

Abstract
The axon initial segment (AIS) is a specialized neuronal subcompartment located at the beginning of the axon that is crucially involved in both the generation of action potentials and the regulation of neuronal polarity. We recently showed that prolonged neuronal depolarization produces a distal shift of the entire AIS structure away from the cell body, a change associated with a decrease in neuronal excitability. Here, we used dissociated rat hippocampal cultures, with a major focus on the dentate granule cell (DGC) population, to explore the signaling pathways underlying activity-dependent relocation of the AIS. First, a pharmacological screen of voltage-gated calcium channels (VGCCs) showed that AIS relocation is triggered by activation of L-type Cav1 VGCCs with negligible contribution from any other VGCC subtypes. Additional pharmacological analysis revealed that downstream signaling events are mediated by the calcium-sensitive phosphatase calcineurin; inhibition of calcineurin with either FK506 or cyclosporin A totally abolished both depolarization- and optogenetically-induced activity-dependent AIS relocation. Furthermore, calcineurin activation is sufficient for AIS plasticity, because expression of a constitutively active form of the phosphatase resulted in relocation of the AIS of DGCs without a depolarizing stimulus. Finally, we assessed the role of calcineurin in other forms of depolarization-induced plasticity. Neither membrane resistance changes nor spine density changes were affected by FK506 treatment, suggesting that calcineurin acts via a separate pathway to modulate AIS plasticity. Together, these results emphasize calcineurin as a vital player in the regulation of intrinsic plasticity as governed by the AIS.
Introduction
Integration of neuronal input into an all-or-nothing action potential output occurs at a specialized structure located within the proximal axon of a neuron, the axon initial segment (AIS) (Clark et al., 2009; Bender and Trussell, 2012; Kole and Stuart, 2012). This highly structured region is composed of a number of scaffolding and extracellular matrix proteins, along with high concentrations of voltage-gated ion channels (Grubb and Burrone, 2010a; Rasband, 2010). As the governor of the major output from a neuron the AIS is well positioned to modulate cell excitability. Accordingly, the precise position and/or length of the AIS within the axon can vary a great deal across different cell types (Kuba et al., 2006; Fried et al., 2009), and modeling studies show that these variations can have a significant impact on the intrinsic excitability of a neuron (Kuba et al., 2006; Wang et al., 2011).
The AIS appears to be a relatively static structure at rest (Hedstrom et al., 2007); however, it also undergoes dynamic changes in its location, length, and viability in response to modulation of external stimuli. Increasing the activity of dissociated hippocampal neurons for 48 h causes a distal relocation of the entire AIS structure in a change associated with decreased neuronal excitability (Grubb and Burrone, 2010b), whereas removal of input from nuclear magnocellularis neurons in the chick auditory system results in an increase in AIS length over a number of days, a neuronal change associated with increased intrinsic excitability (Kuba et al., 2010). Changes at the AIS can also act much more rapidly: in response to ischemic injury, the entire AIS is degraded in a calpain-dependent manner within hours (Schafer et al., 2009).
Therefore, modulating AIS position and length may be an important tool that a neuron can use to control its intrinsic excitability. It seems that a likely source of this control is the ubiquitous signaling molecule calcium: AIS relocation in dissociated hippocampal neurons relies on activation of L-type (Cav1) or T-type (Cav3) voltage-gated calcium channels (VGCCs; Grubb and Burrone, 2010b). Although T-type VGCCs play a key role in modulating action potential dynamics (Contreras, 2006; Bender and Trussell, 2009; Dreyfus et al., 2010; Bender et al., 2012), their activation is only transient. Conversely, L-type VGCCs remain activated during long-term and low-level stimuli (Lipscombe et al., 2004) and have been shown to trigger a number of plasticity mechanisms via a variety of different signaling pathways (Deisseroth et al., 2003; Wheeler et al., 2012). However, the pathways leading to AIS plasticity and the extent to which these are shared with other forms of plasticity are entirely unknown.
Here we investigate the signaling pathways mediating AIS relocation in dissociated hippocampal neurons. We show that AIS movement is mediated by activation of L-type Cav1 VGCCs and that subsequent downstream signaling events require the calcium–calmodulin-activated phosphatase calcineurin. Calcineurin activity is also sufficient for AIS relocation plasticity. However, this mechanism is independent from other forms of depolarization-induced intrinsic and structural plasticity in the same neurons.
Materials and Methods
Dissociated hippocampal culture and transfections.
Dissociated hippocampal neurons were prepared from Wistar rat embryos of either sex dissected in HBSS at embryonic day 18. Hippocampi were digested with trypsin (0.5 mg/ml; 15 min at 37°C; Worthington) before trituration and subsequent plating at 45,000 cells per well on 13 mm glass coverslips precoated with poly-l-lysine at 50 μg/ml (Sigma) and laminin at 40 μg/ml. Neurons were cultured at 37°C with 5% CO2 in Neurobasal medium supplemented with 1% B27, 1% fetal calf serum, and 500 μm Glutamax. Every 3 d thereafter, half of the media was changed with media supplemented with 2% B27 and 500 μm Glutamax. Transfections were performed using Lipofectamine 2000 at either 3 or 7 DIV as stated below. Unless otherwise stated, all cell-culture materials were obtained from Invitrogen.
DNA constructs.
The constitutively active calcineurin (CACaN) construct was a kind gift from Eloise Hudry and Brad Hyman (Massachusetts General Hospital, MA) and has been fully described previously (Wu et al., 2010). Channelrhodopsin-2–YFP (ChR2–YFP) was a kind gift from Karl Deisserorth (Stanford University, CA) and drives neuronal expression of ChR2 using the synapsin promoter. The YFP construct documented here throughout, which is also driven from the synapsin promoter, was created by replacing the ChR2–YFP coding region from ChR2–YFP with YFP alone using the AgeI and BsrgI restriction sites. EGFP–C1 nuclear factor of activated T-cells 3 (NFAT3) (referred to as NFAT–GFP throughout text) was obtained from Addgene (plasmid 10961) and was deposited by Toren Finkel (National Heart, Lung and Blood Institute, Bethesda, MD) (Ichida and Finkel, 2001).
Pharmacology.
All pharmacological experiments were performed at 10 DIV. TTA-P2 [3,5-dichloro-N-1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide], a novel and selective T-type VGCC inhibitor, was developed by and obtained from Merck and Co. All pharmacological agents (drugs) were made up as per the instructions of the manufacturer and added at previously described effective working concentrations to neurons in 50:50 media (containing 50% conditioned and 50% fresh media) for at least 30 min before control or depolarizing treatment. Neurons subjected to either the drug or solvent control were subsequently treated with +10 mm NaCl (control) or depolarized with +10 mm KCl for 48 h before being fixed and immunostained (see below).
Immunostaining.
Neurons were fixed in 4% paraformaldehyde (in 3% sucrose, 60 mm PIPES, 25 mm HEPES, 5 mm EGTA, and 1 mm MgCl2; TAAB Laboratories) for 20 min at room temperature. Cells were permeabilized for 5 min with 0.25% Triton X-100 (Sigma) before blocking for 1 h in 10% goat serum (GS; Sigma). Coverslips were then placed in primary antibody, made in 2% GS, to relevant concentrations: mouse monoclonal anti-ankryin-G (clone N106/36; 1:500; NeuroMab); rabbit polyclonal anti-prox1 (1:1000; Sigma); rat anti-CTIP2 (1:1000; Abcam); mouse monoclonal anti-αCaMKII (1:1000; Millipore); and rabbit anti-calcineurin A (1:500; Abcam) for 1 h 30 min. After subsequent washing steps, coverslips were placed in relevant secondary antibody solution (Invitrogen Alexa Fluor-conjugated antibodies in 2% GS, all at 1:1000) for an additional hour, before further washing and mounting in MOWIOL (Calbiochem).
Imaging and analyses.
All imaging and subsequent analyses were performed blind to experimental group. Neurons with AISs of obvious somatic origin were visualized under epifluorescence and imaged using a laser-scanning confocal microscope (Carl Zeiss LSM 710). Neurons were imaged with appropriate excitation and emission filters, with the pinhole set to 1 A.U. and using a 40× oil-immersion objective (Carl Zeiss). Laser power and gain settings were adjusted to prevent signal saturation. Images were taken with 3× zoom, 512 × 512 pixels (0.138 μm/pixel), and in z-stacks with 0.5 μm steps. z-Stack images were flattened into single maximum intensity projections and imported into MATLAB computer software (MathWorks) for analysis using custom-written functions (Matthew Grubb and Thomas Watkins, King's College London, UK; Grubb and Burrone, 2010b; freely available at www.mathworks.com/matlabcentral/fileexchange/28181-ais-quantification).
In each experimental group, we imaged and analyzed ∼40 neurons for AIS start, maximum, and end positions. This n allowed us to detect a 5 μm shift in AIS end position with 90% power based on mean ± SD for pilot experiments (G*Power; Franz Faul, Kiel University, Kiel, Germany). We drew a line profile along each maximum intensity projection starting at the soma that extended down the axon, through and past the AIS. At each pixel (1 pixel = 0.138 μm) along this profile, fluorescence intensity values were averaged over a 3 × 3 pixel square centered on the pixel of interest. Averaged profiles were then smoothed using a 40-point (∼5 μm) sliding mean and normalized between 1 (maximum smoothed fluorescence, location of the AIS max position) and 0 (minimum smoothed fluorescence). AIS start and end positions were obtained at the proximal and distal axonal positions, respectively, at which the normalized and smoothed profile declined to 0.33. AIS maximum position was taken as the peak fluorescence of each profile.
An AIS movement index (AMI) was calculated for each neuron subjected to both drug and depolarization using its own AIS max position (a) and the mean AIS max position for other treatment groups [drug + 10 mm NaCl (b̄); solvent + 10 mm KCl (c̄); and solvent + 10 mm NaCl (d̄)] as follows:
 |
AMI values calculated for each neuron were then subjected to a single-sample t test versus 1 to assess AIS movement in the drug compared with solvent control. If the mean AMI was greater than or statistically indistinguishable from 1, the AIS was deemed to have moved at least as far in drug as in control; however, if the mean AMI was significantly <1, AIS movement had been at least partially blocked. In parallel analyses, an AMI significantly <1 always matched a significant drug × treatment interaction when performing a nonparametric two-way ANOVA on sample ranks with the same dataset (Akritas, 1990). To establish a full block of AIS movement, the AMI values were further subjected to a single-sample t test versus 0 with appropriate Bonferroni's correction for repeated testing. If the AMI was less than or statistically the same as 0, AIS movement was deemed to be fully blocked.
Calcium imaging.
We loaded 10 DIV dissociated hippocampal neurons with the ratiometric calcium indicator fura-2 AM (5 μm; Invitrogen) in HBSS (see below) for 30 min at room temperature, followed by wash and incubation in phenol red-free Neurobasal medium (Invitrogen) containing either DMSO (control) or 1 μm FK506 [(3S,4R,5S,8R,9E,12S,14S,15R,16S,18R,19R,26aS)-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[(1E)-2-[(1R,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propen-1-yl)-15,19-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)tetrone] treatments (30 min at 37°C). Cells were maintained in a steady gravity-fed flow of phenol red-free Neurobasal medium (34–36°C; maintained with an in-line heater (SH-27B; Harvard Apparatus) and were allowed to equilibrate for 10 min before an imaging protocol began. Single z-axis images were captured for both 340 and 380 nm excitation wavelengths every 5 s using an inverted Olympus IX71 microscope, an Olympus 40× oil-immersion objective, and a CCD camera coupled to Slidebook software (2 × 2 pixel binning). Baseline fluorescence was established during 5 min of perfusion before a +10 mm KCl stimulus was washed in. Fura-2 340/380 ratios were then calculated before and during +10 mm KCl wash-in, using fluorescence intensities averaged across cell body regions of interest and normalized to background fluorescence.
Electrophysiology.
For electrophysiological experiments, pipettes were pulled from borosilicate glass (1.5 mm outer diameter, 1.17 mm inner diameter; Harvard Apparatus) with a resistance of 3–7 MΩ and were filled with an internal solution (for individual experiment details, see below). Recordings were obtained with a HEKA EPC10/2 amplifier coupled to Patchmaster acquisition software. Signals were Bessel filtered at 10 kHz (filter 1) and 2.9 kHz (filter 2), digitized, and sampled at 25–50 kHz (20–40 μs sample interval). Fast capacitance was compensated in the on-cell configuration. After membrane rupture, at a holding voltage of −60 mV, with slow capacitance compensation inactive, we used responses to a 10 mV hyperpolarization step to estimate the series resistance of the recording (<20 MΩ for all cells; from the current peak) and the membrane resistance (Rm; from the steady holding current at the new voltage) and membrane capacitance (from the area under the exponentially decaying current from peak to holding) of the neuron.
For assessment of resting potential at 10 DIV, hippocampal neurons were treated with either DMSO + 10 mm NaCl, DMSO + 10 mm KCl, or 1 μm FK506 + 10 mm KCl for at least 0.5 h before both neurons and media were transferred to the recording chamber. Neurons were then targeted for whole-cell patch clamping with pipettes filled with an internal solution containing the following (in mm): 130 K-gluconate, 10 NaCl, 1 EGTA, 0.133 CaCl2, 2 MgCl2, 10 HEPES, 3.5 MgATP, and 1 NaGTP. Immediately on membrane rupture, resting potential was assessed and recorded at I = 0 in current clamp. Resting potential values and all voltages reported here were subsequently corrected for the appropriate calculated liquid junction potential of approximately −15 mV.
For assessment of L-type VGCC current density at 10 DIV, we blocked all other VGCCs. Neurons were treated with 0.15 μm ω-agatoxin-TK (to block P/Q-type VGCCs), 3 μm ω-conotoxin-GIVA (to block N-type VGCCs), and DMSO, 1 μm FK506, or 1 μm nifedipine for at least 30 min before recording. Because of the slow washout of the VGCC drugs (McCleskey et al., 1987; Sather et al., 1993; Oliveria et al., 2007), ω-agatoxin and ω-conotoxin were not included in recording media, but to ensure that there was little contamination from P/Q/N-type channels, neurons were used within 45 min of the preincubation step. Neurons were recorded in an extracellular HBSS containing the following: 136 mm NaCl, 2.5 mm KCl, 10 mm HEPES, 10 mm d-glucose, 2 mm CaCl2, 1.3 mm MgCl2, 22.5 mm tetraethylammonium (TEA), 0.05 mm nickel (to block R- and T-type VGCCs), 0.01 mm tetrodotoxin, and DMSO, 1 μm FK506, or 1 μm nifedipine. Neurons were targeted for whole-cell patch clamping with pipettes filled with an internal solution containing the following (in mm): 130 Cs-gluconate, 10 NaCl, 1 EGTA, 0.133 CaCl2, 2 MgCl2, 10 HEPES, 3.5 NaATP, 1 NaGTP, and 22.5 TEA. With slow capacitance compensated, in voltage clamp, neurons were subjected to a 250 ms depolarization from a holding voltage of −60 to −40 mV every 5 s. Holding at −60 mV eliminates >80% of R-type currents (Sochivko et al., 2003; Oliveria et al., 2007). L-type current density in each treatment was measured as the average plateau current at −40 mV in the last 10 ms of the stimulus, normalized to cell capacitance.
For assessment of Rm, hippocampal neurons were treated for 48 h in either DMSO + 10 mm NaCl, DMSO + 10 mm KCl, 1 μm FK506 + 10 mm NaCl, or 1 μm FK506 + 10 mm KCl before transfer to HBSS containing the following (in mm): 136 NaCl, 2.5 KCl, 10 HEPES, 10 d-glucose, 2 CaCl2, and 1.3 MgCl2. Dentate granule cells (DGCs) were specifically targeted for whole-cell recordings with pipettes filled with potassium gluconate internal solution (see above). DGCs were identified by searching for cell morphological features: DGCs are small compared with other neuronal types, have a rounded cell body, and have either one or two thick dendrites emanating from their cell body (Lee et al., 2013). Rm was assessed as described above.
Photostimulation.
Dissociated hippocampal neurons were sparsely transfected with ChR2–YFP at 7 DIV. At 10 DIV, neurons were prepared for photostimulation by supplementing their media for at least 2 h with a mixture of antioxidants (3.2 μm glutathione from Thermo Fisher Scientific and 110 μm vitamin C, 100 μm Trolox, 2.3 μm vitamin E, 77 nm superoxide dismutase, and 10 nm catalase all from Sigma), blocking synaptic transmission with 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX; Sigma) and adding DMSO (1:10,000), 1 μm FK506, or 1 μm cyclosporin A (CsA). Four-well plates containing treated media and neurons were then placed on top of blue light-emitting diodes (LEDs) (Luxeon Star) with individual collimators (RS Components), supported by an aluminum heat sink (Fisher Electronik) and driven by direct current/direct current LED drivers (RS Components) by in-house software written by Andrew Lowe (King's College, London, UK). Neurons were stimulated by illumination with individual LED flashes of 5 ms duration. Flashes were given at an average of 1 Hz frequency, but these stimuli were grouped into bursts of five flashes at a frequency of 20 Hz with bursts every 5 s (see Fig. 7A). After 48 h, photostimulated neurons were fixed with 4% paraformaldehyde and immunostained for ankyrin-G and prox1.
Figure 7.

Photostimulation-induced AIS relocation is blocked by calcineurin inhibitors. Top right, Diagram of LED setup and stimulation paradigm. Left, Example images of ChR2-expressing DGCs subjected to 48 h photostimulation in the presence of DMSO, 1 μm FK506, or 1 μm CsA. Scale bar, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1. Bottom right, Mean ± SEM of AIS start (S), maximum (M), and end (E) position for ChR2-expressing DGCs photostimulated in each treatment. Kruskall-Wallis test with Dunn's multiple comparison for start, maximum, and end positions versus DMSO, *p < 0.05; **p < 0.01; ***p < 0.001.
NFAT–GFP.
For NFAT localization experiments, hippocampal neurons were sparsely transfected with NFAT–GFP at 7 DIV and then treated at 10 DIV with either +10 mm NaCl (control) or depolarized with +10 mm KCl for 3 h. Neurons were subsequently fixed and stained for prox1 and the nuclear marker TOPRO-3. NFAT–GFP distribution was assessed in Prox1-positive neurons by tracing around the nucleus of a single-plane confocal image in NIH ImageJ and measuring the mean gray value of nuclear NFAT–GFP and cytoplasmic NFAT–GFP. Then,
 |
CACaN.
To confirm the effectiveness of the CACaN construct, we cotransfected CACaN with NFAT–GFP at 3 DIV, fixed the neurons at 12 DIV, and took single-plane confocal images of construct-expressing neurons.
For assessment of AIS position in neurons expressing CACaN, hippocampal neurons were sparsely transfected at 3 DIV with either synapsin-driven YFP alone (control) or YFP together with CACaN (in a 1:2 ratio). Neurons were then cultured until 12 DIV, when they were fixed and immunostained for prox1 and ankyrin-G.
Dendritic spine analyses.
Dissociated hippocampal neurons were sparsely transfected with synapsin-driven YFP at 7 DIV. At 10 DIV, neurons were treated for 48 h with DMSO + 10 mm NaCl, DMSO + 10 mm KCl, 1 μm FK506 + 10 mm NaCl, or 1 μm FK506 + 10 mm KCl before fixation and immunostaining for prox1. Secondary dendrites from DGCs were imaged with 4× zoom, 1024 × 1024 pixels (0.104 μm/pixel), and in z-stacks with 0.5 μm steps. z-Stacks were imported into NIH ImageJ, in which they were flattened into maximum intensity projections. NeuronJ was subsequently used to measure dendritic length, and spine density was calculated from the number of spines, defined as <5 μm in length, counted in each region.
Statistics.
AIS positional data were analyzed by Prism software (GraphPad Software). Datasets that were non-normal as assessed with a D'Agostino and Pearson omnibus normality test were analyzed using nonparametric tests. All tests were two tailed with α = 0.05 unless otherwise stated.
Results
All excitatory hippocampal neuron subtypes show AIS plasticity
We previously reported that chronic depolarization of dissociated hippocampal neurons leads to an activity-dependent relocation of the entire AIS structure (Grubb and Burrone, 2010b). Because the hippocampus constitutes a heterogeneous population of neurons including many classes of excitatory and inhibitory neurons, we first decided to investigate AIS plasticity in different classes of hippocampal cells using a simple immunostaining protocol (Williams et al., 2011; Lee et al., 2013; see Materials and Methods). Immunostaining for DGCs, CA1 and CA3 pyramidal neurons together with the crucial AIS scaffolding protein ankyrin-G revealed population-specific differences in baseline AIS position and length, which agree well with in vivo data. In particular, although all excitatory neurons have their AIS close to the cell soma, AIS length in DGCs is much shorter than in pyramidal cells (Kress et al., 2010). Interestingly, all major excitatory hippocampal subtypes undergo a significant activity-dependent AIS relocation in response to +10 mm KCl depolarization for 48 h from 10 to 12 DIV (Fig. 1; CA1 start and maximum position, Mann–Whitney U test, p < 0.0001 and end position, p = 0.0014, n = 158; CA3 start, maximum, and end position, Mann–Whitney U test, p < 0.0001, n = 154; DGC start, maximum, and end position, Mann–Whitney U test, p < 0.0001, n = 1391). However, inhibitory neurons expressing the neurotransmitter GABA do not show this type of plasticity (Fig. 1; GABA start position, Mann–Whitney U test, p = 0.092, max position, p = 0.51, and end position, p = 0.85, n = 54; Grubb and Burrone, 2010b).
Figure 1.
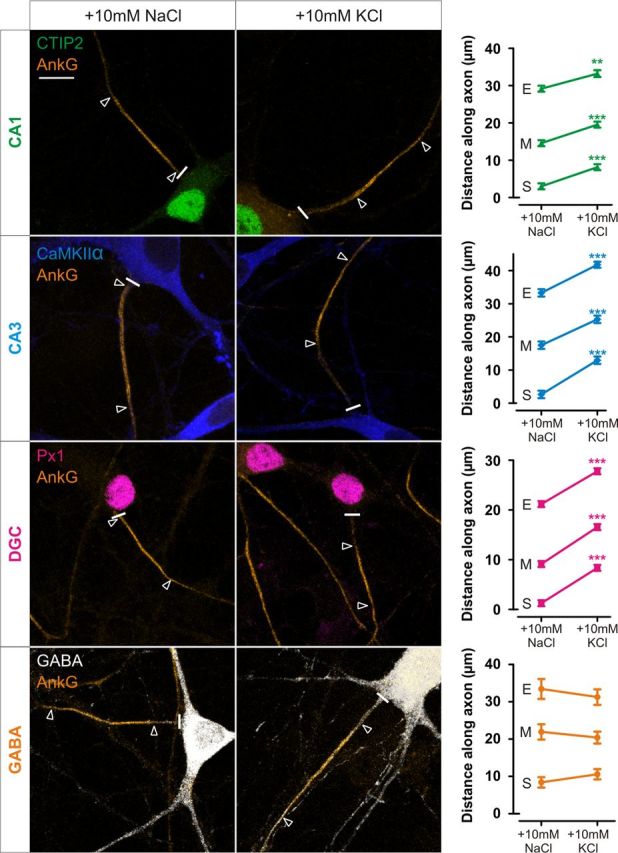
All excitatory hippocampal neuronal subtypes show depolarization-induced AIS plasticity. Left, Example images of DGC, CA1, CA3, and GABAergic neurons after 48 h treatment with NaCl or KCl from 10 DIV. Cell types were determined by combinatorial immunostaining (see Materials and Methods). Scale bars, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1. Right, Plots show mean ± SEM for AIS start (S), maximum (M), and end (E) position for each neuronal subtype in NaCl and KCl treatment conditions. ***p < 0.0001, Mann–Whitney U test for start, maximum, or end position.
Calcium entry through voltage-gated L-type channels triggers AIS relocation
To reduce the variability within the hippocampal population highlighted in Figure 1, we have focused our investigation into the signaling pathways underlying AIS plasticity on a single cell type, the DGC. This neuronal type shows reliable AIS plasticity over numerous experiments (Fig. 1; see below), shows low variability in AIS position (SD for DGC end position = 5.5 μm vs 10.8 μm for the whole hippocampal neuron population), and has unmyelinated axons in vivo as is the case in dissociated culture. DGCs can also be efficiently identified by labeling for prox1, a transcription factor expressed in all DGCs and only in DGCs in dissociated embryonic hippocampal cultures (Williams et al., 2011). This is true despite prox1-expressing interneurons migrating into the cortex and hippocampus from subpallial regions (N. Kessaris, personal communication); no GABA-labeled neurons in our cultures stained positively for prox1 (0 of 100 GABA-expressing neurons). DGCs are also one of the only cell populations in the brain to undergo neurogenesis throughout life (Lledo et al., 2006). However, after culturing neurons in the presence of the thymidine analog BrdU to label dividing cells, no DGCs were BrdU positive (0 of 763 prox1-expressing neurons), suggesting that DGCs are not continuously generated in our culture conditions.
Depolarization-induced relocation of the AIS requires the activation of L-type and/or T-type Ca2+ channels (Grubb and Burrone, 2010b), suggesting that Ca2+ entry to the neuron is the trigger for AIS movement. We used a pharmacological approach to further delineate this signaling cascade in DGCs. A new measure, the AMI, gave a single value for the effect of a particular drug on AIS relocation (see Materials and Methods and Fig. 2A). An AMI value of 1 means that the AIS moves as far in the presence of the drug tested as in the solvent control, whereas an AMI value of 0 means that AIS movement has been totally blocked by the drug tested.
Figure 2.

Only activation of L-type calcium channels is essential for AIS relocation. A, Effects of L- and T-type channel block. Left, Sample images of DGCs treated with DMSO, 1 μm nifedipine, or 10 μm TTA-P2 in NaCl or KCl treatment groups. Right, Mean ± SEM of AIS start (S), maximum (M), and end (E) position for each treatment and subsequent calculation of AMI. B, Example images of DGCs treated with KCl in the presence of 50 μm nickel, cocktail of VGCC inhibitors, 50 μm APV + 10 μm NBQX, or 10 μm ryanodine. C, AMI mean ± SEM for each drug experiment. Brackets denote VGCC subtype blocked in each experiment. ***p < 0.0001, single-sample t test of AMI vs 1. Numbers within bars show the number of cells for each experiment. Scale bars, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1.
As predicted (Grubb and Burrone, 2010b), AIS relocation in DGCs was blocked by treatment with the L-type VGCC antagonist nifedipine (1 μm; Fig. 2A,C; AMI = 0.20 ± 0.09, single-sample t test, p vs 1 < 0.0001, p vs 0 = 0.028, n = 39). At this concentration, nifedipine does not block T-type channels (Lee et al., 2006). Interestingly, treatment with either the novel and specific T-type antagonist TTA-P2 (10 μm) or a low concentration of nickel (50 μm) that blocks only T- and R-type calcium channels (Magee and Johnston, 1995; Bender and Trussell, 2009) failed to prevent AIS movement (Fig. 2A–C; TTA-P2, AMI = 1.33 ± 0.12, n = 40; nickel, AMI = 1.28 ± 0.13, n = 39). This suggests that T-type VGCCs are not in fact required for depolarization-induced AIS plasticity.
To confirm that the major source of calcium for depolarization-induced AIS relocation comes through L-type VGCCs, we simultaneously inhibited all other VGCCs. Concurrently blocking T-type, P/Q-type, N-type, and R-type channels with TTA-P2 (10 μm), ω-agatoxin-TK (0.15 μm), ω-conotoxin-GIVA (3 μm), and SNX-482 (0.5 μm), respectively, failed to prevent AIS movement (Fig. 2B,C; AMI = 0.75 ± 0.13, single-sample t test, p vs 1 = 0.077, n = 39). There was a nonsignificant trend toward a lower magnitude of movement in this mixed-block treatment; this may suggest a small contribution from a combination of these channels but could also be attributable to nonspecific combinatorial effects of these drugs over 48 h. Regardless, L-type channels are by far the most important voltage-gated carrier of the depolarization-induced signal for AIS relocation.
Chronic depolarization could theoretically exert its effects through enhanced synaptic transmission, including signaling through calcium-permeable postsynaptic receptors (Berridge et al., 2000). However, this does not contribute to depolarization-induced AIS relocation, because simultaneous treatment with both NBQX (10 μm) and APV (50 μm) failed to block AIS plasticity (Fig. 2B,C; AMI = 0.96 ± 0.19, single-sample t test, p vs 1 = 0.81, n = 29). Finally, we also ruled out amplification of the L-type calcium signal by release from internal stores. Blockade of ryanodine receptors and IP3 receptors with 10 μm ryanodine or 20 μm 2-aminoethoxydiphenyl borate (2-APB), respectively, failed to prevent AIS movement (Fig. 2B,C; ryanodine, AMI = 1.36 ± 0.05, n = 38; 2-APB, AMI = 1.07 ± 0.17, n = 29).
Together, these results suggest that calcium entry though L-type VGCCs is the only trigger for AIS relocation and that this signal is not amplified by internal stores.
Calcium signaling pathways downstream of the L-type channel
Calcium influx results in numerous signaling cascades, controlling many cellular processes and different forms of neuronal plasticity. Interestingly, calcium influx is important in another form of AIS plasticity; after ischemic injury, activation of calpain leads to rapid degradation of the AIS (Schafer et al., 2009). However, calpain is not involved in depolarization-induced AIS relocation. Blocking calpain activity with the cell-permeable inhibitor MDL-28170 (N-[N-[(Phenylmethoxy)carbonyl]-L-va-phenylalaninal]), at a concentration known to inhibit excitotoxicity or injury-induced AIS loss in primary hippocampal cultures (10 μm; Brorson et al., 1995; Jordán et al., 1997; Schafer et al., 2009) failed to inhibit AIS plasticity (Fig. 3; AMI = 1.00 ± 0.17, n = 38). Therefore, we focused on possible calcium signaling events downstream of the L-type VGCC.
Figure 3.
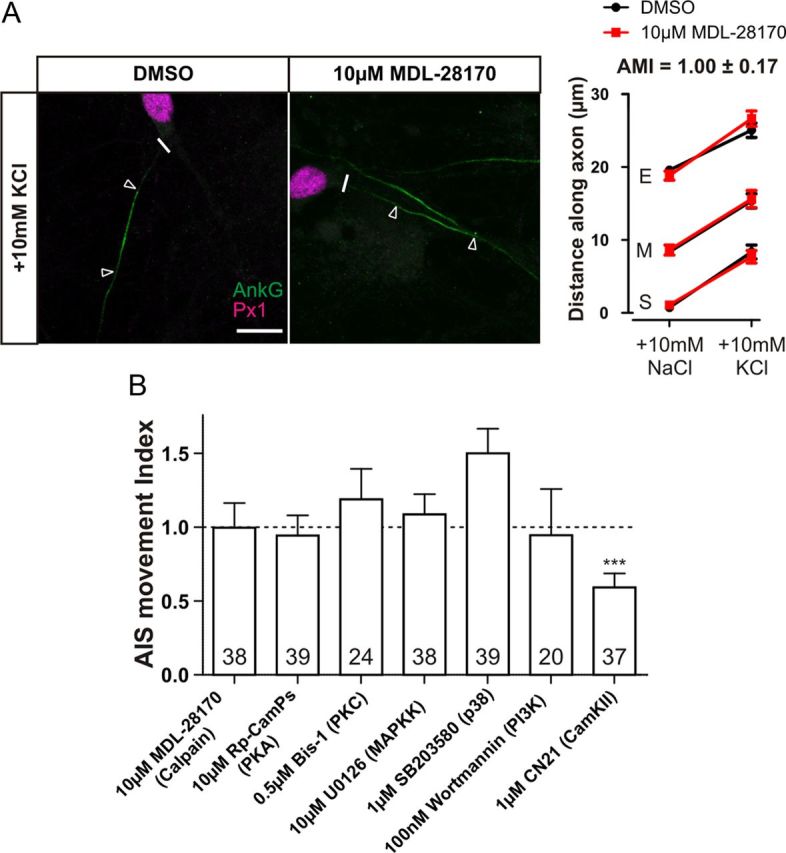
Screening possible calcium-activated targets downstream of L-type channels. A, Calpain is not involved in AIS relocation. Left, Example images of DGCs after 48 h depolarization in the presence of DMSO or 10 μm MDL-28170. Scale bar, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1. Right, Mean ± SEM of AIS start (S), maximum (M), and end (E) position for each treatment and subsequent calculation of AMI. B, AMI mean ± SEM for possible downstream calcium-activated targets. ***p = 0.0001, single-sample t test of AMI vs 1. Numbers within bars show the number of cells for each experiment.
Neither blocking protein kinase A with 10 μm Rp-cAMPs (Fig. 3B; AMI = 0.94 ± 0.14, single-sample t test, p vs 1 = 0.69, n = 39), protein kinase C with 0.5 μm bisindoylmaleimide-1 (Bis-1) (Fig. 3B; AMI = 1.19 ± 0.21, n = 24), mitogen-activated protein kinase kinase with 10 μm U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] (Fig. 3B; AMI = 1.08 ± 0.14, n = 38), the p38 MAPK pathway with 1 μm SB203580 [4-[5-(4-Fluorophenyl)-2-[4-(methylsphenyl]-1H-imidazol-4-yl]pyridine] (Fig. 3B; AMI = 1.50 ± 0.17, n = 39), nor phosphoinositide 3-kinase with 100 nm wortmannin (Fig. 3B; AMI = 0.95 ± 0.31, single-sample t test, p vs 1 = 0.86, n = 20) caused any block in AIS relocation, suggesting that these L-type VGCC targets are not involved in AIS plasticity. However, specifically inhibiting CaMKII with 1 μm tat-CN21 (Vest et al., 2007) caused a partial but not total block of AIS relocation (Fig. 3B; AMI = 0.59 ± 0.10, single-sample t test, p vs 1 = 0.0001, p vs 0 < 0.0001, n = 37), implying that CaMKII may contribute to AIS plasticity downstream of the L-type VGCC.
Calcineurin activation is necessary for AIS relocation
Calcineurin is a phosphatase involved in neuronal plasticity, often in processes that downregulate neuronal activity in excitatory neurons (Zeng et al., 2001; Flavell et al., 2006). To test the possibility that calcineurin is involved in AIS plasticity, we blocked its activity using the immunosuppressant drug FK506 (1 μm). Inhibiting calcineurin with this drug totally abolished depolarization-induced ankyrin-G relocation in DGCs (Fig. 4A; AMI = 0.11 ± 0.06, single-sample t test, p vs 1 < 0.0001, p vs 0 = 0.07, n = 118). It also completely blocked AIS movement in CA1 and CA3 pyramidal cells (Fig. 4C; CA1, AMI = −0.36 ± 0.18, n = 40; CA3, AMI = 0.22 ± 0.14, single-sample t test, p vs 1 < 0.0001, p vs 0 = 0.11, n = 37), showing that this effect is maintained throughout the population of hippocampal excitatory neurons. FK506 continued to block AIS relocation in DGCs even at much lower concentrations (100 and 10 nm), close to the IC50 for the drug (Fig. 4B; 100 nm FK506, AMI = 0.02 ± 0.12, single-sample t test, p vs 1 < 0.0001, p vs 0 = 0.88, n = 40; 10 nm FK506, AMI = 0.25 ± 0.11, single-sample t test, p vs 1 < 0.0001, p vs 0 = 0.04, n = 30). Together, these results indicate that activation of the phosphatase calcineurin is critical for depolarization-induced AIS plasticity in all hippocampal excitatory subtypes.
Figure 4.
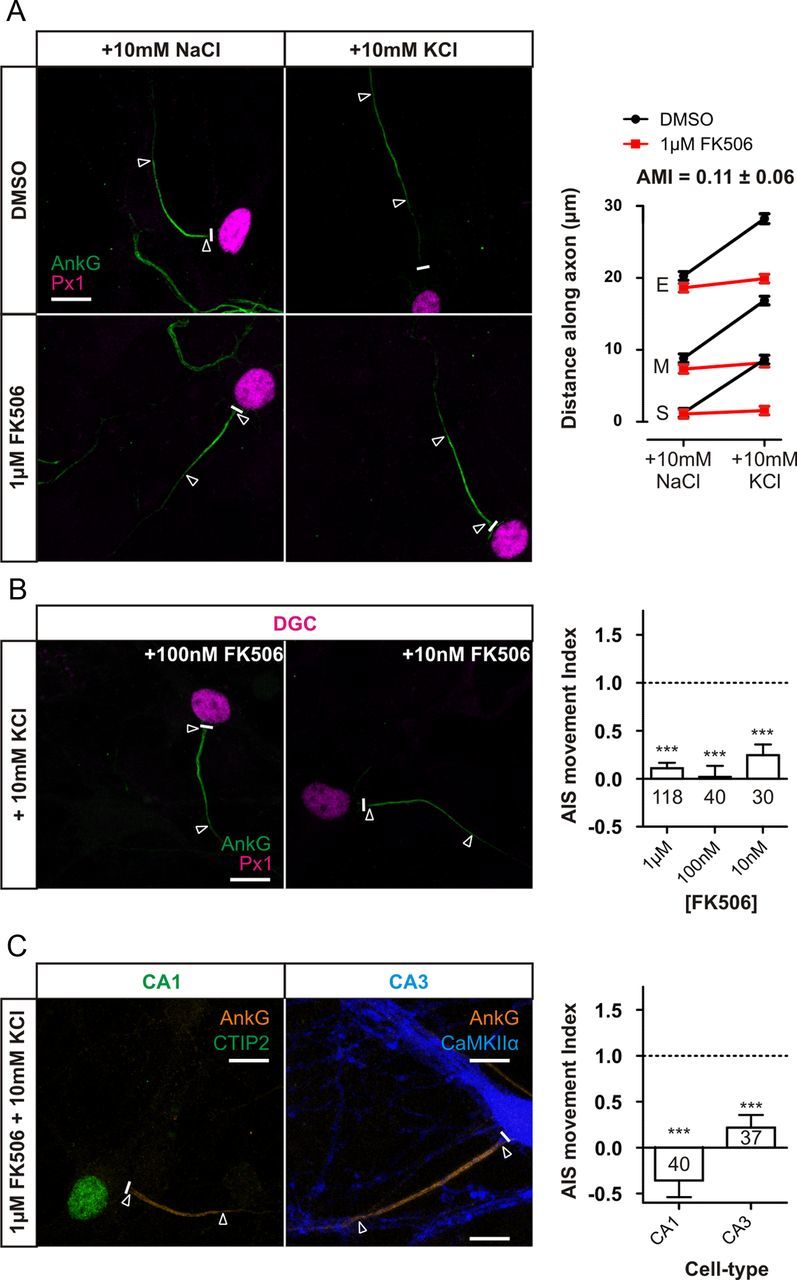
Calcineurin inhibition with FK506 totally blocks AIS relocation in all excitatory cell types. A, FK506 at 1 μm totally inhibits DGC AIS relocation. Left, Example images of DGCs treated with DMSO or 1 μm FK506 in NaCl or KCl treatment groups. Right, Mean ± SEM of AIS start (S), maximum (M), and end (E) for each treatment and subsequent calculation of AMI. B, FK506 inhibits AIS relocation even at low concentrations. Left, Example images of DGCs depolarized for 48 h in the presence of either 100 or 10 nm FK506. Right, AMI mean ± SEM values for DGCs after FK506 treatment. C, FK506 at 1 μm blocks AIS relocation in other excitatory cell types. Left, Example images of CA1 and CA3 neurons depolarized for 48 h in the presence of 1 μm FK506. Right, AMI mean ± SEM values for CA1 and CA3 neurons after FK506 treatment. ***p < 0.0001, single-sample t test of AMI vs 1. Numbers within bars show the number of cells for each experiment. Scale bars, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1.
A number of important control experiments ensured that calcineurin signaling is indeed required for depolarization-induced AIS relocation. First, treatment of DGC neurons with another classical calcineurin inhibitor, CsA (1 μm), totally blocked AIS relocation (Fig. 5A,C; AMI = −0.17 ± 0.07, n = 39). To confirm that both FK506 and CsA were acting on calcineurin in our neurons, we made use of a sparsely expressed NFAT–GFP construct. NFAT is a transcription factor that, when inactive, is located in the cytosol but on direct activation by calcineurin shuttles into the nucleus (Graef et al., 1999). Importantly, inhibiting calcineurin with either 1 μm FK506 or 1 μm CsA prevented translocation of NFAT–GFP to the nucleus after 3 h of +10 mm KCl stimulation in our cultures (Fig. 5B; one-way ANOVA with Tukey's multiple comparison test, DMSO + 10 mm KCl, p < 0.0001 vs DMSO + 10 mm NaCl, 1 μm FK506 + 10 mm KCl, and 1 μm CsA + 10 mm KCl, n = 75).
Figure 5.
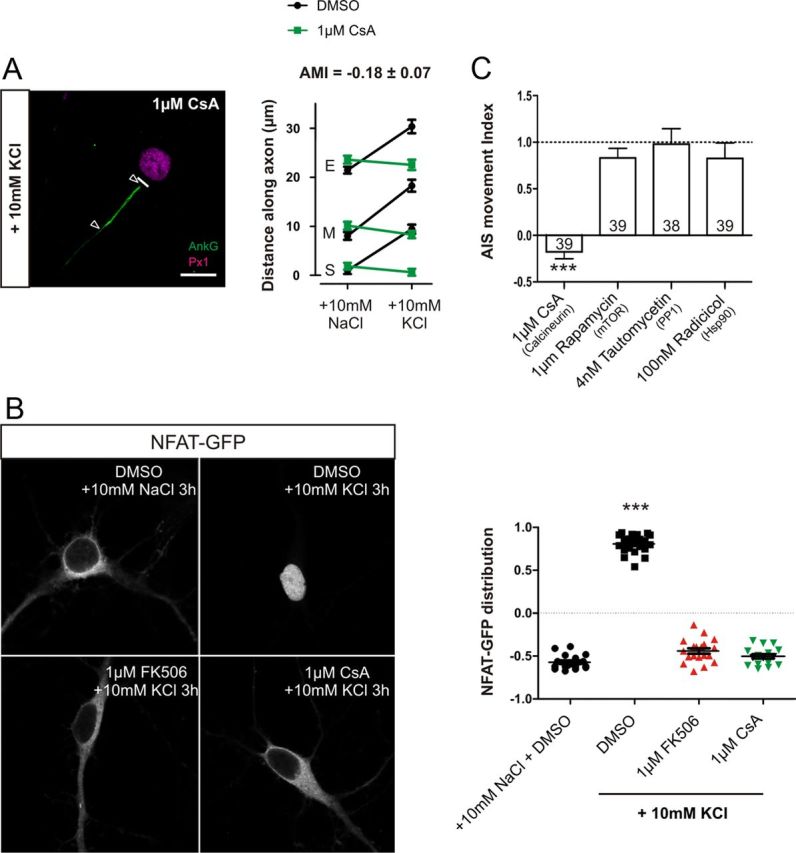
Inhibition of AIS relocation by FK506 is not a result of nonspecific effects. A, Calcineurin inhibition by CsA totally blocks AIS movement. Left, Example image of a DGC depolarized for 48 h in the presence of 1 μm CsA. Scale bar, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1. Right, Mean ± SEM of AIS start (S), maximum (M), and end (E) position for each treatment and subsequent calculation of AMI. B, Both 1 μm FK506 and 1 μm CsA block nuclear translocation of NFAT–GFP after 3 h depolarization. Left, Sample images of NFAT–GFP localization from each treatment group. Right, NFAT–GFP nucleus/cytoplasm distribution in each treatment group. ***p < 0.0001, one-way ANOVA with Tukey's multiple comparison test. C, AMI mean ± SEM values for pharmacological experiments used to test calcineurin inhibitor secondary effects. ***p < 0.0001, single-sample t test of AMI vs 1. Numbers within bars show the number of cells for each experiment.
Both FK506 and CsA work indirectly by binding and sequestering immunophilins—FK506 binding protein (FKBP12) and cyclophillin, respectively—required for calcineurin activity. FKBP12 is also an important component of the mammalian target of rapamycin signaling pathway. Importantly, however, the highly specific inhibitor of FKBP12, rapamycin (1 μm), had no effect on AIS relocation in DGCs (Fig. 5C; AMI = 0.83 ± 0.10, single-sample t test, p vs 1 = 0.1, n = 39). Both FK506 and CsA can also act by inhibiting the Hsp90 stress response pathway (Owens-Grillo et al., 1995); however, blockade of Hsp90 with radicicol (100 nm) had no effect on AIS movement (Fig. 5C; AMI = 0.83 ± 0.17, single-sample t test, p vs 1 = 0.30, n = 39). Finally, calcineurin can act through activation of another phosphatase, PP1, via inhibition of inhibitor-1 (Groth et al., 2003). However, specific block of PP1 with tautomycetin (4 nm) also failed to prevent AIS movement (Fig. 5C; AMI = 0.98 ± 0.17, single-sample t test, p vs 1 = 0.90, n = 38). By ruling out the off-target effects of these drugs, these results show that calcineurin activation is necessary for AIS plasticity.
Calcineurin acts downstream of L-type channels
The stimuli used in these experiments result in a depolarized membrane potential, calcium influx through L-type calcium channels, and AIS relocation (Grubb and Burrone, 2010b). Given the reported influence of calcineurin signaling on both leak potassium channels (Czirják et al., 2004; Li et al., 2011) and on L-type calcium channels themselves (Norris et al., 2002; Oliveria et al., 2007), it is entirely feasible that blocking calcineurin could in fact exert its effects on AIS movement via upstream mechanisms, on membrane depolarization, and/or calcium influx. To tease apart these possibilities, we studied high-potassium-evoked changes in both membrane potential and calcium influx in the presence or absence of FK506.
After treatment with 10 mm KCl, the mean resting potential of neurons was depolarized by 15.3 to −42.7 mV. This depolarization was the same in neurons treated with 1 μm FK506 for 48 h, illustrating that the membrane potential change is not dependent on calcineurin signaling (Fig. 6A; one-way ANOVA with Tukey's multiple comparison test, DMSO + 10 mm KCl vs FK506 + 10 mm KCl, p > 0.05, n = 27).
Figure 6.

Calcineurin effects are downstream of L-type VGCCs. A, Calcineurin activity does not affect KCl-induced depolarization. Resting membrane potential of neurons treated for 48 h with DMSO (black) or 1 μm FK506 (red) in NaCl or KCl treatment groups. ***p < 0.0001; ns, non-significant; one-way ANOVA with Tukey's multiple comparison test. B, Left, Mean ± SEM of normalized 340/380 ratio of fura-2 ratiometric dye after depolarization with KCl. Right, Mean plateau value of normalized 340/380 ratio between 18 and 20 min after wash-in. Points show individual cells treated with DMSO (black) or FK506 (red). Bars show mean ± SEM of all cells in each treatment group. C, Calcineurin activity does not affect calcium influx through L-type VGCCs in neurons depolarized to −40 mV. Left, Example traces of isolated L-type VGCC currents in each treatment group. Right, Plateau current density of isolated L-type VGCCs at −40 mV during the last 10 ms of the depolarizing step in each treatment group. Points show mean of two to five repeats from individual neurons. Bars show mean ± SEM of all cells recorded. ***p < 0.0001, one-way ANOVA with Tukey's multiple comparison test.
We then imaged the ratiometric calcium indicator fura-2 to study calcium influx into neurons for 20 min after a +10 mm KCl stimulus. Despite a lower response to the initial stimulus, the plateau phase of the calcium response, which consists of calcium influx mostly through L-type VGCCs (data not shown), was the same in neurons treated with either DMSO or 1 μm FK506 (Fig. 6B; Mann–Whitney U test, p = 0.83, n = 32). This suggests that long-term calcium entry is unaffected by calcineurin signaling. However, to test whether calcineurin has any effect on signaling specifically through L-type VGCCs (Norris et al., 2002; Oliveria et al., 2007), we used a mixture of inhibitors to isolate these channels specifically (see Materials and Methods) and then used whole-cell patch-clamp recordings to measure L-type current densities under different treatment conditions. Importantly, when neurons were stepped to and held at −40 mV, the membrane potential imposed by 10 mm KCl, there was no difference in L-type current density between DMSO- and FK506-treated neurons (Fig. 6C; one-way ANOVA with Tukey's multiple comparison test, DMSO + 10 mm KCl vs FK506 + 10 mm KCl, p > 0.05, n = 28). Treatment with 1 μm nifedipine blocked the majority of this current, showing that the channels were reasonably well isolated with this protocol (Fig. 6C).
Together, blocking calcineurin has no effect on the membrane depolarization or calcium influx induced by +10 mm KCl treatment. We therefore conclude that the calcineurin activity crucial for AIS relocation is downstream of the L-type VGCC.
Calcineurin inhibitors also block photostimulation-induced AIS relocation
Thus far, we have used chronic depolarization as a simple paradigm to induce AIS relocation, but it is important to show that the calcineurin pathway is also involved in a more physiologically relevant method of inducing this type of AIS plasticity. Chronic 48 h photostimulation of ChR2-expressing neurons with bursts of blue light has been shown previously to cause AIS relocation (Grubb and Burrone, 2010b). A slightly adapted protocol (see Materials and Methods) caused distal relocation of the AIS of ChR2-expressing DGCs in DMSO control conditions, but this was completely blocked by treatment with either 1 μm FK506 or 1 μm CsA (Fig. 7; Kruskall-Wallis with Dunn's post hoc test, photostimulation + DMSO vs either photostimulation + 1 μm FK506: start, p < 0.001; maximum, p < 0.01; end, p < 0.01; or photostimulation + 1 μm CsA: start, p < 0.001; maximum, p < 0.05; end, p < 0.05; n = 76). Therefore, AIS relocation is reliant on calcineurin signaling whether induced by depolarization or patterned chronic photostimulation.
Calcineurin activation is sufficient for AIS relocalization
We have shown that calcineurin signaling downstream of the L-type VGCC is necessary for AIS relocation. To assess whether calcineurin signaling is also sufficient for AIS plasticity, we used a constitutively active form of the phosphatase (CACaN). CACaN activity was confirmed by the nuclear localization of cotransfected NFAT–GFP without the need for depolarization (nuclear localization observed in 15 of 15 neurons; Fig. 8A). In DGCs cotransfected with CACaN and YFP at 3 DIV, AISs labeled by ankyrin-G immunostaining at 12 DIV were significantly more distal than those transfected with YFP alone (Fig. 8B; YFP + CACaN vs YFP alone, start, maximum, and end position, Mann–Whitney U test, p < 0.0001, n = 104). These data show that calcineurin activation is also sufficient for AIS plasticity.
Figure 8.

Calcineurin activation is sufficient for AIS relocation. A, CACaN expression causes nuclear translocation of NFAT–GFP. Left, Example image of a 12 DIV neuron expressing NFAT–GFP alone. Right, Example image of a 12 DIV neuron coexpressing NFAT–GFP and CACaN. Scale bar, 10 μm. B, CACaN expression causes AIS relocation. Left, Example images of DGCs at 12 DIV after 3 DIV transfection of neurons with YFP (top) or YFP + CACaN (bottom). Scale bar, 10 μm. Thick white line denotes axon start, and white triangles illustrate AIS location. AnkG, Ankyrin-G; Px1, prox1. Right, Mean ± SEM of AIS start (S), maximum (M), and end (E) position for each group. ***p < 0.0001, Mann–Whitney U test for start, maximum, or end position.
Calcineurin is not localized to the AIS
Many proteins that regulate AIS structure, function, and development are concentrated within the AIS region (Grubb and Burrone, 2010a; Rasband, 2010). Because calcineurin is both necessary and sufficient for AIS relocation, we wanted to assess whether there was a specific concentration of the enzyme at the AIS. Immunostaining for the catalytic subunit of calcineurin (calcineurin A) brightly labeled DGCs transfected with CACaN, showing that the antibody does recognize a calcineurin epitope (Fig. 9A). Untransfected cells showed nuclear concentration of the phosphatase, with diffuse expression throughout the neurites but no AIS concentration of calcineurin in DGCs treated with +10 mm NaCl, 3 h of depolarization (+10 mm KCl) or after 48 h depolarization and full AIS relocation (Fig. 9B). These data suggest that, although its enzymatic activity affects AIS position, there is no specific localization of calcineurin at the AIS.
Figure 9.
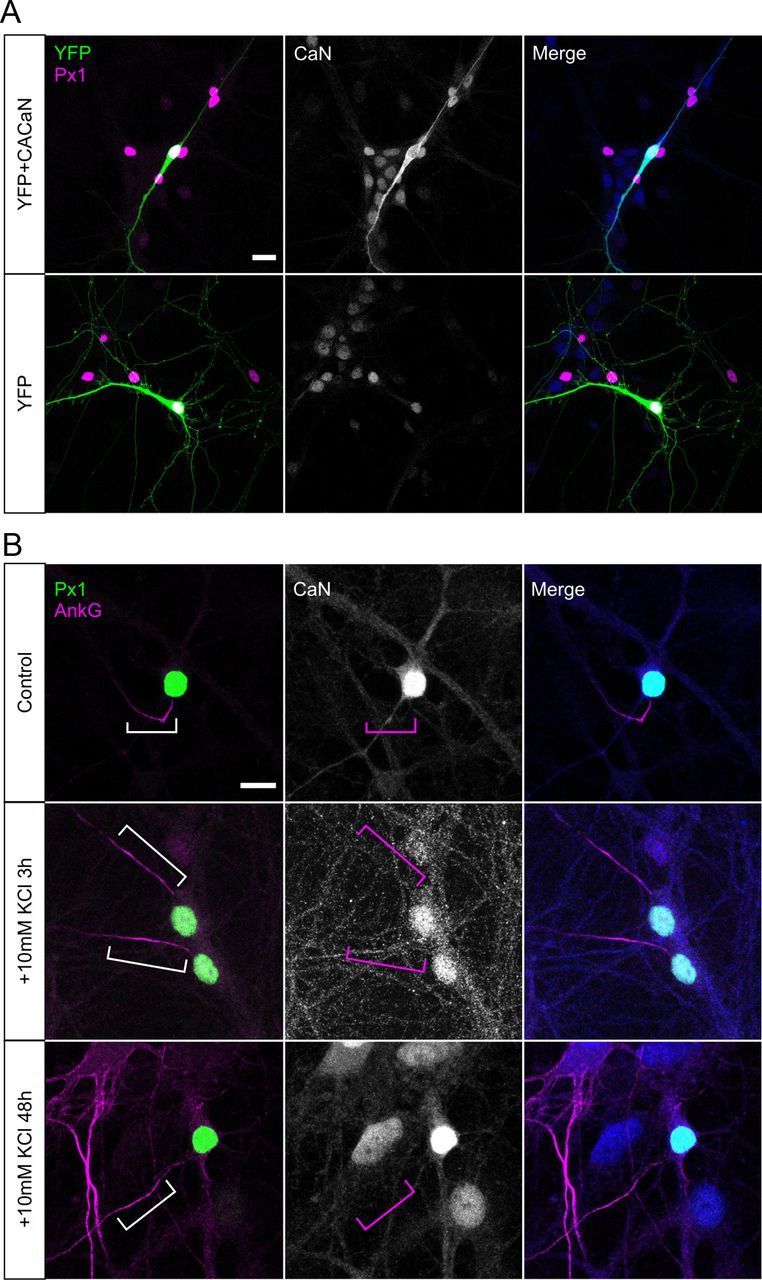
Calcineurin is not concentrated at the AIS. A, Example DGCs transfected with either CACaN + YFP (top) or YFP alone (bottom), labeled for calcineurin A. Scale bar, 20 μm. B, Example DGCs in control conditions (top) or depolarized with KCl for 3 h (middle) or 48 h (bottom). Scale bar, 10 μm. Brackets outline AIS location; note the lack of calcineurin staining in this region in all treatment groups. AnkG, Ankyrin-G; Px1, prox1; CaN, calcineurin.
Calcineurin is not crucial for other forms of depolarization-induced plasticity
Depolarization-induced AIS relocation is a form of structural plasticity in neurons that may act to alter intrinsic excitability. We finally wanted to examine whether other forms of intrinsic and structural plasticity brought about by our chronic depolarization stimulus were also regulated by calcineurin signaling.
Rm in cultured hippocampal neurons has been shown to decrease after chronic depolarization (Grubb and Burrone, 2010b; O'Leary et al., 2010). We targeted DGCs for whole-cell voltage-clamp recordings (see Materials and Methods and Fig. 10A) after 48 h depolarization from 10 or 11 DIV in the presence or absence of 1 μm FK506. As predicted, in DMSO, DGCs treated with +10 mm KCl showed decreased Rm compared with neurons treated with +10 mm NaCl. Interestingly, DGCs treated with FK506 had increased Rm in both control and depolarizing treatments, but Rm still decreases in response to chronic depolarization. This led to significant effects of treatment and drug, but no interaction between the two factors (Fig. 10A; two-way ANOVA on ranks, effect of treatment, p < 0.001; drug, p = 0.025; interaction, p = 0.133, n = 231). Therefore, Rm changes induced by depolarization with +10 mm KCl are not reliant on calcineurin signaling (Fig. 10A).
Figure 10.

Calcineurin does not regulate +10 mm KCl depolarization-induced plasticity of membrane resistance or dendritic spine density. A, Membrane resistance. Left, Example DGC expressing YFP. Px1, Prox1. Middle, Example image of a DGC (white arrow) targeted for whole-cell patch-clamp recordings. Scale bars, 20 μm. Right, Mean ± SEM of membrane resistance in DGCs treated with NaCl or KCl in the presence of either DMSO (black) or 1 μm FK506 (red). B, Dendritic spine density. Example images of secondary dendrites of YFP-expressing DGCs treated with NaCl or KCl in the presence of either DMSO or 1 μm FK506. Right, Mean ± SEM of dendritic spine density in each treatment group. Two-way ANOVA, *p < 0.05, ***p < 0.001.
Dendritic spine density can also be modulated by strong activity in a calcineurin-dependent manner (Halpain et al., 1998; Kurz et al., 2008; Tian et al., 2010). Therefore, we decided to investigate the effects of our low-level depolarizing stimulus on the dendritic spines of DGCs. We sparsely transfected neurons with YFP at 7 DIV to reveal their morphology and measured DGC spine density from secondary dendrites after 48 h treatment with +10 mm NaCl or +10 mm KCl. Interestingly we also observed an activity-dependent decrease in spine density (Fig. 10B). However, although there was a trend toward increased baseline spine density with calcineurin blockade, a similar level of spine density loss was induced by depolarization in both DMSO-treated and FK506-treated neurons (Fig. 10B; two-way ANOVA on ranks, effect of treatment, p < 0.0001; drug, p = 0.11; interaction, p = 0.43, n = 180). Whereas previous studies that used much stronger depolarization have shown calcineurin to be important in regulating spine density (Halpain et al., 1998; Kurz et al., 2008; Tian et al., 2010), calcineurin signaling is not required for the effect of +10 mm KCl on DGC spine density.
Together, these results suggest that AIS plasticity is governed by calcineurin in a mechanism separate from other forms of depolarization-induced intrinsic and structural plasticity.
Discussion
We have shown that AIS relocation plasticity occurs in all excitatory (DGC, CA1, and CA3) but not inhibitory hippocampal subtypes and is governed by the activation of L-type Cav1 VGCCs. The phosphatase calcineurin acts downstream of the L-type VGCC, totally blocking both depolarization and optogenetically induced AIS plasticity when inhibited, and inducing it when constitutively active. However, some other forms of depolarization-induced plasticity are independent of this signaling pathway.
An L-type → calcineurin signaling pathway for AIS relocation
The long-term nature of activity-dependent AIS plasticity makes it an obvious target for control by Cav1 L-type VGCCs and calcineurin signaling. L-type VGCCs have been shown in many instances to couple electrical activity to different forms of long-term neuronal plasticity, including activity-dependent decreases in intrinsic excitability (Wu et al., 2008; O'Leary et al., 2010). They are ideally suited to this role: L-type VGCCs are predominantly located somatodendritically (Westenbroek et al., 1990; Hell et al., 1993; Pravettoni et al., 2000; Obermair et al., 2004), can be activated by low-level depolarization, remain activated during long-term stimulation (Lipscombe et al., 2004), and recruit a number of signaling molecules that convey nuclear changes through the activation of transcription factors (Deisseroth et al., 2003; Wheeler et al., 2012).
Calcineurin is one such molecule, a phosphatase that is extremely sensitive to small changes in local calcium and whose activity faithfully tracks [Ca2+] increases (Kd of ∼0.05 μm; Stemmer and Klee, 1994; Graupner and Brunel, 2007; Forbes et al., 2012). Although recent data suggest that calpain can activate calcineurin in certain systems (Silverman-Gavrila et al., 2013), the lack of effect of calpain blockade here (Fig. 3) suggests that this indirect mechanism of calcium-dependent calcineurin activation is unlikely to operate in our neurons. Calcineurin-dependent pathways have been linked to plastic changes in synaptic strength and intrinsic excitability in various systems. For example, calcineurin is crucial for hippocampal long-term depression (Zeng et al., 2001), for activity-dependent Kv channel declustering in hippocampal pyramidal neurons (Misonou et al., 2004), for developmental synaptic and morphological changes in Xenopus tectal neurons (Schwartz et al., 2009), and for long-term potentiation (LTP) and increased excitability in cerebellar Purkinje cells (Schonewille et al., 2010). A pool of calcineurin is tethered directly to the L-type VGCC through interactions with A-kinase anchoring proteins (AKAPs; Oliveria et al., 2007). Calcineurin may therefore become activated within the L-type VGCC microdomain at low calcium influx levels and faithfully follow calcium changes to modulate onward signaling controlling AIS position.
Other than calcineurin, only inhibition of one other calcium-dependent pathway had any effect on AIS plasticity: selective block of CaMKII partially decreased AIS relocation (Fig. 3). This is intriguing because calcineurin and CaMKII signaling usually act antagonistically in the control of calcium-dependent processes (Lisman and Zhabotinsky, 2001; Wen et al., 2004; Graupner and Brunel, 2007; Gao et al., 2012). Perhaps CaMKII plays a supplementary role in AIS plasticity downstream of calcineurin signaling (Greer and Greenberg, 2008) or is involved in local cytoskeletal alterations necessary for physically relocating AIS components (Hund et al., 2010).
A non-local signal for AIS plasticity
However, the calcineurin-based signal for AIS relocation is unlikely to act locally within the structure itself. Although the AIS has some of the machinery required for activity-dependent plasticity, including VGCCs (Bender and Trussell, 2009; Yu et al., 2010) and a structure specialized for calcium-induced calcium release (Sánchez-Ponce et al., 2011), selective individual or combined block of the VGCC subtypes found at the AIS does not prevent AIS relocation (Fig. 2; Grubb and Burrone, 2010b), nor does pharmacological inhibition of release from internal calcium stores (Fig. 2). Furthermore, L-type VGCCs are crucial for activity-dependent AIS relocation but have never been functionally localized to the AIS (Bender and Trussell, 2009); instead, these channels are known to predominantly distribute in the soma and proximal dendrites of hippocampal neurons (Hell et al., 1993; Pravettoni et al., 2000; Obermair et al., 2004). In addition, we show here that calcineurin is needed for relocation of the AIS but is not at all concentrated at the structure (Fig. 9). These considerations are most consistent with the trigger for AIS relocation being somatodendritic and away from its site of final effect, as is the case for some other forms of long-term plasticity (Ibata et al., 2008).
Possible downstream targets of calcineurin
The structure of calcineurin confers specificity to a relatively small number of downstream targets (Klee et al., 1998; Huai et al., 2002), many of which are transcription factors. NFATc4 (also known as NFAT3) is a major target expressed in the hippocampus (Graef et al., 1999) and is important for neurite outgrowth, synaptogenesis, and neuronal development (Nguyen and Di Giovanni, 2008). Interestingly, like NFATc4 (Fig. 4; Graef et al., 1999), TORC1 (transducer of regulated cAMP response element-binding protein 1), a mediator of dendritic growth, translocates from the cytosol to the nucleus during calcineurin-mediated dephosphorylation (Li et al., 2009). Moreover, this translocation is dependent on L-type VGCCs (Li et al., 2009). Other direct targets of calcineurin include myocyte enhancer factor 2 (MEF2), a transcription factor involved in cell survival (Mao et al., 1999) but also shown to regulate activity-dependent spine density through NMDA and L-type VGCCs (Flavell et al., 2006). Calcineurin also mediates dephosphorylation of retinoblastoma, releasing a repressor complex that allows calcium responsive transactivator (CREST)-mediated transcription (Qiu and Ghosh, 2008). Acting via any of these transcription factors, somatic calcineurin activation could lead to transcriptional changes, producing axonally concentrated proteins needed for local AIS movement mechanisms.
Because the AIS is also strongly associated with a distinct cytoskeleton, including a high density of actin (Nakada et al., 2003) and fasciculated microtubules (Palay et al., 1968), it would seem logical that its relocation would require cytoskeletal remodeling. Therefore, it is interesting to note that calcineurin can influence actin remodeling through activation of slingshot, a pathway described in growth cone turning (Wen et al., 2007) and AMPA receptor insertion (Yuen and Yan, 2009). How it might do this in the context of AIS relocation, however, given that it is not concentrated at the structure (Fig. 9), is not immediately obvious. Somatically activated calcineurin could diffuse to the AIS, where it might induce changes in actin dynamics at low concentrations. Alternatively, calcineurin could act somatically on cytoskeletal modifying proteins, which if trafficked to the AIS could influence AIS relocation. In future experiments, it will be interesting to test whether any of the above targets are involved in AIS relocation downstream of calcineurin activation.
Multiple pathways to long-term plasticity
We have shown here that calcineurin governs AIS relocation in dissociated cultures of the hippocampus. However, this does not suggest that all neuronal subtypes show this type of plasticity. In fact, it is becoming increasingly evident that different neuronal types are equipped to use different methods of regulation at their AIS, including relocation, length changes, and ion channel modification (Kuba et al., 2010; Grubb et al., 2011; Kaphzan et al., 2011; Baalman et al., 2013). It will be very interesting to see whether calcineurin is at the heart of all changes at the AIS or whether this phosphatase solely regulates AIS relocation. Another intriguing question for future studies to ask is why inhibitory neurons in the hippocampus do not show AIS relocation? It is a possibility that these cells have low densities of L-type VGCCs, low expression of calcineurin, or a lack of AKAP-dependent calcineurin localization to L-type channels. Alternatively, these cells may require an altogether different trigger for AIS movement.
Another crucial question is how ongoing activity changes lead to different forms of plasticity in the same type of neuron. Differences in the subcellular location and kinetics of various signaling pathways enable distinct spatiotemporal activity alterations to produce unique plastic outcomes (Berridge et al., 2000). However, a given neuronal response is not always produced by a unique stimulus activating a dedicated pathway. Sometimes the same signaling pathway can mediate multiple forms of plasticity; calcineurin, for example, is vital for both synaptic and morphological development in Xenopus tectal neurons (Schwartz et al., 2009) and is required for both LTP and increased intrinsic excitability in cerebellar Purkinje cells (Schonewille et al., 2010). In other cases, the same alteration in activity can give rise to multiple forms of neuronal plasticity, not all dependent on the same signaling pathway. This was the case here, in which chronic moderate depolarization produced calcineurin-dependent relocation of the AIS but calcineurin-independent changes in membrane resistance and spine density in cultured DGCs (Fig 10). Interestingly, homeostatic synaptic plasticity induced by chronic optogenetic photostimulation is also independent of calcineurin signaling and instead relies on the activation of CaMKIV (Ibata et al., 2008; Goold and Nicoll, 2010). It appears, then, that the pathways to long-term plasticity are modular to at least some degree, lacking a single overarching “program” that governs neuronal responses to chronic activity changes.
Therapeutic control of AIS position
The relevance of activity-dependent AIS relocation under physiological conditions is still under debate. Computational models suggest that there may not be a simple monotonic relationship between AIS position and excitability (Kuba et al., 2006; Kress et al., 2010; Wang et al., 2011; Baranauskas et al., 2013). In addition, unlike length changes, relocation of the AIS has yet to be reported under any conditions in vivo. Nevertheless, if the AIS is an important regulator of neuronal output, it is likely that mutations in the proteins involved in positioning or maintaining the AIS would lead to neurological abnormalities. Indeed, the ankyrin-G and L-type VGCC genes contain specific risk loci for disorders, including schizophrenia and bipolar disorder (O'Donovan et al., 2009). Moreover, in a mouse model of the neurodevelopmental disorder Angelman syndrome, hippocampal neurons had a longer AIS when compared with wild-type mice. This change resulted in higher neuronal excitability, and these mice showed seizures and impaired motor function comparable with this disease in humans (Kaphzan et al., 2011). Although this field is in its infancy, it is likely that many more disorders associated with abnormalities in AIS location and/or size will be found. Thus, understanding the molecular mechanisms governing these types of plasticity may give insights into treatments for particular neurological diseases (Gründemann and Häusser, 2010). It will be interesting to see whether inhibition of calcineurin or its following downstream pathway will be able to regulate AIS position in such disorders.
Footnotes
This work was supported by a Wellcome Trust Career Development Fellowship (M.S.G.), Medical Research Council four-year PhD studentships (M.D.E., R.P.S., A.S.D.), a University of California, Irvine Minority Health International Research Training studentship (S.L.), and a Dravet Syndrome United Kingdom summer studentship (T.B.K.W.). We thank Annisa Chand for assistance with dissociated hippocampal cultures, Phillip Gordon-Weeks, Britta Eickholt, and Miles Houslay for provision of pharmacological agents and advice, Karl Deisseroth (ChR2–YFP), Eloise Hudry, and Brand Hyman (CACaN) for providing DNA constructs, Andrew Lowe and Jana Mukanowa for construction and setup of the LED system, and finally Juan Burrone for comments on this manuscript.
V.N.U. and J.J.R. are employees of Merck and Co. and may own stock and/or stock options in the company. M.D.E., R.P.S., A.S.D., S.L., T.B.K.W., and M.S.G. declare no competing financial interests.
References
- Akritas MG. The rank transform method in some two factor designs. J Am Stat Assoc. 1990;85:73–78. [Google Scholar]
- Baalman KL, Cotton RJ, Rasband SN, Rasband MN. Blast wave exposure impairs memory and decreases axon initial segment length. J Neurotrauma. 2013 doi: 10.1089/neu.2012.2478. doi: 10.1089/neu.2012.2478. Advance online publication. Retrieved March 3, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranauskas G, David Y, Fleidervish IA. Spatial mismatch between the Na+ flux and spike initiation in axon initial segment. Proc Natl Acad Sci U S A. 2013;10:4051–4056. doi: 10.1073/pnas.1215125110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61:259–271. doi: 10.1016/j.neuron.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO. The physiology of the axon initial segment. Annu Rev Neurosci. 2012;35:249–265. doi: 10.1146/annurev-neuro-062111-150339. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Uebele VN, Renger JJ, Trussell LO. Control of firing patterns through modulation of axon initial segment T-type calcium channels. J Physiol. 2012;590:109–118. doi: 10.1113/jphysiol.2011.218768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Brorson JR, Marcuccilli CJ, Miller RJ. Delayed antagonism of calpain reduces excitotoxicity in cultured neurons. Stroke. 1995;26:1259–1266. doi: 10.1161/01.STR.26.7.1259. discussion 1267. [DOI] [PubMed] [Google Scholar]
- Clark BD, Goldberg EM, Rudy B. Electrogenic tuning of the axon initial segment. Neuroscientist. 2009;15:651–668. doi: 10.1177/1073858409341973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D. The role of T-channels in the generation of thalamocortical rhythms. CNS Neurol Disord Drug Targets. 2006;5:571–585. doi: 10.2174/187152706779025526. [DOI] [PubMed] [Google Scholar]
- Czirják G, Tóth ZE, Enyedi P. The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J Biol Chem. 2004;279:18550–18558. doi: 10.1074/jbc.M312229200. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/S0959-4388(03)00076-X. [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J Neurosci. 2010;30:99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Forbes EM, Thompson AW, Yuan J, Goodhill GJ. Calcium and cAMP levels interact to determine attraction versus repulsion in axon guidance. Neuron. 2012;74:490–503. doi: 10.1016/j.neuron.2012.02.035. [DOI] [PubMed] [Google Scholar]
- Fried SI, Lasker AC, Desai NJ, Eddington DK, Rizzo JF., 3rd Axonal sodium-channel bands shape the response to electric stimulation in retinal ganglion cells. J Neurophysiol. 2009;101:1972–1987. doi: 10.1152/jn.91081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, van Beugen BJ, De Zeeuw CI. Distributed synergistic plasticity and cerebellar learning. Nat Rev Neurosci. 2012;13:619–635. doi: 10.1038/nrn3312. [DOI] [PubMed] [Google Scholar]
- Goold CP, Nicoll RA. Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron. 2010;68:512–528. doi: 10.1016/j.neuron.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 1999;401:703–708. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- Graupner M, Brunel N. STDP in a bistable synapse model based on CaMKII and associated signaling pathways. PLoS Comput Biol. 2007;3:e221. doi: 10.1371/journal.pcbi.0030221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Groth RD, Dunbar RL, Mermelstein PG. Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun. 2003;311:1159–1171. doi: 10.1016/j.bbrc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Building and maintaining the axon initial segment. Curr Opin Neurobiol. 2010a;20:481–488. doi: 10.1016/j.conb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010b;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Shu Y, Kuba H, Rasband MN, Wimmer VC, Bender KJ. Short- and long-term plasticity at the axon initial segment. J Neurosci. 2011;31:16049–16055. doi: 10.1523/JNEUROSCI.4064-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann J, Häusser M. Neuroscience: a plastic axonal hotspot. Nature. 2010;465:1022–1023. doi: 10.1038/4651022a. [DOI] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci. 1998;18:9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178:875–886. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huai Q, Kim HY, Liu Y, Zhao Y, Mondragon A, Liu JO, Ke H. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc Natl Acad Sci U S A. 2002;99:12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME, Mohler PJ. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–3519. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. J Biol Chem. 2001;276:3524–3530. doi: 10.1074/jbc.M004275200. [DOI] [PubMed] [Google Scholar]
- Jordán J, Galindo MF, Miller RJ. Role of calpain- and interleukin-1 beta converting enzyme-like proteases in the beta-amyloid-induced death of rat hippocampal neurons in culture. J Neurochem. 1997;68:1612–1621. doi: 10.1046/j.1471-4159.1997.68041612.x. [DOI] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J Neurosci. 2011;31:17637–17648. doi: 10.1523/JNEUROSCI.4162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73:235–247. doi: 10.1016/j.neuron.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Kress GJ, Dowling MJ, Eisenman LN, Mennerick S. Axonal sodium channel distribution shapes the depolarized action potential threshold of dentate granule neurons. Hippocampus. 2010;20:558–571. doi: 10.1002/hipo.20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Ishii TM, Ohmori H. Axonal site of spike initiation enhances auditory coincidence detection. Nature. 2006;444:1069–1072. doi: 10.1038/nature05347. [DOI] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Moore BJ, Henderson SC, Campbell JN, Churn SB. A cellular mechanism for dendritic spine loss in the pilocarpine model of status epilepticus. Epilepsia. 2008;49:1696–1710. doi: 10.1111/j.1528-1167.2008.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Queenan BN, Rozeboom AM, Bellmore R, Lim ST, Vicini S, Pak DT. Mossy fiber-CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron. 2013;77:99–114. doi: 10.1016/j.neuron.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Kaku T, Takebayashi S, Uchino T, Miyamoto S, Hadama T, Perez-Reyes E, Ono K. Actions of mibefradil, efonidipine and nifedipine block of recombinant T- and L-type Ca channels with distinct inhibitory mechanisms. Pharmacology. 2006;78:11–20. doi: 10.1159/000094900. [DOI] [PubMed] [Google Scholar]
- Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21:91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang C, Takemori H, Zhou Y, Xiong Z-Q. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J Neurosci. 2009;29:2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/S0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol. 1995;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- McCleskey EW, Fox AP, Feldman DH, Cruz LJ, Olivera BM, Tsien RW, Yoshikami D. Omega-conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc Natl Acad Sci U S A. 1987;84:4327–4331. doi: 10.1073/pnas.84.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Nakada C, Ritchie K, Oba Y, Nakamura M, Hotta Y, Iino R, Kasai RS, Yamaguchi K, Fujiwara T, Kusumi A. Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol. 2003;5:626–632. doi: 10.1038/ncb1009. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Di Giovanni S. NFAT signaling in neural development and axon growth. Int J Dev Neurosci. 2008;26:141–145. doi: 10.1016/j.ijdevneu.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Landfield PW. Calcineurin enhances L-type Ca(2+) channel activity in hippocampal neurons: increased effect with age in culture. Neuroscience. 2002;110:213–225. doi: 10.1016/S0306-4522(01)00574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci. 2004;19:2109–2122. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- O'Donovan MC, Craddock NJ, Owen MJ. Genetics of psychosis; insights from views across the genome. Hum Genet. 2009;126:3–12. doi: 10.1007/s00439-009-0703-0. [DOI] [PubMed] [Google Scholar]
- O'Leary T, van Rossum MC, Wyllie DJ. Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. J Physiol. 2010;588:157–170. doi: 10.1113/jphysiol.2009.181024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens-Grillo JK, Hoffmann K, Hutchison KA, Yem AW, Deibel MR, Jr, Handschumacher RE, Pratt WB. The cyclosporin A-binding immunophilin CyP-40 and the FK506-binding immunophilin hsp56 bind to a common site on hsp90 and exist in independent cytosolic heterocomplexes with the untransformed glucocorticoid receptor. J Biol Chem. 1995;270:20479–20484. doi: 10.1074/jbc.270.35.20479. [DOI] [PubMed] [Google Scholar]
- Palay SL, Sotelo C, Peters A, Orkand PM. The axon hillock and the initial segment. J Cell Biol. 1968;38:193–201. doi: 10.1083/jcb.38.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravettoni E, Bacci A, Coco S, Forbicini P, Matteoli M, Verderio C. Different localizations and functions of L-type and N-type calcium channels during development of hippocampal neurons. Dev Biol. 2000;227:581–594. doi: 10.1006/dbio.2000.9872. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Ghosh A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron. 2008;60:775–787. doi: 10.1016/j.neuron.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- Sánchez-Ponce D, DeFelipe J, Garrido JJ, Muñoz A. In vitro maturation of the cisternal organelle in the hippocampal neuron's axon initial segment. Mol Cell Neurosci. 2011;48:104–116. doi: 10.1016/j.mcn.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Sather WA, Tanabe T, Zhang JF, Mori Y, Adams ME, Tsien RW. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel alpha 1 subunits. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-T. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, Hosy E, Hoebeek FE, Elgersma Y, Hansel C, De Zeeuw CI. Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron. 2010;67:618–628. doi: 10.1016/j.neuron.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz N, Schohl A, Ruthazer ES. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron. 2009;62:655–669. doi: 10.1016/j.neuron.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Silverman-Gavrila LB, Praver M, Mykles DL, Charlton MP. Calcium, calpain, and calcineurin in low-frequency depression of transmitter release. J Neurosci. 2013;33:1975–1990. doi: 10.1523/JNEUROSCI.3092-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochivko D, Chen J, Becker A, Beck H. Blocker-resistant Ca2+ currents in rat CA1 hippocampal pyramidal neurons. Neuroscience. 2003;116:629–638. doi: 10.1016/S0306-4522(02)00777-7. [DOI] [PubMed] [Google Scholar]
- Stemmer PM, Klee CB. Dual calcium ion regulation of calcineurin by calmodulin and calcineurin B. Biochemistry. 1994;33:6859–6866. doi: 10.1021/bi00188a015. [DOI] [PubMed] [Google Scholar]
- Tian X, Kai L, Hockberger PE, Wokosin DL, Surmeier DJ. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol Cell Neurosci. 2010;44:94–108. doi: 10.1016/j.mcn.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, Davies KD, O'Leary H, Port JD, Bayer KU. Dual mechanism of a natural CaMKII inhibitor. Mol Biol Cell. 2007;18:5024–5033. doi: 10.1091/mbc.E07-02-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang H, Yu L, Chen Y. Role of axonal sodium-channel band in neuronal excitability. Phys Rev E Stat Nonlin Soft Matter Phys. 2011;84 doi: 10.1103/PhysRevE.84.052901. 052901. [DOI] [PubMed] [Google Scholar]
- Wen Z, Guirland C, Ming GL, Zheng JQ. A CaMKII/calcineurin switch controls the direction of Ca(2+)-dependent growth cone guidance. Neuron. 2004;43:835–846. doi: 10.1016/j.neuron.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Wen Z, Han L, Bamburg JR, Shim S, Ming GL, Zheng JQ. BMP gradients steer nerve growth cones by a balancing act of LIM kinase and Slingshot phosphatase on ADF/cofilin. J Cell Biol. 2007;178:107–119. doi: 10.1083/jcb.200703055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Wilke SA, Daggett A, Davis E, Otto S, Ravi D, Ripley B, Bushong EA, Ellisman MH, Klein G, Ghosh A. Cadherin-9 regulates synapse-specific differentiation in the developing hippocampus. Neuron. 2011;71:640–655. doi: 10.1016/j.neuron.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Surmeier DJ, Disterhoft JF. Coupling of L-type Ca2+ channels to KV7/KCNQ channels creates a novel, activity-dependent, homeostatic intrinsic plasticity. J Neurophysiol. 2008;100:1897–1908. doi: 10.1152/jn.90346.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Maureira C, Liu X, McCormick D. P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J Neurosci. 2010;30:11858–11869. doi: 10.1523/JNEUROSCI.2651-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Yan Z. Dopamine D4 receptors regulate AMPA receptor trafficking and glutamatergic transmission in GABAergic interneurons of prefrontal cortex. J Neurosci. 2009;29:550–562. doi: 10.1523/JNEUROSCI.5050-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/S0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]