

Abstract
Endothelial cell (EC) barrier disruption induced by inflammatory agonists such as thrombin leads to potentially lethal physiological dysfunction such as alveolar flooding, hypoxemia, and pulmonary edema. Thrombin stimulates paracellular gap and F-actin stress fiber formation, triggers actomyosin contraction, and alters EC permeability through multiple mechanisms that include protein kinase C (PKC) activation. We previously have shown that the ezrin, radixin, and moesin (ERM) actin-binding proteins differentially participate in sphingosine-1 phosphate-induced EC barrier enhancement. Phosphorylation of a conserved threonine residue in the COOH-terminus of ERM proteins causes conformational changes in ERM to unmask binding sites and is considered a hallmark of ERM activation. In the present study we test the hypothesis that ERM proteins are phosphorylated on this critical threonine residue by thrombin-induced signaling events and explore the role of the ERM family in modulating thrombin-induced cytoskeletal rearrangement and EC barrier function. Thrombin promotes ERM phosphorylation at this threonine residue (ezrin Thr567, radixin Thr564, moesin Thr558) in a PKC-dependent fashion and induces translocation of phosphorylated ERM to the EC periphery. Thrombin-induced ERM threonine phosphorylation is likely synergistically mediated by protease-activated receptors PAR1 and PAR2. Using the siRNA approach, depletion of either moesin alone or of all three ERM proteins significantly attenuates thrombin-induced increase in EC barrier permeability (transendothelial electrical resistance), cytoskeletal rearrangements, paracellular gap formation, and accumulation of phospho-myosin light chain. In contrast, radixin depletion exerts opposing effects on these indexes. These data suggest that ERM proteins play important differential roles in the thrombin-induced modulation of EC permeability, with moesin promoting barrier dysfunction and radixin opposing it.
Keywords: thrombin, ERM, PKC, phosphorylation, endothelial cells, barrier dysfunction, cytoskeleton
the pulmonary vascular endothelium serves as a semiselective barrier between circulating blood and surrounding tissues. Endothelial cell (EC) barrier integrity is therefore critical to tissue and organ function. Disruption of the endothelial barrier by inflammatory mediators such as thrombin, histamine, and LPS leads to potentially lethal physiological dysfunction such as hypoxemia, atherosclerosis, and pulmonary edema, a hallmark of acute lung injury and its more severe form, acute respiratory distress syndrome (12). Therefore, the preservation of vascular EC barrier integrity has the potential for profound clinical impact. Multiple studies have demonstrated that inflammation-induced EC barrier dysfunction involves cytoskeletal rearrangement, contraction of EC, and intercellular gap formation, leading to increased paracellular permeability (18, 20, 55, 65). Thrombin, a multifunctional serine protease, proteolytically cleaves and activates PAR1, a member of a unique class of G protein-coupled receptors activated by proteolytic cleavage of their extracellular NH2-terminal domains and expressed at the surfaces of EC (43, 66). Thrombin can transactivate PAR2 through PAR1 in cultured human umbilical vein endothelial cells (48). PAR1 and PAR2 activate heterotrimeric G proteins Gq, G12/13, and Gi, all of which are involved in permeability regulation (42). Activation of Gq mobilizes Ca2+ and activates PKC, RhoA, and EC contraction, resulting in endothelial barrier disruption.
The widely distributed ERM family of membrane-associated proteins (ezrin, radixin, moesin) regulates the structure and function of specific domains of the cell cortex (reviewed in Refs. 2, 14, 46). The ERM proteins are actin-binding linkers that connect the actin cytoskeleton to the plasma membrane. This linker function makes ERM proteins essential for many fundamental cellular processes including cell adhesion, determination of cell shape, motility, cytokinesis, and integration of membrane transport with signaling pathways (14, 47, 71). The three ERM proteins share a high level of amino acid identity (70–85%) (14), and prior to activation exist in an autoinhibited conformation in which the actin-binding COOH-terminal tail binds and masks the NH2-terminal FERM domain (band 4.1, ezrin, radixin, moesin homology domains) (50). The activation state of ERM proteins is tightly regulated by phosphorylation events. Binding of the protein to membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2) (15) and subsequent phosphorylation of a conserved COOH-terminal threonine (Thr567 in ezrin, Thr564 in radixin, Thr558 in moesin) (21, 41, 50) are believed to disrupt the intramolecular association, thus unmasking sites for interactions with other proteins. In addition, phosphorylation of ezrin on other residues may be required to direct specific targeted effects in cells (29, 36, 57). Several kinases have been implicated in regulating ERM protein function through phosphorylation of the COOH-terminal threonine residue (3, 10, 35, 40, 59, 67). However, the identity of kinases that directly phosphorylate ERM in many cells remains to be clearly defined (14, 29).
ERM proteins also associate with cytoplasmic signaling molecules in cellular processes that require membrane cytoskeletal reorganization. ERM proteins appear to act both downstream and upstream of the Rho family of GTPases, which regulates remodeling of the actin cytoskeleton (14, 29). However, information is limited concerning the possible role of ERM proteins in the remodeling of endothelial cytoskeleton in response to different agonists. Koss and coworkers (35) demonstrated that ERM proteins are phosphorylated on COOH-terminal threonine residues by TNF-α-induced signaling events and likely play important roles in modulating the cytoskeletal changes and permeability increases in human pulmonary microvascular EC. We previously have shown that PKC isoforms are required for ERM phosphorylation in human pulmonary EC induced by the potent barrier protective factor, platelet-derived phospholipid sphingosine-1 phosphate (S1P) (1). Furthermore, we previously demonstrated that ERM proteins, despite their structural similarities and reported functional redundancy, differentially modulate S1P-induced changes in lung EC cytoskeleton and permeability (1). In the present study, we explored the potential involvement of ERM proteins in modulating thrombin-induced cytoskeletal rearrangement and EC barrier function.
MATERIALS AND METHODS
Reagents.
Thrombin was obtained from Sigma (St. Louis, MO). Antibodies (Ab) were obtained as follows: mouse monoclonal Ab against β-tubulin (Covance, Berkeley, CA), rabbit polyclonal di-phospho-myosin light chain (di-phospho-MLC) and rabbit polyclonal phospho-ezrin (Thr567)/radixin (Thr564)/moesin (Thr558) Ab (Cell Signaling, Danvers, MA), ezrin-specific mouse monoclonal Ab (Invitrogen Life Technologies, Carlsbad, CA), rabbit monoclonal anti-radixin Ab (Sigma), mouse monoclonal anti-moesin Ab (BD Biosciences, San Jose, CA), mouse monoclonal anti-thrombin receptor WEDE15 blocking Ab (Beckman Coulter, Indianapolis, IN), mouse monoclonal thrombin R (ATAP2) blocking Ab (Santa Cruz Biotechnology, Santa Cruz, CA), Texas red phalloidin and Alexa 488- or Alexa 594-conjugated secondary Ab (Molecular Probes, Eugene, OR). ROCK inhibitors Y-27632 and H-1152; PKC inhibitors Ro-31-7549, bisindolylmaleimide I, and Go 6976; and p38 kinase inhibitor SB203580 were purchased from Calbiochem (San Diego, CA); Ca2+ chelator BAPTA-AM was obtained from Sigma, phosphoinositide 3-kinase (PI3K) inhibitor LY294002 PAR1-selective agonist TFLLR-NH2, PAR2-selective agonist SLIGRL-NH2, and reversed amino acid sequence control peptides RLLFT-NH2 and LRGILS-NH2 were obtained from TOCRIS (Bristol, UK). Unless specified, biochemical reagents were obtained from Sigma.
Cell culture.
Human pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (Walkersville, MD) and were utilized at passages 5–9.
Measurement of TER.
Cellular barrier properties were measured by using an electrical cell substrate impedance sensing system (ECIS) (Applied Biophysics, Troy, NY). HPAEC were seeded onto plates with small gold electrodes (10–4 cm2) and measurements of transendothelial electrical resistance (TER) across confluent HPAEC monolayers were performed as previously described (6, 19, 65).
Real-time quantitative RT-PCR.
Endogenous transcript levels of ezrin (EZR), radixin (RDX), and moesin (MSN) in HPAEC were measured in a 384-well PCR plate with an ABI 7900 HT Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Total RNA (1 μg) were first reverse transcribed by using Superscript II Reverse Transcriptase (Invitrogen Life Technologies) and random hexamer primers (Applied Biosystems) to generate cDNA. Quantitative real-time PCR was then performed by using the Assay-on-Demand system [Hs00185574_m1 (EZR); Hs00267954_m1 (RDX); Hs00792607_mH (MSN)] from Applied Biosystems according to the manufacturer's protocol. Purity and specificity of all products were confirmed by omitting the reverse transcriptase or template. Analysis of results is based on the average of triplicates. The standard curve method was used for relative quantitation of target gene expression. Further information on the method can be found on User Bulletin no. 2 on the ABI website.
Depletion of specific EC proteins via siRNA.
To reduce the content of individual EC proteins, cultured EC were treated with specific siRNA duplexes, which guide sequence-specific degradation of the homologous mRNA (13). The following validated siRNAs were obtained from Qiagen (Valencia, CA) in ready-to-use, desalted, and duplexed form: duplex of sense 5′-CACCGUGGGAUGCUCAAAGdTdT-3′ and antisense 5′-CUUUGAGCAUCCCACGGUGdTdT-3′ siRNA was used for targeting sequences that are part of the coding region for Homo sapiens ezrin: 5′-AACACCGTGGGATGCTCAAAG-3′, duplex of sense 5′-GAAAUAACCCAGAGACUCUdTdT-3′ and antisense 5′-AGAGUCUCUGGGUUAUUUCdTdT-3′ was used for targeting sequences that are part of the coding region for Homo sapiens radixin: 5′-AAGAAATAACCCAGAGACTCT-3′, and duplex of sense 5′-GGGAUGUCAACUGACCUAAdTdT-3′ and antisense 5′-UUAGGUCAGUUGACAUCCCdTdG-3′ was used for targeting sequences that are part of the coding region for Homo sapiens moesin: 5′-CAGGGATGTCAACTGACCTAA-3′. Duplex of sense 5′-AGAGCUAAG-UAGAUGUGUAdTdT-3′ and antisense 5′-UACACAUCUACUUAGCUCUdTdG-3′ siRNA was used for targeting sequences that are part of the coding region for Homo sapiens PKCβI: 5′-CAAGAGCTAAGTAGATGTGTA-3′, duplex of sense 5′-GAAGCAUGACAGCAUUAAA dTdT-3′ and antisense 5′-UUUAAUGCUGUCAUGCUUCdCdG-3′ was used for targeting sequences that are part of the coding region for Homo sapiens PKCζ: 5′-CGGAAGCATGACAGCATTAAA-3′, duplex of sense 5′-CUCUACCGUGCCACGUUUUdTdT-3′ and antisense 5′-AAAACGUGGCACGGUAGAGdTdT-3′ was used for targeting sequences that are part of the coding region for Homo sapiens PKCδ: 5′-AACTCTACCGTGCCACGTTTT-3′, duplex of sense 5′-CAAGAAGUGUAUUGAUAAAdTdT-3′ and antisense 5′-UUUAUCAAUACACUUCUUGdTdG-3′ was used for targeting sequences that are part of the coding region for Homo sapiens PKCθ: 5′-CACAAGAAGTGTATTGATAAA-3′, duplex of sense 5′-CGGAAACACCCGUACCUUAdTdT-3′ and antisense 5′-UAAGGUACGGGUGUUUCCGdTdG-3′ was used for targeting sequences that are part of the coding region for Homo sapiens PKCε: 5′-CACGGAAACACCCGTACCTTA-3′. Silencer select predesigned siRNA duplex (Life Technologies, Grand Island, NY) of sense 5′-CCCGUAACCUAAUUCCUAUdTdT-3′ and antisense 5′-AUAGGAAUUAGGUUACGGGdCdC-3′ was used for targeting sequences that are part of the coding region for Homo sapiens PKCγ: 5′-GGCCCGTAACCTAATTCCTAT-3′. Nonspecific, nontargeting AllStars siRNA duplex (Qiagen) was used as negative control treatment. HPAEC were grown to 70% confluence, and the transfection of siRNA (final concentration 50 nM) was performed by using DharmaFECT1 transfection reagent (Dharmacon Research, Lafayette, CO) according to manufacturer's protocol. Forty-eight hours posttransfection cells were harvested and used for experiments. Additional control experiments using EC transfections with fluorescently labeled nonspecific RNA showed that this protocol allowed us to achieve 90–100% transfection efficiency.
Plasmid constructs.
Moesin constructs (wild-type and phosphorylation-deficient mutant) were prepared as we have previously described (9).
Immunofluorescent staining.
EC were plated on glass coverslips, grown to 70% confluence, and transfected with siRNA followed by stimulation with thrombin. Then cells were fixed in 3.7% formaldehyde solution in PBS for 10 min at 4°C, washed three times with PBS, permeabilized with 0.2% Triton X-100 in PBS-Tween (PBST) for 30 min at room temperature and blocked with 2% BSA in PBST for 30 min. Incubation with antibody of interest was performed in blocking solution for 1 h at room temperature followed by staining with either Alexa 488- or Alexa 594-conjugated secondary Ab (Molecular Probes). Actin filaments were stained with Texas red-conjugated phalloidin (Molecular Probes) for 1 h at room temperature. After immunostaining, the glass slides were analyzed by using a Nikon video-imaging system (Nikon Instech, Japan) consisting of a phase-contrast inverted microscope Nikon Eclipse TE2000 connected to Hamamatsu (Hamamatsu Photonics, Japan) digital camera and image processor. The images were recorded and processed with Adobe Photoshop 6.0.
Immunoblotting.
Protein extracts were separated by 4–15% gradient SDS-PAGE, transferred to nitrocellulose or polyvinylidene difluoride membranes (30 V for 18 h or 100 V for 1.5 h) and reacted with Ab that recognizes ezrin, moesin, radixin, or other Ab of interest as indicated for individual experiments. The level of phosphorylated ERM was examined by using a single Ab that recognizes any of the three ERM proteins only when they are phosphorylated on the threonine residue: ezrin (Thr567)/radixin (Thr564)/moesin (Thr558) (Cell Signaling). Immunoreactive proteins were detected with the enhanced chemiluminescent detection system (ECL) according to the manufacturer's directions (Amersham, Little Chalfont, UK). Intensities of immunoreactive protein bands were quantified by use of ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Statistical analysis.
Results are expressed as means ± SD of three to six independent experiments. We performed statistical comparison among treatment groups by unpaired Student's t-test or by randomized-design two-way analysis of variance followed by the Newman-Keuls post hoc test for multiple groups. Results with P < 0.05 were considered statistically significant.
RESULTS
Expression of ezrin, radixin, and moesin in HPAEC.
The mRNA expression profiles of individual ERMs were analyzed for confluent human pulmonary EC. RT-PCR analysis reveals differential expression with highest relative expression of moesin and lowest expression of radixin (Fig. 1).
Fig. 1.

Relative quantity of moesin (MSN), ezrin (EZR), and radixin (RDX) (i.e., ERM) mRNA in human pulmonary artery endothelial cells (HPAEC). Total RNA was isolated from HPAEC and quantitative real-time reverse transcriptase-polymerase chain reaction (qRT-PCR) was performed by using target gene specific primers and probes and the relative amounts were expressed by standard curve method as described in materials and methods. Each value represents the mean of triplicates.
Thrombin induces threonine phosphorylation of ERM via a PKC-mediated pathway.
To elucidate the effect of thrombin on phosphorylation of ERM at its critical COOH-terminal threonine, confluent human pulmonary EC were stimulated with thrombin (0.5 U/ml), and threonine phosphorylation then was evaluated by Western blot analysis utilizing phospho-specific ERM antibody (phospho-ezrin Thr567/radixin Thr564/moesin Thr558). Thrombin induced the sustained threonine phosphorylation of ERM, which reached maximum levels by 5 min and remained elevated for at least 120 min (Fig. 2).
Fig. 2.

Time-dependent effects of thrombin on threonine phosphorylation of ERM. A: confluent HPAEC were treated either with control vehicle or thrombin (0.5 U/ml) for the indicated times, and phosphorylated ERM [phospho-ezrin (Thr567)/radixin (Thr564)/moesin (Thr558)] was detected via immunoblotting. B: bar graph represents relative densitometry. Data are presented as fold changes in phosphorylated ERM over vehicle-treated control and expressed as means ± SE from 3 independent experiments. *P < 0.05 vs. unstimulated control.
Because several kinases, including members of PKC, ROCK, GRK2, p38, Mst4, and LOK, have been reported to phosphorylate the regulatory COOH-terminal threonine residue of ERM proteins in various systems (3, 10, 35, 40, 59, 67), experiments were performed to determine the signaling mechanisms leading to ERM phosphorylation in pulmonary EC upon thrombin treatment. The role of different kinases was examined by using specific pharmacological inhibitors: PKC-specific inhibitor Ro-31-7549, p38 MAPK inhibitor SB203580, Rho-associated protein kinase (ROCK) inhibitor Y-27632, PI3K inhibitor LY294002, and chelator of intracellular Ca2+ BAPTA (Fig. 3). In our experiments, pretreatment with Ro-31-7549 effectively prevented the increase in ERM phosphorylation induced by thrombin (Fig. 3A), suggesting that this increase is PKC dependent. Pretreatment with BAPTA, a chelator of intracellular Ca2+, partially inhibited ERM phosphorylation (Fig. 3A). We next explored whether additional signaling pathways previously reported to participate in ERM regulation are involved in thrombin-induced ERM phosphorylation. Pretreatment with Y-27632 did not exert a significant effect on ERM threonine phosphorylation. At the same time, phosphorylation of myosin light chain (MLC), regulated by the ROCK/myosin phosphatase-dependent signaling pathway, was significantly attenuated in EC preincubated with Y-27632 (Fig. 3A). Because of the critical role of Rho activation, through its downstream effector ROCK, in thrombin-induced EC barrier dysfunction, we next studied how the inhibition of ROCK affects ERM phosphorylation using the more potent and selective cell-permeable pharmacological ROCK inhibitor H-1152. Pretreatment with either H-1152 or Y-27632 did not exert significant effects on ERM threonine phosphorylation after thrombin (Fig. 3B). In addition, preincubation of HPAEC with the pharmacological inhibitor of p38 MAPK SB203580 did not significantly affect ERM phosphorylation (Fig. 3, A and B).
Fig. 3.

Thrombin-induced ERM phosphorylation requires activation of PKC. HPAEC were pretreated with either control vehicle or the following inhibitors: PKC inhibitors Ro-31-7549 (10 μM, A and C) for 30 min, bisindolylmaleimide (BIM, 1 μM, C) for 30 min, Go 6976 (1 μM, C) for 1 h, Ca2+ chelator BAPTA-AM (25 μM, A) for 1 h, p38 kinase inhibitor SB203580 (20 μM, A and B) for 30 min, Rho kinase inhibitors Y-27632 (10 μM, A and B) for 1 h and H-1152 (3 μM, B) for 1 h, phosphoinositide 3-kinase inhibitor LY294002 (10 μM, A) for 1 h. EC were then stimulated with EBM-2 medium alone or thrombin (0.5 U/ml) for the indicated time. Phosphorylation of ERM proteins and myosin light chain (MLC) were analyzed by immunoblotting of cell lysates with phospho-ERM (as in Fig. 2) or di-phospho-myosin light chain kinase (di-phospho-MLC) (Thr18/Ser19) specific Abs. GAPDH or β-actin Abs were used as a normalization control. Rearranged lanes from the same blot are outlined by vertical dotted line. Results of scanning densitometry of Western blots are shown as fold changes of ERM or MLC phosphorylation relative to vehicle treated EC stimulated by thrombin. Results are representative of 3–6 independent experiments. Values are means ± SE. *Significantly different from cells treated with vehicle (P < 0.05); **significantly different from cells stimulated with thrombin (P < 0.05).
The role of PKC isoforms was examined by two alternative approaches: pretreatment of EC with PKC-specific pharmacological inhibitors and using isoform-specific siRNAs. We utilized three PKC-specific pharmacological inhibitors that have different IC50 values for different PKC isoforms, bisindolylmaleimide I (BIM), Go 6976, and Ro-31-7549. Bis I, Ro-31-7549, and Go 6970 are all competitive inhibitors for the ATP-binding site of PKC (39, 63, 69). BIM inhibits the conventional PKC isoforms α, βI, βII, and γ (activated by phosphatidylserine, diacylglycerol, and Ca2+) with similar potency (IC50 = 10 nM) (63), and the unconventional isoforms δ and ε (require phosphatidylserine and diacylglycerol but are Ca2+ independent), and the atypical isoform ζ (requires only phosphatidylserine), to a lesser extent (39). In contrast to BIM, Ro-31-7549 has slight selectivity for the α isoform (IC50 = 53 nM) but also affects βI, βII, ε, and γ (69). Go 6970 inhibits Ca2+-dependent PKC isoforms α and βI (39). In our experiments, pretreatment with Ro-31-7549, BIM, and Go 6970 effectively suppressed ERM phosphorylation induced by thrombin (Fig. 3C). These pharmacological PKC inhibitor data combined with the BAPTA data suggest that multiple PKC isoforms may be required. Furthermore, our experiments demonstrate that incubation with Go 6976 significantly inhibited phosphorylation of MLC at Thr18 and Ser19 induced by thrombin (Fig. 3C).
To better characterize the PKC isoforms involved in ERM phosphorylation after thrombin, activation of individual PKC kinases was explored by use of isoform-specific phospho-antibodies. Thrombin stimulation (0.5 U/ml) of HPAEC significantly increased phosphorylation of PKCβ (Thr500) (Fig. 4A), PKCγ (Thr514) (Fig. 4B), PKCε (Ser729) (Fig. 4C), PKCζ (Thr410) (Fig. 4D), PKCθ (Thr538) (Fig. 4E), and PKCδ (Tyr311) (Fig. 4F). Previous studies have indicated that these PKC isoforms play important roles in thrombin- and other inflammatory agonists (TNF-α, IL-1β, VEGF, hypoxia)-induced modulation of endothelial permeability (16, 38, 44, 51, 53, 56, 61, 62, 70) and therefore were selected for further experimentation using isoform-specific siRNA. We first validated that these siRNAs efficiently inhibit their respective targets: 65 ± 7% depletion of PKCβ, 97 ± 2.2% depletion of PKCδ, 75.5 ± 2.4% depletion of PKCθ, 92 ± 2.5% depletion of PKCε, and 65 ± 2.5% depletion of PKCγ (Fig. 5; see also Ref. 1). It is important to note the limitations of this siRNA approach since it does not provide 100% protein suppression and therefore some, albeit reduced, protein function likely remains. EC were transfected with siRNA for PKCβI, PKCγ, PKCε, PKCζ, PKCθ, or PKCδ and then stimulated with thrombin to determine the effects on ERM threonine phosphorylation. Depletion of individual PKCγ, PKCε, PKCζ, PKCθ, or PKCδ isoforms significantly reduced ERM phosphorylation after thrombin (Fig. 6, A and B). To further clarify the involvement of PKC activity in thrombin-induced ERM and MLC phosphorylation, we pursued simultaneous depletion of several PKC isoforms (pan-PKC) via siRNA. Combined depletion of several PKC isoforms markedly reduced ERM phosphorylation after thrombin (Fig. 6, C and D), suggesting possible cooperative regulation in this phosphorylation response. Moreover, downregulation of pan-PKC significantly attenuates thrombin-induced MLC di-phosphorylation (Fig. 6, C and E), suggesting that multiple PKC isoforms may be involved. Taken together, these data indicate that multiple PKC isoforms (conventional, unconventional, and atypical) likely participate in thrombin-induced ERM and MLC phosphorylation. One limitation of these data is that combined silencing of multiple PKC isoforms may result in some nonspecific effects, e.g., nonspecific activation or/and inhibition of other signaling pathways. Therefore, further studies utilizing alternative approaches (e.g., isoform-specific PKC peptide inhibitors) will be needed to verify these findings.
Fig. 4.
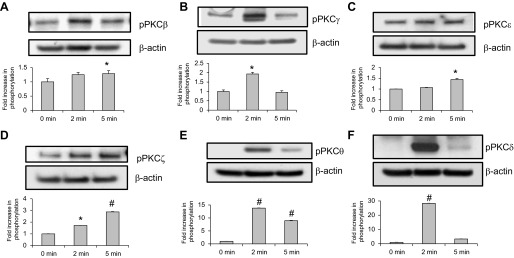
Effects of thrombin on phosphorylation of PKC isoforms in HPAEC. Confluent HPAEC were treated either with control vehicle or thrombin (0.5 U/ml) for the indicated times, and phosphorylated PKCβ (A), PKCγ (B), PKCε (C), PKCζ (D), PKCθ (E), and PKCδ (F) were detected via immunoblotting. Bar graphs represent relative densitometry of fold changes in phosphorylated PKC isoforms after thrombin relative to vehicle-treated control and expressed as means ± SE from 3 independent experiments. *Significantly different from cells treated with EBM-2 (P < 0.05); #significantly different from cells treated with EBM-2 (P < 0.01).
Fig. 5.

Depletion of PKC isoforms by siRNA. PKCε (A) and PKCγ (B) depletion were induced by specific siRNA duplexes and assessed for silencing effects by immunoblotting with appropriate antibody (Ab), compared with treatment with nonspecific (ns) siRNA. Immunoblotting with β-actin Ab was used as a normalization control. Rearranged lanes from the same blot are outlined by vertical dotted line. Quantitative analysis of protein expression was performed by scanning densitometry and expressed in relative density units (RDU). Results are means ± SE for 3 independent experiments. #Significant difference (P < 0.01) compared with cells treated with ns siRNA.
Fig. 6.

Depletion of PKC isoforms inhibits thrombin-induced ERM and MLC phosphorylation. Confluent HPAEC were incubated with nonspecific and PKCβI-, PKCζ-, PKCθ-, PKCδ-, PKCγ-, and PKCε-specific siRNA (A) or with nonspecific, combinations of PKCδ- and PKCε-specific, combinations of PKCγ-, PKCδ-, and PKCε-specific, and combinations of PKCβI-, PKCθ-, and PKCζ-specific siRNAs (C) as described in materials and methods and then stimulated by thrombin (0.5 U/ml, 5 min) or vehicle. Total lysates were analyzed by immunoblotting for phospho-ERM or di-phospho-MLC (Thr18/Ser19). Immunoblotting with β-tubulin Ab was used as a normalization control. Rearranged lanes from the same blot are outlined by vertical dotted line. B, D, and E: bar graphs represent relative densitometry of fold changes in phosphorylated ERM and MLC after thrombin relative to vehicle-treated control. Results are means ± SE of 4 independent experiments. *Significantly different from cells treated with ns siRNA without thrombin (P < 0.01); #significantly different from cells treated with ns siRNA without thrombin (P < 0.05). **Significantly different from cells treated with ns siRNA and thrombin (P < 0.05).
Effects of ERM depletion on thrombin-induced ERM threonine and MLC phosphorylation.
Numerous studies have reported a critical role for activation of the contractile apparatus in specific models of agonist-induced EC barrier dysfunction (reviewed in Ref. 12). A key contractile event in thrombin-induced barrier dysfunction is the phosphorylation of regulatory MLC, catalyzed by Ca2+/calmodulin-dependent MLC kinase (MLCK) (60, 72) and regulated by small GTPase Rho. There are no antibodies currently available that specifically differentiate among the COOH-terminal phospho-threonines of individual ERM proteins. Therefore, to assess the contribution of individual proteins in total ERM threonine phosphorylation and their roles in MLC phosphorylation after thrombin, we used siRNAs targeting ezrin, radixin, and moesin. We first validated that these siRNAs efficiently and specifically inhibited their respective targets (1). Pulmonary EC were then transfected with nonspecific siRNA, or siRNA for moesin or radixin, or siRNA for the combination of ezrin, radixin, and moesin (pan-ERM), and then stimulated with thrombin. Pan-ERM and MLC phosphorylation was evaluated by Western blot analysis utilizing phospho-specific ERM antibody and phospho-myosin light chain 2 (Thr18/Ser19) antibody (di-phospho-MLC). In contrast to cells transfected with nonspecific RNA, depletion of moesin alone or downregulation of pan-ERM significantly reduced time-dependent ERM and MLC phosphorylation after thrombin (Fig. 7, A and D). Thrombin-induced ERM and MLC phosphorylation was partially attenuated by depletion of ezrin (Fig. 7C). In contrast, depletion of radixin did not have any significant effect on MLC phosphorylation after thrombin (Fig. 7B). Radixin siRNA partially, but significantly, reduced ERM threonine phosphorylation (Fig. 7B). Taken together, these data indicate that in pulmonary EC thrombin induces primarily threonine phosphorylation of moesin, followed by ezrin, and then radixin. These data also suggest that activated moesin and to a lesser degree ezrin, but not radixin, may play a role in MLC phosphorylation induced by thrombin via the ROCK/myosin phosphatase signaling pathway.
Fig. 7.

Effects of ERM depletion on thrombin-induced ERM and MLC phosphorylation. Confluent HPAEC were incubated with nonspecific, moesin- (A), radixin- (B), or ezrin-specific (C) or combined siRNAs for ezrin, radixin, and moesin (D) as described in materials and methods then stimulated by thrombin (0.5 U/ml, 5 min) or vehicle. Total lysates were analyzed by immunoblotting with phospho-ERM or di-phospho-MLC (Thr18/Ser19) Abs. Immunoblotting with β-tubulin Ab was used as a normalization control. Bar graphs represent relative densitometry of fold changes in phosphorylated ERM and MLC after thrombin relative to vehicle-treated control. Results are means ± SE of 3 independent experiments. *P < 0.05, compared with corresponding pretreatment vehicle control.
Role of ERM in thrombin-induced lung EC hyperpermeability.
To evaluate the functional involvement of individual ERM proteins in thrombin-induced EC barrier dysfunction, we measured changes in TER, a highly sensitive in vitro assay of permeability, in lung EC treated with nonspecific siRNA or those treated with siRNA for ezrin, radixin, or moesin (either singly or in combination). Comparing the data expressed as normalized resistance (Fig. 8) to the time course of ERM threonine phosphorylation (Fig. 2) demonstrates that the increase in ERM phosphorylation is highly correlated with the onset of thrombin-induced hyperpermeability. Depletion of individual ERM proteins does not affect basal permeability. However, the thrombin-induced decrease in TER is markedly attenuated in EC transfected with moesin or pan-ERM-specific siRNA (Fig. 8, A, D, and E) compared with cells transfected with nonspecific RNA duplexes. The initial thrombin-induced decrease in TER was attenuated by siRNA depletion of ezrin, but this intervention markedly enhanced the recovery above baseline in the later stage (Fig. 8, C and E). In contrast, depletion of radixin slightly augments the decrease in TER during the early phase and attenuates the later recovery phase after thrombin (Fig. 8, B and E) compared with agonist-stimulated cells transfected with nonspecific RNA. These data clearly indicate differential roles for individual ERM proteins in mediating thrombin-induced lung EC hyperpermeability.
Fig. 8.

Effects of ERM depletion on thrombin-induced endothelial barrier hyperpermeability. EC grown in chambers on gold microelectrodes were transfected with siRNA for moesin (A), radixin (B), ezrin (C), or combined siRNAs for ezrin, radixin, and moesin (D) or treated with ns siRNA, as described in materials and methods and used for transendothelial electrical resistance (TER) measurements. At time = 0, cells were stimulated with thrombin (0.5 U/ml) or vehicle control. Shown are pooled data of 5 independent experiments. Bar graph (E) depicts pooled TER data (n = 5) as maximal value of normalized TER elevation above baseline achieved within 30 min ± SE. *Significantly different from cells treated with ns siRNA reagent without thrombin (P < 0.05); **significantly different from control cells stimulated with thrombin (P < 0.05).
Role of PARs in thrombin-induced ERM phosphorylation.
In human pulmonary EC, activation of PAR1 and PAR2 promotes PKC and RhoA activation and triggers the endothelial barrier-disruptive response (7, 22, 37, 43). We next utilized PAR1-specific blocking antibodies ATAP2 and WEDE15, and PAR1 (TFLLR-NH2) and PAR2 (SLIGRL-NH2)-selective agonist peptides, to determine the roles of these receptors in thrombin-induced threonine phosphorylation of ERM. HPAEC preincubation with both ATAP2 and WEDE15, either singly or in combination, significantly decreased ERM phosphorylation after thrombin relative to controls (Fig. 9). Stimulation of EC with both TFLLR-NH2 and SLIGRL-NH2 augmented the phospho-ERM signal (Fig. 10) compared with cells treated with control peptides. Interestingly, incubation of EC with a combination of TFLLR-NH2 and SLIGRL-NH2 markedly enhanced ERM phosphorylation, compared with cells treated with these two agonists alone (Fig. 10), suggesting that both PAR1 and PAR2 may have additive or synergistic effects in this phosphorylation response.
Fig. 9.

Effects of PAR1 blocking antibodies on thrombin-induced ERM phosphorylation. A: HPAEC were pretreated for 1 h with either control vehicle or the PAR1 blocking Abs ATAP2 (25 μg/ml) or WEDE15 (25 μg/ml) or with combination of ATAP2 and WEDE15, then stimulated by thrombin (0.5 U/ml, 5 min) or vehicle. Total lysates were analyzed by immunoblotting for phospho-ERM. Immunoblotting with β-actin Ab was used as a normalization control. B: bar graph represents relative densitometry of fold changes in phosphorylated ERM after thrombin relative to vehicle-treated control. Results are means ± SE of 4 independent experiments. *Significantly different from cells treated with ns siRNA without sphingosine-1 phosphate (S1P) (P < 0.05); #significantly different from cells treated with ns siRNA without thrombin (P < 0.01). **Significantly different from cells treated with ns siRNA and thrombin (P < 0.05).
Fig. 10.

Effects of PAR1- and PAR2-selective agonists on thrombin-induced ERM phosphorylation. A: EC were pretreated for 5 min with thrombin (0.5 U/ml), PAR1-selective agonist TFLLR-NH2 (50 μM), PAR2-selective agonist SLIGRL-NH2 (50 μM), or combination of TFLLR-NH2 and SLIGRL-NH2. Pretreatment with vehicle and reversed amino acid sequence peptides RLLFT-NH2 and LRGILS-NH2 used as controls. B: bar graph represents relative densitometry of fold changes in phosphorylated ERM after thrombin, TFLLR-NH2, or SLIGRL-NH2 relative to vehicle-treated control. Results are means ± SE of 3 independent experiments. *P < 0.05, compared with corresponding pretreatment controls.
Involvement of ERM in thrombin-induced EC cytoskeletal remodeling.
Compromised barrier function induced by thrombin is tightly associated with actin cytoskeletal rearrangements, F-actin stress fiber formation, increased MLCK-catalyzed MLC phosphorylation spatially colocalized with stress fibers, actomyosin contraction, opening of paracellular gaps, and hyperpermeability (12, 43). Within this context, the distribution of phosphorylated ERM in EC before and after thrombin treatment was examined via immunofluorescence. Before thrombin treatment, minimal phosphorylated ERM was observed in the cytoplasm of pulmonary EC. In some areas of quiescent monolayers, phospho-ERM localized in the spikelike structures overlapping the cell-cell contact areas (Fig. 11, image a, arrow 1). Stimulation with thrombin induced an increase in the amount of phosphorylated ERM, consistent with immunoblotting studies (Fig. 2). During the early response (5 min after initial stimulation), phosphorylated ERM was localized primarily at the cell periphery and spikelike areas (Fig. 11, image b, arrows 1 and 2) with a smaller amount of phospho-ERM also observed in the cytoplasm (Fig. 11, image b). In contracting EC (15 min after initial challenge) the phospho-ERM signal markedly increased in the peripheral cytoplasmic areas (Fig. 11, image c). During partial restoration phases (60 and 120 min after the initial challenge), phosphorylated ERM primarily localized in cytoplasmic areas and spikelike structures that were originally seen in quiescent cells (Fig. 11, images d and e), but the overall level of phosphorylated ERM appeared slightly elevated compared with baseline. These dynamic changes in phosphorylated ERM localization during the phases of active cell contraction and partial barrier restoration indicate that ERM proteins may play a role in both processes. The observation that phosphorylated ERM proteins were primarily concentrated along the EC periphery upon thrombin treatment led us to examine the role of these proteins in modulating endothelial cytoskeletal rearrangements that occur during thrombin-induced EC hyperpermeability.
Fig. 11.

Distribution of phospho-ERM in EC after thrombin. EC grown on glass coverslips and treated with 0.5 U/ml thrombin for indicated time (B–E) or nontreated control cells (A) were subjected to immunofluorescent staining with anti-phospho-ERM Ab. The phospho-ERM signal is very weak in quiescent monolayers and is evident only in spikelike structures in cell-cell border areas (A, arrow 1). Threonine-phosphorylated ERM proteins predominantly localized to the periphery of ECs following thrombin stimulation (5–15 min, B and C, arrow 2) and also are detectable in peripheral spikelike structures (B, arrow 1). After 1–2 h phosphorylated ERM localized in spikelike structures characteristic of quiescent cells and in cytoplasm (D and E). Images are representative of 3 independent experiments. Scale bar = 10 μm.
In the next series of experiments we analyzed the effect of ERM depletion on the human endothelial actin cytoskeleton. EC were transfected with nonspecific RNA duplex oligonucleotides (Fig. 12) or ERM-specific siRNA (Fig. 12) followed by thrombin challenge (15 min, 0.5 U/ml). Double immunofluorescent staining using Texas red phalloidin to visualize F-actin and di-phospho-MLC antibody to detect phosphorylated MLC was performed. Unstimulated EC transfected with ezrin-, radixin-, or moesin-specific siRNAs, either individually or in combination, demonstrated no significant differences in the organization of actin cytoskeleton and levels of MLC phosphorylation compared with control EC exposed to nonspecific RNA (Fig. 12, A, images a–n). However, thrombin stimulation of EC treated with nonspecific RNA induced robust F-actin stress fiber and gap formation and accumulation of di-phospho-MLC (Fig. 12, B, images c and d), which were nearly abolished by the combination of siRNAs for ezrin/radixin/moesin (pan-ERM depletion), or by siRNA for moesin alone. In contrast, radixin depletion alone slightly enhanced stress fiber formation and MLC phosphorylation (Fig. 12, B, images k and l). We next explored the effects of overexpression of a phosphorylation-deficient mutant of moesin (Thr558Ala) on EC cytoskeletal organization. Scanning densitometry (Fig. 13A) demonstrated relatively equivalent expression of the wild-type and mutant moesin in EC. Monolayers overexpressing mutant moesin exhibited decreased F-actin stress fibers after thrombin and prominent cortical actin compared with EC overexpressing wild-type moesin (Fig. 13, B, images c and g). These data together demonstrate that ERM proteins are downstream targets of thrombin-induced signaling mechanisms and play an essential and differential role in the regulation of the endothelial actomyosin cytoskeleton.
Fig. 12.
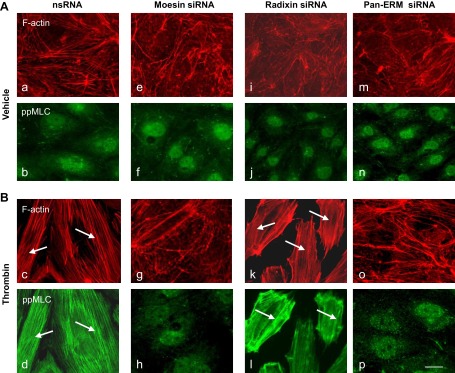
Effects of ERM depletion on thrombin-induced cytoskeletal remodeling. HPAEC grown on glass coverslips were incubated with siRNA to ezrin, radixin, moesin, or combination of siRNAs to all 3 proteins, or treated with nonspecific siRNA duplex as described in materials and methods followed by thrombin treatment (0.5 U/ml, 5 min). EC were subjected to double immunofluorescent staining with Texas red phalloidin to visualize F-actin (A and B, top) and anti-pp-MLC Ab (A and B, bottom). Incubation with siRNA to moesin (g and h) and combined siRNAs to ezrin, radixin, and moesin (o and p) almost completely abolishes thrombin-induced F-actin stress fiber and gap formation and MLC phosphorylation compared with control (nsRNA) incubation (c and d, arrows). In contrast, pretreatment with siRNA to radixin slightly enhances the thickness of stress fibers and MLC phosphorylation (k and l, arrows) compared with incubation with nsRNA. Bar = 10 μM. Images are representative of 3 independent experiments.
Fig. 13.

Effects of overexpression of the phosphorylation-deficient mutant of moesin (Thr558Ala) on thrombin-induced cytoskeletal remodeling. A: EC were transfected with empty vector (control), V5 (a peptide GKPIPNPLLGLDST, recognized by an antibody)-tagged wild-type (WT), or mutant (Mut) moesin, which were then detected via immunoblotting with V5 Ab. Results of scanning densitometry of Western blots are shown as % of moesin relative to control. Immunoblotting with β-actin Ab was used as a normalization control. B: after transfection with vectors expressing moesin (wild-type or mutant) tagged with V5, EC were grown on glass coverslips as described in materials and methods followed by thrombin treatment (0.5 U/ml, 5 min). EC were subjected to double immunofluorescent staining with Texas red phalloidin to visualize F-actin (B and C, top) and V5 Ab (B and C, bottom). Overexpression with mutant moesin abolishes thrombin-induced F-actin stress fibers and induces cortical actin formation (C, image g) compared with EC overexpressed with wild-type moesin (C, image c). Arrow indicates cell transfected with mutant moesin EC (C, image g). Images are representative of 3 independent experiments. Scale bar = 10 μm.
DISCUSSION
Prior work has revealed that the procoagulant serine protease thrombin induces endothelial barrier compromise through G protein-coupled Ca2+ mobilization; MLCK, PKC, and RhoA activation, which produces cytoskeletal rearrangement; dissociation of endothelial cell-cell junctions; as well as cytoskeleton contraction resulting in paracellular hyperpermeability (12, 27, 33, 34, 43, 45, 66). Although ERM proteins act as signal transducers for agonists that induce cytoskeletal remodeling (14), the role of ERM in barrier regulation by thrombin is unknown. Moesin is the most expressed ERM protein in several types of EC (4, 24, 30, 35). Here we demonstrate that moesin is the most abundant ERM expressed in HPAEC as measured by mRNA content, with radixin being the least expressed. We previously have shown that the angiogenic sphingolipid S1P induces ERM phosphorylation on a conserved threonine residue critical for ERM activation via a pathway that requires PKC, Rac1, Rho A, and p38 MAPK (1). We also have previously demonstrated that phosphorylation of ERM in response to the microtubule disruptor 2-methoxyestradiol (2ME) occurs in a p38/PKC-dependent manner (9). We therefore explored whether the ERM family of proteins plays a role in modulating the thrombin-induced endothelial barrier response. Our data demonstrate that thrombin increases ERM phosphorylation at a critical regulatory threonine site in HPAEC monolayers and strongly suggest important roles for the ERM proteins in mediating endothelial barrier dysfunction by thrombin. This phosphorylation requires thrombin-induced signaling pathways that include activation of PKC isoforms, but it is not regulated in a RhoA/ROCK- or p38-dependent manner. Our results differ from those in our (1, 9), and others' previous studies (23, 35, 68), in which the phosphorylation of ERM was PKC, RhoA/ROCK, and p38 dependent in response to S1P, PKC and RhoA/ROCK dependent in response to 2ME and TNF-α, and RhoA/ROCK dependent in response to advanced glycation end products (AGE). Together, these observations suggest that ERM may be phosphorylated at the critical COOH-terminal threonine site by different upstream pathways that can vary from endothelium to endothelium and from stimulus to stimulus.
We and others previously have demonstrated that several PKC isoforms (conventional, unconventional, and atypical) are likely to phosphorylate the COOH-terminal threonine residue of ERM proteins (1, 35, 52, 67). Our data now demonstrate that PKC isoforms ζ, γ, ε, θ, and δ, which previously have been demonstrated to play roles in inflammation-induced changes in endothelial permeability (16, 44, 53, 56, 61, 62, 70), participate in thrombin-induced ERM phosphorylation at the COOH-terminal threonine site in human pulmonary EC (Fig. 6). Moreover, our data indicate that these isoforms may synergistically regulate both ERM threonine and MLC phosphorylation (Fig. 6, C and D). One of the key events in the signaling cascade triggered by thrombin binding to PAR receptors is Ca2+-dependent activation of PKC and Ca2+/calmodulin-dependent MLCK, which phosphorylates MLC (12, 37). RhoA and its effector ROCK indirectly regulate MLCK activation and MLC phosphorylation and therefore also mediate endothelial hyperpermeability in response to thrombin (8, 49, 64). The RhoA/ROCK pathway is one of the downstream signals activated by PKC (8). Data demonstrating that ERM are phosphorylated by PKC in response to thrombin prompted us to examine the time-dependent phosphorylation status of individual ERM and whether ERM play a role in MLC phosphorylation after thrombin. The effects of ERM siRNA treatment, either singly or in combination, on the phosphorylated ERM and MLC phosphorylation before or after thrombin treatment were examined. We discovered that all three proteins were phosphorylated; however, knockdown of moesin alone or all three ERM proteins together significantly reduced thrombin-induced ERM and MLC phosphorylation (0–90 min). In contrast, depletion of radixin did not have any significant effect on MLC phosphorylation while slightly reducing ERM threonine phosphorylation after thrombin. Depletion of ezrin partially attenuated thrombin-induced ERM and MLC phosphorylation. Importantly, these data demonstrate differential roles for the ERM proteins in response to thrombin, despite their structural similarities, and reported functional redundancy. These data also clearly suggest that moesin and to a lesser degree ezrin, but not radixin, are critically involved in MLC phosphorylation after thrombin.
The underlying mechanism through which moesin and ezrin regulate MLC phosphorylation remains to be determined. ERM proteins are known to activate the Rho signaling in cell adhesion regulation via association with Rho regulator Rho GDP-dissociation inhibitor (GDI) (58). Our data indicate that ERM may be upstream of MLC phosphorylation. Depletion of PKC isoforms (Fig. 6, A–D) indicated that PKC-dependent moesin and ezrin phosphorylation on regulatory COOH-terminal threonine is involved in thrombin-induced RhoA activation and subsequent increased MLC phosphorylation. In addition, phosphorylation of moesin or/and ezrin at sites other then COOH-terminal threonine may contribute to this activation. For example, several additional sites have been reported for ezrin (threonine 235, tyrosines 145 and 353) (36, 73), but the functional roles of phosphorylation at these sites are still unclear. Interestingly, as we recently have been reported (31), each ERM protein has a distinct binding ability toward the subunits (CSIβ and MYPT) of MLC phosphatase (MLCP), a type 1 protein phosphatase (PPase 1) that regulates reversible phosphorylation of the MLC in intercellular gap formation and barrier dysfunction of EC. Our data demonstrated that the catalytical subunit CSIβ preferably bound to moesin, whereas the band that corresponds to ezrin detected in CSIβ immunoprecipitates is faint. In contrast, radixin did not bind to CSIβ but strongly interacted with MLCP targeting subunit MYPT I. It is possible that, in addition to RhoA activation after thrombin, moesin may inhibit MLCP function, leading to increased MLC phosphorylation. Our prior results indicated that MLCP plays an important role in barrier protection in EC (31). Radixin may regulate MLCP activation through binding to MYPT I in S1P-induced barrier enhancement. Future studies will be needed to clarify the link between PKC/ERM and Rho/ROCK/MLCP pathways in thrombin-induced formation of stress fibers and increased endothelial permeability.
We next examined the role of ERM in thrombin-induced cytoskeletal changes. Thrombin induces translocation of phosphorylated ERM from the cytoplasm to EC periphery during the early stages of cell contraction and loss of monolayer integrity (Fig. 11), which is consistent with previous studies (1, 9, 35). Most importantly, our data again demonstrate differential roles for the ERM proteins in response to thrombin. Moesin exerted a particularly prominent and essential role in the promotion of EC barrier dysfunction by thrombin, whereas radixin appears to have opposing effects. This observation is consistent with recent reports describing moesin involvement in increased permeability induced by hypoxia and truncation of monocyte chemoattractant protein 1 in the blood-brain barrier (25, 74) and AGE in human microvascular EC (23, 68). Prior data obtained in knockout mice lacking individual ERM proteins largely support the functional redundancy of the three ERM proteins (11, 32, 54). However, recent studies demonstrate differential biological functioning of these proteins. For example, ezrin and moesin have distinct and critical functions in the T cell cortex during immunological synapse formation (28). Moreover, ezrin, but not moesin, is phosphorylated on tyrosine in EGF-challenged human A431 cells despite tyrosine 145 conservation in both proteins (17). In addition, moesin has a nonredundant function in lymphocyte homeostasis (26). Our recent findings also support the distinct biological roles of these proteins in agonist-mediated EC barrier responses (1).
The observation that phosphorylated ERM is mostly localized to the peripheral area in EC undergoing contraction after thrombin stimulation led us to examine the role of these proteins in modulating permeability increases. We evaluated the role of ERM in thrombin-induced hyperpermeability by measuring the TER in ERM depleted EC. The depletion of moesin alone or triple ERM siRNA knockdown significantly attenuated the increase in permeability after thrombin (Fig. 8, A, D, and E). Ezrin knockdown also attenuated the decrease in TER induced by thrombin, but to a lesser degree then moesin (Fig. 8, C and E). In contrast, radixin depletion leads to a slight increase in permeability during the early phase and delayed recovery during the later phase of thrombin-mediated decreases in TER (Fig. 8, B and E). These results suggest that ERM are differentially involved in the development of thrombin-induced permeability with moesin and ezrin promoting barrier permeability during the phase of active contraction (5–60 min of treatment with thrombin). In contrast, despite the lower level of expression compared with moesin and ezrin, radixin exerted a particularly prominent and essential role in the promotion of EC barrier function in the restoration phase (60–120 min of treatment).
In EC, thrombin-induced activation of PAR1 receptor initiate signaling through Gq- and G12/13-coupled Ca2+ mobilization, PKC and RhoA activation, and MAPK signaling (5, 7, 27). In addition, thrombin transactivates PAR2 through PAR1 (48). We next used PAR1 (TFLLR-NH2), PAR2 (SLIGRL-NH2)-selective agonists, and the PAR1-specific blocking antibodies ATAP2 and WEDE15 to determine the role of PARs in thrombin-induced threonine phosphorylation of ERM. Our data indicate that thrombin primarily induces ERM threonine phosphorylation in pulmonary EC by combined activation of both PAR1 and PAR2.
Conclusion.
The present study demonstrates that thrombin induces PKC-dependent ERM phosphorylation on a critical threonine residue (ezrin Thr567, radixin Thr564, moesin Thr558) and translocation of phosphorylated ERM to the EC periphery. ERM phosphorylation is mediated by the combined actions of PAR1 and PAR2. Thrombin-induced barrier dysfunction in pulmonary endothelium is associated with remodeling of the actin cytoskeleton that increases permeability. ERM proteins are critically involved in the barrier-disruptive response induced in the endothelium by thrombin and may modulate cytoskeletal changes and barrier hyperpermeability via such intermediate signaling events as PKC-mediated RhoA/ROCK-dependent signaling. Our data demonstrate that depletion of either moesin alone, or of all three ERM proteins, attenuates thrombin-induced F-actin cytoskeleton rearrangement, paracellular gap formation, MLC phosphorylation, and decrease in TER. In contrast, radixin depletion has the opposite effect on barrier function. On the basis of these results and our prior data (1), we propose the following model of ERM-dependent signaling in thrombin- and S1P-treated EC (Fig. 14): thrombin treatment induces PAR1- and PAR2-mediated COOH-terminal threonine phosphorylation primarily of moesin and ezrin by PKC and PIP2. Recently it has been reported that phosphorylation of the RhoA activator guanine nucleotide exchange factor p115RhoGEF by PKC-α mediates TNF-α-induced RhoA activation and subsequent barrier dysfunction in mouse brain microvascular EC (51). We hypothesize that PKC isoforms may simultaneously phosphorylate both moesin and ezrin and p115RhoGEF. Activated moesin and ezrin may displace RhoGDI from RhoA, allowing p115RhoGEF-mediated RhoA activation by GDP-to-GTP exchange. Increased intracellular concentration of Ca2+ activates MLCK. RhoA and MLCK phosphorylate MLC, allowing EC contraction and reorganization of the cytoskeleton, resulting in endothelial barrier dysfunction. In contrast, S1P induces S1PR1-mediated radixin activation (primarily), resulting in Rac1 activation. Activated Rac1 via its downstream target PAK1 induces actomyosin remodeling, including formation of a prominent cortical actin rim, which stabilizes cell-cell junctions, peripheral accumulation of phosphorylated MLC, and disappearance of central stress fibers, resulting in endothelial barrier enhancement. Thus, despite their structural similarities and reported functional redundancy, the ERM proteins differentially modulate thrombin-induced changes in lung EC cytoskeleton and permeability. These results advance our mechanistic understanding of EC barrier regulation, identify the ERM family as potential targets for therapeutic manipulation in this clinically important physiological process, and extend previous knowledge about the involvement of PKC and ERM in endothelial barrier regulation.
Fig. 14.

Proposed model of ERM-dependent signaling in thrombin- and S1P-challenged lung endothelium (see explanation in Conclusion).
GRANTS
This work was supported by National Heart Lung, and Blood Institute Grants HL58064, HL88144, and HL101902.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
D.M.A., S.M.D., J.G.G., and A.D.V. conception and design of research; D.M.A., N.M., K.-M.K., S.F.M., and A.K. performed experiments; D.M.A., N.M., K.-M.K., S.F.M., A.K., and J.G.G. analyzed data; D.M.A., S.M.D., N.M., S.F.M., J.G.G., and A.D.V. interpreted results of experiments; D.M.A., N.M., K.-M.K., S.F.M., and A.K. prepared figures; D.M.A. and A.D.V. drafted manuscript; D.M.A., S.M.D., J.G.G., and A.D.V. edited and revised manuscript; J.G.G. and A.D.V. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are grateful to Drs. D. Fulton and S. Black from Georgia Health Sciences University for help with moesin mutant preparation.
REFERENCES
- 1. Adyshev DM, Moldobaeva NK, Elangovan VR, Garcia JG, Dudek SM. Differential involvement of ezrin/radixin/moesin proteins in sphingosine 1-phosphate-induced human pulmonary endothelial cell barrier enhancement. Cell Signal 23: 2086–2096, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arpin M, Chirivino D, Naba A, Zwaenepoel I. Emerging role for ERM proteins in cell adhesion and migration. Cell Adh Migr 5: 199–206, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Belkina NV, Liu Y, Hao JJ, Karasuyama H, Shaw S. LOK is a major ERM kinase in resting lymphocytes and regulates cytoskeletal rearrangement through ERM phosphorylation. Proc Natl Acad Sci USA 106: 4707–4712, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berryman M, Franck Z, Bretscher A. Ezrin is concentrated in the apical microvilli of a wide variety of epithelial cells whereas moesin is found primarily in endothelial cells. J Cell Sci 105: 1025–1043, 1993 [DOI] [PubMed] [Google Scholar]
- 5. Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JG, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J 18: 1879–1890, 2004 [DOI] [PubMed] [Google Scholar]
- 6. Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res 67: 64–77, 2004 [DOI] [PubMed] [Google Scholar]
- 7. Bogatcheva NV, Garcia JG, Verin AD. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry 67: 75–84, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Bogatcheva NV, Verin AD. The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc Res 76: 202–207, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bogatcheva NV, Zemskova MA, Gorshkov BA, Kim KM, Daglis GA, Poirier C, Verin AD. Ezrin, radixin, and moesin are phosphorylated in response to 2-methoxyestradiol and modulate endothelial hyperpermeability. Am J Respir Cell Mol Biol 45: 1185–1194, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cant SH, Pitcher JA. G protein-coupled receptor kinase 2-mediated phosphorylation of ezrin is required for G protein-coupled receptor-dependent reorganization of the actin cytoskeleton. Mol Biol Cell 16: 3088–3099, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Doi Y, Itoh M, Yonemura S, Ishihara S, Takano H, Noda T, Tsukita S. Normal development of mice and unimpaired cell adhesion/cell motility/actin-based cytoskeleton without compensatory up-regulation of ezrin or radixin in moesin gene knockout. J Biol Chem 274: 2315–2321, 1999 [DOI] [PubMed] [Google Scholar]
- 12. Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001 [DOI] [PubMed] [Google Scholar]
- 13. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 11: 276–287, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, Arpin M. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol 164: 653–659, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fleegal MA, Hom S, Borg LK, Davis TP. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am J Physiol Heart Circ Physiol 289: H2012–H2019, 2005 [DOI] [PubMed] [Google Scholar]
- 17. Franck Z, Gary R, Bretscher A. Moesin, like ezrin, colocalizes with actin in the cortical cytoskeleton in cultured cells, but its expression is more variable. J Cell Sci 105: 219–231, 1993 [DOI] [PubMed] [Google Scholar]
- 18. Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol 163: 510–522, 1995 [DOI] [PubMed] [Google Scholar]
- 19. Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108: 689–701, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW, 2nd, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol 128: 96–104, 1986 [DOI] [PubMed] [Google Scholar]
- 21. Gautreau A, Louvard D, Arpin M. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J Cell Biol 150: 193–203, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grand RJ, Turnell AS, Grabham PW. Cellular consequences of thrombin-receptor activation. Biochem J 313: 353–368, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo X, Wang L, Chen B, Li Q, Wang J, Zhao M, Wu W, Zhu P, Huang X, Huang Q. ERM protein moesin is phosphorylated by advanced glycation end products and modulates endothelial permeability. Am J Physiol Heart Circ Physiol 297: H238–H246, 2009 [DOI] [PubMed] [Google Scholar]
- 24. Hayashi K, Yonemura S, Matsui T, Tsukita S. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J Cell Sci 112: 1149–1158, 1999 [DOI] [PubMed] [Google Scholar]
- 25. Hicks K, O'Neil RG, Dubinsky WS, Brown RC. TRPC-mediated actin-myosin contraction is critical for BBB disruption following hypoxic stress. Am J Physiol Cell Physiol 298: C1583–C1593, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirata T, Nomachi A, Tohya K, Miyasaka M, Tsukita S, Watanabe T, Narumiya S. Moesin-deficient mice reveal a non-redundant role for moesin in lymphocyte homeostasis. Int Immunol 24: 705–717, 2012 [DOI] [PubMed] [Google Scholar]
- 27. Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem 278: 28793–28798, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Ilani T, Khanna C, Zhou M, Veenstra TD, Bretscher A. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol 179: 733–746, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ivetic A, Ridley AJ. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology 112: 165–176, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson MW, Miyata H, Vinters HV. Ezrin and moesin expression within the developing human cerebrum and tuberous sclerosis-associated cortical tubers. Acta Neuropathol (Berl) 104: 188–196, 2002 [DOI] [PubMed] [Google Scholar]
- 31. Kim KM, Csortos C, Czikora I, Fulton D, Umapathy NS, Olah G, Verin AD. Molecular characterization of myosin phosphatase in endothelium. J Cell Physiol 227: 1701–1708, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S, Tsukita S. Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol 166: 559–570, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Knezevic N, Roy A, Timblin B, Konstantoulaki M, Sharma T, Malik AB, Mehta D. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol 27: 6323–6333, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Konstantoulaki M, Kouklis P, Malik AB. Protein kinase C modifications of VE-cadherin, p120, and β-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am J Physiol Lung Cell Mol Physiol 285: L434–L442, 2003 [DOI] [PubMed] [Google Scholar]
- 35. Koss M, Pfeiffer GR, 2nd, Wang Y, Thomas ST, Yerukhimovich M, Gaarde WA, Doerschuk CM, Wang Q. Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and modulate permeability increases in human pulmonary microvascular endothelial cells. J Immunol 176: 1218–1227, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Krieg J, Hunter T. Identification of the two major epidermal growth factor-induced tyrosine phosphorylation sites in the microvillar core protein ezrin. J Biol Chem 267: 19258–19265, 1992 [PubMed] [Google Scholar]
- 37. Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med 11: e19, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Z, Liu YH, Xue YX, Liu LB, Wang P. Signal mechanisms underlying low-dose endothelial monocyte-activating polypeptide-II-induced opening of the blood-tumor barrier. J Mol Neurosci 48: 291–301, 2012 [DOI] [PubMed] [Google Scholar]
- 39. Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem 268: 9194–9197, 1993 [PubMed] [Google Scholar]
- 40. Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol 140: 647–657, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matsui T, Yonemura S, Tsukita S, Tsukita S. Activation of ERM proteins in vivo by Rho involves phosphatidyl-inositol 4-phosphate 5-kinase and not ROCK kinases. Curr Biol 9: 1259–1262, 1999 [DOI] [PubMed] [Google Scholar]
- 42. McLaughlin JN, Shen L, Holinstat M, Brooks JD, Dibenedetto E, Hamm HE. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem 280: 25048–25059, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Minshall RD, Vandenbroucke EE, Holinstat M, Place AT, Tiruppathi C, Vogel SM, van Nieuw Amerongen GP, Mehta D, Malik AB. Role of protein kinase Czeta in thrombin-induced RhoA activation and inter-endothelial gap formation of human dermal microvessel endothelial cell monolayers. Microvasc Res 80: 240–249, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moy AB, Blackwell K, Kamath A. Differential effects of histamine and thrombin on endothelial barrier function through actin-myosin tension. Am J Physiol Heart Circ Physiol 282: H21–H29, 2002 [DOI] [PubMed] [Google Scholar]
- 46. Neisch AL, Fehon RG. Ezrin, Radixin and Moesin: key regulators of membrane-cortex interactions and signaling. Curr Opin Cell Biol 23: 377–382, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ng T, Parsons M, Hughes WE, Monypenny J, Zicha D, Gautreau A, Arpin M, Gschmeissner S, Verveer PJ, Bastiaens PI, Parker PJ. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J 20: 2723–2741, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. O'Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem 275: 13502–13509, 2000 [DOI] [PubMed] [Google Scholar]
- 49. Patil SB, Bitar KN. RhoA- and PKC-α-mediated phosphorylation of MYPT and its association with HSP27 in colonic smooth muscle cells. Am J Physiol Gastrointest Liver Physiol 290: G83–G95, 2006 [DOI] [PubMed] [Google Scholar]
- 50. Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101: 259–270, 2000 [DOI] [PubMed] [Google Scholar]
- 51. Peng J, He F, Zhang C, Deng X, Yin F. Protein kinase C-α signals P115RhoGEF phosphorylation and RhoA activation in TNF-α-induced mouse brain microvascular endothelial cell barrier dysfunction. J Neuroinflammation 8: 28, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pietromonaco SF, Simons PC, Altman A, Elias L. Protein kinase C-theta phosphorylation of moesin in the actin-binding sequence. J Biol Chem 273: 7594–7603, 1998 [DOI] [PubMed] [Google Scholar]
- 53. Rigor RR, Beard RS, Jr, Litovka OP, Yuan SY. Interleukin-1β-induced barrier dysfunction is signaled through PKC-θ in human brain microvascular endothelium. Am J Physiol Cell Physiol 302: C1513–C1522, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saotome I, Curto M, McClatchey AI. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell 6: 855–864, 2004 [DOI] [PubMed] [Google Scholar]
- 55. Schaphorst KL, Pavalko FM, Patterson CE, Garcia JG. Thrombin-mediated focal adhesion plaque reorganization in endothelium: role of protein phosphorylation. Am J Respir Cell Mol Biol 17: 443–455, 1997 [DOI] [PubMed] [Google Scholar]
- 56. Sonobe Y, Takeuchi H, Kataoka K, Li H, Jin S, Mimuro M, Hashizume Y, Sano Y, Kanda T, Mizuno T, Suzumura A. Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase Cepsilon-dependent manner. J Biol Chem 284: 31834–31842, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Srivastava J, Elliott BE, Louvard D, Arpin M. Src-dependent ezrin phosphorylation in adhesion-mediated signaling. Mol Biol Cell 16: 1481–1490, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takahashi K, Sasaki T, Mammoto A, Takaishi K, Kameyama T, Tsukita S, Takai Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J Biol Chem 272: 23371–23375, 1997 [DOI] [PubMed] [Google Scholar]
- 59. ten Klooster JP, Jansen M, Yuan J, Oorschot V, Begthel H, Di Giacomo V, Colland F, de Koning J, Maurice MM, Hornbeck P, Clevers H. Mst4 and ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell 16: 551–562, 2009 [DOI] [PubMed] [Google Scholar]
- 60. Tinsley JH, De Lanerolle P, Wilson E, Ma W, Yuan SY. Myosin light chain kinase transference induces myosin light chain activation and endothelial hyperpermeability. Am J Physiol Cell Physiol 279: C1285–C1289, 2000 [DOI] [PubMed] [Google Scholar]
- 61. Tinsley JH, Teasdale NR, Yuan SY. Involvement of PKCδ and PKD in pulmonary microvascular endothelial cell hyperpermeability. Am J Physiol Cell Physiol 286: C105–C111, 2004 [DOI] [PubMed] [Google Scholar]
- 62. Titchenell PM, Lin CM, Keil JM, Sundstrom JM, Smith CD, Antonetti DA. Novel atypical PKC inhibitors prevent vascular endothelial growth factor-induced blood-retinal barrier dysfunction. Biochem J 446: 455–467, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamell L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem 266: 15771–15781, 1991 [PubMed] [Google Scholar]
- 64. van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res 87: 335–340, 2000 [DOI] [PubMed] [Google Scholar]
- 65. Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG. Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am J Physiol Lung Cell Mol Physiol 281: L565–L574, 2001 [DOI] [PubMed] [Google Scholar]
- 66. Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64: 1057–1068, 1991 [DOI] [PubMed] [Google Scholar]
- 67. Wald FA, Oriolo AS, Mashukova A, Fregien NL, Langshaw AH, Salas PJ. Atypical protein kinase C (iota) activates ezrin in the apical domain of intestinal epithelial cells. J Cell Sci 121: 644–654, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang J, Liu H, Chen B, Li Q, Huang X, Wang L, Guo X, Huang Q. RhoA/ROCK-dependent moesin phosphorylation regulates AGE-induced endothelial cellular response. Cardiovasc Diabetol 11: 7, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J 294: 335–337, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Willis CL, Meske DS, Davis TP. Protein kinase C activation modulates reversible increase in cortical blood-brain barrier permeability and tight junction protein expression during hypoxia and posthypoxic reoxygenation. J Cereb Blood Flow Metab 30: 1847–1859, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu KL, Khan S, Lakhe-Reddy S, Jarad G, Mukherjee A, Obejero-Paz CA, Konieczkowski M, Sedor JR, Schelling JR. The NHE1 Na+/H+ exchanger recruits ezrin/radixin/moesin proteins to regulate Akt-dependent cell survival. J Biol Chem 279: 26280–26286, 2004 [DOI] [PubMed] [Google Scholar]
- 72. Wysolmerski RB, Lagunoff D. Regulation of permeabilized endothelial cell retraction by myosin phosphorylation. Am J Physiol Cell Physiol 261: C32–C40, 1991 [DOI] [PubMed] [Google Scholar]
- 73. Yang HS, Hinds PW. Increased ezrin expression and activation by CDK5 coincident with acquisition of the senescent phenotype. Mol Cell 11: 1163–1176, 2003 [DOI] [PubMed] [Google Scholar]
- 74. Yao Y, Tsirka SE. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood-brain barrier disruption. J Cell Sci 124: 1486–1495, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]