCN channels in CA1 pyramidal neurons and HCN channels could be regulated by NO signaling. In the following studies, NO/Sp or l-NNA was applied during the perfusion of Hb on CA1 pyramidal neurons before the excitatory effect of Hb evaluated. As indicated in Figure 7, A and B, the inhibition of Hb on HCN currents was attenuated after application of NO/Sp, accompanied by a decreased spike firing of recording cells. The amplitude of HCN currents at −120 mV was reduced to 45.7 ± 4.1% during the perfusion of Hb (10 μm). After incubation with 100 μm NO/Sp for ∼10 min, the amplitude of HCN currents recovered to 71.4 ± 6.9% (p < 0.01, n = 8; Fig. 7F,G, left). Accordingly, the action potential firing of recording cells decreased from 54.6 ± 8.5 to 34.7 ± 6.7 action potential per minute at resting potential (p < 0.01, n = 8; Fig. 7E, left). Conversely, the inhibition of Hb on HCN currents was enhanced moderately after application of 100 μm l-NNA (from 42.8 ± 3.6% to 33.2 ± 4.9% at −120 mV, p < 0.05, n = 8; Fig. 7D,F,G, right), but the spike firing of recording cells did not significantly increase during application of l-NNA (Fig. 7C).
Figure 7.
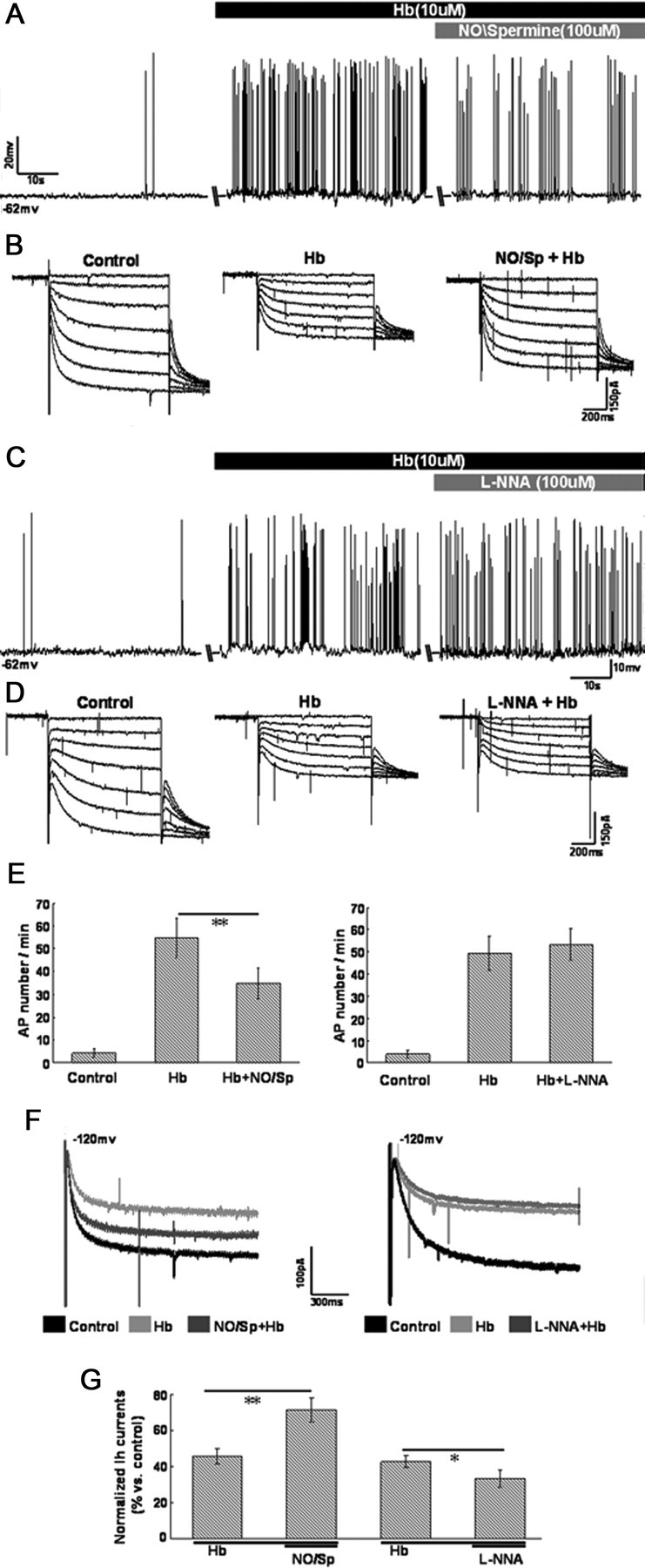
Changes of neuronal excitability and HCN currents when application of NO/Spermine and l-NNA during the perfusion of Hb in CA1 pyramidal neurons. A, C, Changes of spike firing when application of NO/Spermine (100 μm) and l-NNA (100 μm) during perfusion of Hb (10 μm) at resting potential. B, D, Changes of HCN currents when application of NO/Spermine and l-NNA during perfusion of Hb. E, Summary of the comparison of AP number per minute between control, Hb, Hb + NO/Sp (left) and control, Hb, Hb + l-NNA (right). F, Amplification of changes of HCN currents at −120 mV in B and D. G, Bar graph summarizes the statistical data of comparison of normalized HCN amplitude between Hb, Hb + NO/Sp, and Hb, Hb + l-NNA, respectively. The amplitude of HCN currents at −120 mV in baseline was integrated as 100%. n = 8 cells per group.
HCN1 expression in hippocampus CA1 region in rat SAH model
The mortality rate in the SAH group was 38% (15 of 39 rats). None of the sham-operated control animals died during experiment. Filament puncture induced extensive bleeding which was particularly pronounced on the ipsilateral side, around the Circle of Willis and along the ventral brainstem (Fig. 8A). At 24 h post-hemorrhage, the SAH score was 13 ± 2 out of a possible 18 in the SAH groups. Despite the rapid vanishing of subarachnoid blood, a significant amount of blood clot could still be observed at 72 h after SAH. The macroscopic coronary sections displayed that the endogenous blood was densely distributed within the cerebral ventricles surrounding the hippocampus tissues at 24 h after SAH, and disappeared moderately at 72 h after SAH (Fig. 8B). H&E staining showed dense red blood clots distributed within the cerebral ventricles near hippocampus tissue at 24 and 72 h after SAH (Fig. 8C). Furthermore, the immune-positive results of Hb (rabbit monoclonal anti-HbA2, Epitomics) demonstrated Hb penetrated into hippocampus tissue around cerebral ventricles after SAH. Especially, the distribution of Hb in hippocampus tissue at 72 h is more extensive than that at 24 h after SAH (Fig. 8D).
Figure 8.
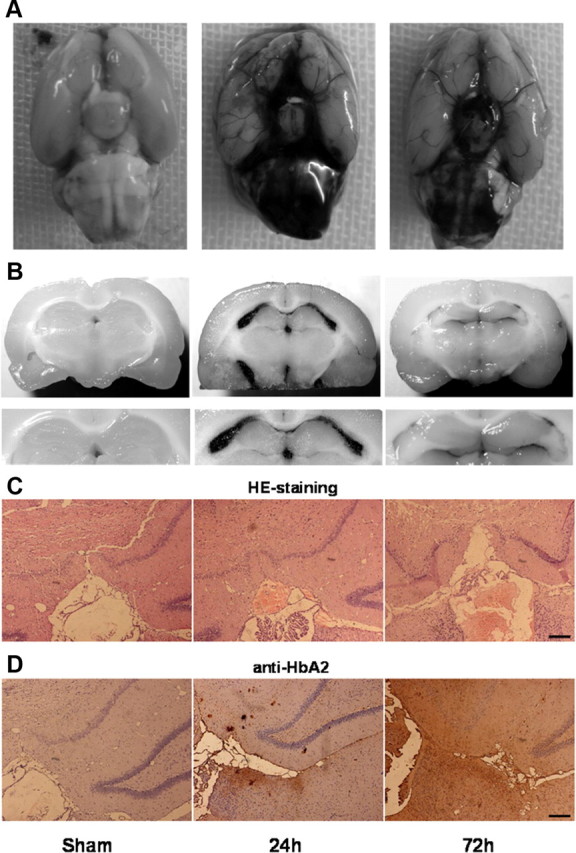
The distribution of blood and hemoglobin in rat hippocampus tissue after SAH. A, Representative photos of SAH induced by endovascular perforation of the right internal carotid artery. Accumulated blood clots were seen around the basal surface of brain 24 h post-hemorrhage. Then, blood clots diminished slowly over 72 h following hemorrhage. B, Macroscopic photos of coronary sections at hippocampus level. Endogenous blood was densely distributed within the cerebral ventricles surrounding the hippocampus tissues at 24 h after SAH, and disappeared moderately at 72 h after SAH. C, H&E staining showing dense red blood clots distributed within the cerebral ventricles near hippocampus tissue at 24 and 72 h after SAH. D, Immunohistochemistry results of HbA2 in hippocampus tissue around cerebral ventricles at 24 and 72 h after SAH. Bar, 100 μm.
Western blot for HCN1 protein expression in hippocampus CA1 region was observed. As indicated in Figure 9B, HCN1 protein expression in SAH group was significantly reduced compared with control group, and the decrease at the 72 h post-SAH point was more pronounced (p < 0.01, n = 6 rats per group). HCN1 protein expression in sham group has only slightly decreased (p > 0.05, n = 6 rats).
To further assess the expression levels of HCN1 subunits quantitatively after SAH, the real-time quantitative PCR for HCN1 mRNA was performed. Similar to the results of Western blot, HCN1 mRNA expression reduced obviously in SAH group. As indicated in Figure 9C, HCN1 mRNA expression reduced by 60.7% ± 5.4% at 24 h post-SAH point and 81.2% ± 4.1% at 72 h post-SAH point, respectively (p < 0.01, n = 6 rats per group).
Discussion
This study has obtained evidences that Hb induced neural hyperexcitability and inhibited HCN currents in rat CA1 pyramidal neurons. In addition, the function of HCN channels could be regulated by NO signaling, and increased HCN currents which was induced by enhancing NO signaling, could effectively attenuate the excitatory effect of Hb on CA1 pyramidal neurons. Furthermore, the expression of HCN1 channels in rat CA1 region decreased both in Hb-perfusion slice and in animal SAH model.
Hb, a major component of hemolysis in the subarachnoid clot, is considered as the main culprit for a series of pathological changes after SAH (Koźniewska et al., 2006; Pluta et al., 2009). However, previous publications on the effect of Hb focused primarily on cerebral vasospasm, but not on neuronal activity. In fact, the results from our study and other reports demonstrated that subarachnoid blood distributed rapidly over the entire brain and penetrated into the deeper layers of the cortex, including hippocampus tissues (Turner et al., 1998; Lee et al., 2009) and the process of hemolysis starts ∼16 h after SAH (Macdonald and Wei, 1991). Thus, Hb can directly play its excitotoxicity role on neurons at early stage after SAH. In the present study, we simulated post-hemolysis environment of SAH by perfusion of Hb-containing artificial CSF in vitro. Because of the concentration of Hb in the blood clot after SAH is in the high micromolar range (Macdonald and Weir, 1991; Wickman et al., 2003), a concentration of 10 μm Hb was used in our experiments. The results indicated that Hb induced fast spike firing of CA1 pyramidal neurons, and simultaneously, HCN channels were inhibited by Hb.
In hippocampus CA1 sector and neocortex, HCN channels are important determinants of neuronal excitability both in physiological and in pathological conditions. For example, blockade of HCN channels increases the activity of prefrontal cortex pyramidal neurons (Li et al., 2010); in hippocampal CA1, the excitability of pyramidal neurons has been confirmed to be selectively reduce by upregulation of HCN currents (Poolos et al., 2002). Furthermore in some disease models, changes of HCN channels induce hyper neuronal excitability. For example, HCN channels were significantly downregulated in the rat pilocarpine model of epilepsy, with loss of channel expression in CA1 pyramidal neurons. These molecular changes of HCN channels increased neuronal excitability and might be associated with both the process of epileptogenesis and maintenance of the epileptic state (Jung et al., 2007). In rat transient cerebral ischemia model, the neuronal excitability of CA1 pyramidal cells was significantly increased after ischemia, which was also associated with a reduction of HCN currents (Fan et al., 2008).
In the current study, the RT-PCR profiling showing that hippocampus CA1 region expressed high levels of HCN1 mRNA which is consistent with a previous study (Day et al., 2005). The electrophysiological features observed in this study were similar to a reported that channels dominated by HCN1 subunits were rapidly gating and had relatively depolarized activation voltage dependence (Chen et al., 2001). During perfusion of Hb, the HCN currents evoked by hyperpolarizing steps in voltage clamp exhibited similar kinetic tendency to the currents that are blocked by the HCN channel blocker CsCl, and the amplitude of HCN currents at −120 mV level was decreased by Hb. After washout of Hb, the neural activity and HCN currents of CA1 pyramidal neurons recovered to the normal level simultaneously. Additionally, changes of neuronal excitability in CA1 region were also directly observed when HCN channels were blocked by CsCl, which are similar to the results from prefrontal cortex pyramidal neurons (Li et al., 2010). CA1 pyramidal neurons displayed fast firing and the membrane potential vigorously fluctuated when HCN channels were blocked. On this condition, perfusion of Hb failed to further enhance the neuronal activity of recording neurons.
The above-mentioned electrophysiological observations after Hb perfusion are supported by the molecular studies on the expression level of HCN1 subunits in CA1 region in slices and in SAH animal model. First, in CA1 slices, the reduction of HCN1 protein level occurred as early as 1 h after Hb perfusion. In the rat SAH model, the protein and mRNA levels of HCN1 in hippocampal CA1 region were remarkably reduced in the acute phases of SAH (up to −72 h). This time course of HCN1 channel changing is earlier than that of cerebral vasospasm occurring, and closely followed by the time when cortical spreading depolarization begin to appear (Dreier et al., 2006).
Additionally, it is generally accepted that the scavenging effect of Hb on NO signaling contributes to a series of pathological changes after SAH (Sehba et al., 2000; Koźniewska et al., 2006; Pluta et al., 2009) and the level of NO determines the neural susceptibility for cortical spreading depolarization after SAH (Petzold et al., 2008). In the current study, the scavenging effect of Hb on NO was observed. The level of NO released from CA1 slices was significantly decreased during Hb perfusion. Interestingly, HCN channel had already been reported to be an important downstream target for NO signaling in many brain areas. For example, exogenous NO modified oscillatory activity in thalamocortical relay neurons by acting on HCN channels (Pape and Mager, 1992); in mesencephalic neurons, NO reversibly depolarized the membrane and reduced the firing threshold by a presumed action on HCN channels (Pose et al., 2003); and in deep cerebellar nuclei neurons, NO enhanced HCN currents (Wilson and Garthwaite, 2010). In hippocampus CA1 region, it was recently reported that presynaptic NO/cGMP signaling facilitated glutamate release via HCN channels (Neitz et al., 2011). In the present study, the regulative role of NO on HCN channels was further confirmed in hippocampus CA1 pyramidal neurons. Enhancing NO signaling by perfusion of NO/Sp could increase HCN current amplitude, and conversely, HCN currents were decreased when endogenous NOS was inhibited by l-NNA. Accordingly, the neuronal excitability of recording cells were attenuated or enhanced during the application of NO/Sp or l-NNA, respectively. Furthermore, application of NO/Sp attenuated the excitatory effect of Hb on recording cells, and synchronously, inhibition by Hb on HCN channels were diminished after NO/Sp application. These results revealed a functional interaction between Hb and HCN channels after SAH. We speculate that Hb, released from blood clot after SAH, exhausted the NO signaling, thus inhibited HCN channels, and consequently induced or facilitated the formation of neuronal hyperexcitability in hippocampus CA1 region after SAH.
However, there are several limitations in this study. First, we tested Hb in artificial CSF to simulate the environment after aneurysmal SAH, and we did not study other potential factors in hemolysate (such as iron, hemin). Second, we did not directly study the relationship between HCN channels and neuronal hyperexcitability in the rat SAH model. Nonetheless, the present results implied that the change of HCN channels may be a novel process involved in the formation of neuronal hyperexcitability after SAH, and may provide new therapeutic clues in patients with SAH.
Footnotes
This work was supported by grants from the National Natural Science Foundation of China (No. 30973101, 81000506, and 30900466) and the International Collaboration Foundation of Chongqing (No. 2011GZ0040). We are grateful to Prof. Z. Qian (Hong Kong Polytechnic University, Hong Kong) for useful discussions and critically reading our manuscript.
References
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Bosche B, Graf R, Ernestus RI, Dohmen C, Reithmeier T, Brinker G, Strong AJ, Dreier JP, Woitzik J. Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol. 2010;67:607–617. doi: 10.1002/ana.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26:1341–1353. doi: 10.1038/sj.jcbfm.9600283. [DOI] [PubMed] [Google Scholar]
- Canals S, Makarova I, López-Aguado L, Largo C, Ibarz JM, Herreras O. Longitudinal depolarization gradients along the somatodendritic axis of CA1 pyramidal cells: a novel feature of spreading depression. J Neurophysiol. 2005;94:943–951. doi: 10.1152/jn.01145.2004. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci. 2005;25:8776–8787. doi: 10.1523/JNEUROSCI.2650-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci. 1996;16:169–185. doi: 10.1523/JNEUROSCI.16-01-00169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17:439–447. doi: 10.1038/nm.2333. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann TN, Sarrafzadeh A, Willumsen L, Hartings JA, Sakowitz OW, Seemann JH, Thieme A, Lauritzen M, Strong AJ. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, Tolias C, Oliveira-Ferreira AI, Fabricius M, Hartings JA, Vajkoczy P, Lauritzen M, Dirnagl U, Bohner G, Strong AJ. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain. 2009;132:1866–1881. doi: 10.1093/brain/awp102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Deng P, Wang YC, Lu HC, Xu ZC, Schulz PE. Transient cerebral ischemia increases CA1 pyramidal neuron excitability. Exp Neurol. 2008;212:415–421. doi: 10.1016/j.expneurol.2008.04.032. [DOI] [PubMed] [Google Scholar]
- George MS, Abbott LF, Siegelbaum SA. HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K(+) channels. Nat Neurosci. 2009;12:577–584. doi: 10.1038/nn.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning EC, Meng X, Fisher M, Sotak CH. Visualization of cortical spreading depression using manganese-enhanced magnetic resonance imaging. Magn Reson Med. 2005;53:851–857. doi: 10.1002/mrm.20438. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Bleeding in the brain: Killer waves of depolarization in subarachnoid bleed. Nat Med. 2009;15:1131–1132. doi: 10.1038/nm1009-1131. [DOI] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koźniewska E, Michalik R, Rafałowska J, Gadamski R, Walski M, Frontczak-Baniewicz M, Piotrowski P, Czernicki Z. Mechanisms of vascular dysfunction after subarachnoid hemorrhage. J Physiol Pharmacol. 2006;57(Suppl 11):145–160. [PubMed] [Google Scholar]
- Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Lee JY, Sagher O, Keep R, Hua Y, Xi G. Comparison of experimental rat models of early brain injury after subarachnoid hemorrhage. Neurosurgery. 2009;65:331–343. doi: 10.1227/01.NEU.0000345649.78556.26. [DOI] [PubMed] [Google Scholar]
- Li B, Chen F, Ye J, Chen X, Yan J, Li Y, Xiong Y, Zhou Z, Xia J, Hu Z. The modulation of orexin A on HCN currents of pyramidal neurons in mouse prelimbic cortex. Cereb Cortex. 2010;20:1756–1767. doi: 10.1093/cercor/bhp241. [DOI] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Lovelock CE, Rinkel GJ, Rothwell PM. Time trends in outcome of subarachnoid hemorrhage: population-based study and systematic review. Neurology. 2010;74:1494–1501. doi: 10.1212/WNL.0b013e3181dd42b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Yi B, Tao G, Li M, Chen Z, Tang W, Zhang JH, Feng H. Adenosine A3 receptor agonist reduces early brain injury in subarachnoid haemorrhage. Neuroreport. 2010;21:892–896. doi: 10.1097/WNR.0b013e32833dbd13. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22:971–982. doi: 10.1161/01.str.22.8.971. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic lh normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/9158. [DOI] [PubMed] [Google Scholar]
- Martin W, Villani GM, Jothiananda D, Furchgott RF. Blockade of endothelium-dependent and glyceryl trinitrate-induced relaxation of rabbit aorta by certain ferrous hemoproteins. J Pharmacol Exp Ther. 1985;233:679–685. [PubMed] [Google Scholar]
- McCormick WF, Nofzinger JD. Saccular intracranial aneurysms: an autopsy study. J Neurosurg. 1965;22:155–159. doi: 10.3171/jns.1965.22.2.0155. [DOI] [PubMed] [Google Scholar]
- Neitz A, Mergia E, Eysel UT, Koesling D, Mittmann T. Presynaptic nitric oxide/cGMP facilitates glutamate release via hyperpolarization-activated cyclic nucleotide-gated channels in the hippocampus. Eur J Neurosci. 2011;33:1611–1621. doi: 10.1111/j.1460-9568.2011.07654.x. [DOI] [PubMed] [Google Scholar]
- Nieuwkamp DJ, Setz LE, Algra A, Linn FH, de Rooij NK, Rinkel GJ. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol. 2009;8:635–642. doi: 10.1016/S1474-4422(09)70126-7. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Nusser Z. Variability in the subcellular distribution of ion channels increases neuronal diversity. Trends Neurosci. 2009;32:267–274. doi: 10.1016/j.tins.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Oviedo H, Reyes AD. Variation of input-output properties along the somatodendritic axis of pyramidal neurons. J Neurosci. 2005;25:4985–4995. doi: 10.1523/JNEUROSCI.0562-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Mager R. Nitric oxide controls oscillatory activity in thalamocortical neurons. Neuron. 1992;9:441–448. doi: 10.1016/0896-6273(92)90182-d. [DOI] [PubMed] [Google Scholar]
- Petzold GC, Haack S, von Bohlen Und Halbach O, Priller J, Lehmann TN, Heinemann U, Dirnagl U, Dreier JP. Nitric oxide modulates spreading depolarization threshold in the human and rodent cortex. Stroke. 2008;39:1292–1299. doi: 10.1161/STROKEAHA.107.500710. [DOI] [PubMed] [Google Scholar]
- Pluta RM, Hansen-Schwartz J, Dreier J, Vajkoczy P, Macdonald RL, Nishizawa S, Kasuya H, Wellman G, Keller E, Zauner A, Dorsch N, Clark J, Ono S, Kiris T, Leroux P, Zhang JH. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009;31:151–158. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Pose I, Sampogna S, Chase MH, Morales FR. Mesencephalic trigeminal neurons are innervated by nitric oxide synthase-containing fibers and respond to nitric oxide. Brain Res. 2003;960:81–89. doi: 10.1016/s0006-8993(02)03776-9. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Schievink WI, Riedinger M, Jhutty TK, Simon P. Racial disparities in subarachnoid hemorrhage mortality: Los Angeles County, California, 1985–1998. Neuroepidemiology. 2004;23:299–305. doi: 10.1159/000080096. [DOI] [PubMed] [Google Scholar]
- Sehba FA, Schwartz AY, Chereshnev I, Bederson JB. Acute decrease in cerebral nitric oxide levels after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2000;20:604–611. doi: 10.1097/00004647-200003000-00018. [DOI] [PubMed] [Google Scholar]
- Shin M, Chetkovich DM. Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J Biol Chem. 2007;282:33168–33180. doi: 10.1074/jbc.M703736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke. 2010;41:1783–1790. doi: 10.1161/STROKEAHA.110.586537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron. 2007;56:1076–1089. doi: 10.1016/j.neuron.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CP, Bergeron M, Matz P, Zegna A, Noble LJ, Panter SS, Sharp FR. Heme oxygenase-1 (HO-1) is induced in glia throughout the brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab. 1998;18:257–273. doi: 10.1097/00004647-199803000-00004. [DOI] [PubMed] [Google Scholar]
- Vajkoczy P, Meyer B, Weidauer S, Raabe A, Thome C, Ringel F, Breu V, Schmiedek P. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9–17. doi: 10.3171/jns.2005.103.1.0009. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. a2A-Adrenoceptors strengthen working memory networks by inhibiting cAMP HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Wickman G, Lan C, Vollrath B. Functional roles of the rho/rho kinase pathway and protein kinase C in the regulation of cerebrovascular constriction mediated by hemoglobin: relevance to subarachnoid hemorrhage and vasospasm. Circ Res. 2003;92:809–816. doi: 10.1161/01.RES.0000066663.12256.B2. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Role of dendritic synapse location in the control of action potential output. Trends Neurosci. 2003;26:147–154. doi: 10.1016/S0166-2236(03)00035-3. [DOI] [PubMed] [Google Scholar]
- Wilson GW, Garthwaite J. Hyperpolarization-activated ion channels as targets for nitric oxide signalling in deep cerebellar nuclei. Eur J Neurosci. 2010;31:1935–1945. doi: 10.1111/j.1460-9568.2010.07226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]