

Abstract
VIP1 (VirE2 interacting protein 1), initially discovered as a host protein involved in Agrobacterium-plant cell DNA transfer, is a transcription factor of the basic leucine-zipper (bZIP) domain family that regulates several defence-related genes in Arabidopsis. We have developed assays to assess VIP1 binding to its DNA target in vitro and transcriptional activation efficiency in planta. Several point mutations in the VIP1 response element VRE affected the VIP1 activity, and a strong correlation between VIP1-VRE binding and transcriptional activation levels was observed. Promoter activation by VIP1 was influenced by bacterial and plant proteins known to interact with VIP1 during Agrobacterium infection, i.e., VirE2, VirF and VIP2. VirF, an F-box protein, strongly decreased VIP1 transcriptional activation ability, but not its binding to VRE in vitro, most likely by triggering proteasomal degradation of VIP1. Finally, activation of a VRE-containing promoter was observed in dividing cells, probably resulting from activation of endogenous VIP1.
VIP1 (VirE2 interacting protein 1) is a plant basic leucine zipper (bZIP) domain transcription factor initially identified as an interactor of the virulence protein E2 (VirE2) of Agrobacterium1. VirE2 plays an important role during Agrobacterium-mediated genetic transformation of host cells by packaging the bacterial transferred DNA (T-DNA) into a nucleoprotein transfer complex (T-complex) and interacting with VIP1 in the course of several critical events of the infection process1,2,3,4. Specifically, VIP1 is thought (i) to enhance the entry of the T-complex into the host cell nucleus via VIP1 interactions with the importin alpha-dependent nuclear import machinery1,5,6; (ii) to mediate the T-complex targeting to the host chromatin via VIP1 interactions with core histones3,7; and (iii) to present the T-complex to the proteasomal degradation machinery for uncoating via VIP1 interactions with the bacterial and/or host F-box proteins, VirF and VBF, respectively4,8,9.
Whereas it appears that Agrobacterium has evolved to exploit VIP1 for infection purposes, the natural function of VIP1 is informed from several recent studies showing its involvement in responses to different types of biotic and abiotic stresses10,11,12. For example, VIP1 has been shown to participate in defence signalling, and, particularly, to act as substrate for the mitogen-activated protein kinase (MAPK) MPK3. When phosphorylated by MPK3, VIP1–which normally partitions between the cell cytoplasm and the nucleus–becomes largely nuclear11, presumably allowing it to activate its target genes. Interestingly, enhancement of VIP1 nuclear uptake is also involved in transcriptional regulation of plant osmosensory signalling genes CYP707A1 and CYP707A310. In the MPK3 pathway, the VIP1 target genes include Trxh8 and MYB4412. The promoters of these latter genes, as well as others that respond to activation by the MPK3 pathway, were shown to contain a DNA hexamer motif that acts as the VIP1 response element (VRE)12. VIP1 specifically binds to VRE, and strongly enhances expression of a synthetic promoter harbouring multiple VRE copies12.
Here, we analysed in further detail the VIP1-VRE interaction and identified VRE nucleotides important for VIP1 binding and promoter activation. We then showed that major Agrobacterium effector proteins known to interact with VIP1 can modulate its VRE transcriptional activation ability.
Results
Activation of a VRE-containing promoter by VIP1 in planta
Functional studies of the VIP1-VRE interaction require a simple and reliable system for detection of VIP1-mediated transcriptional activation directly in living plant tissues. We developed such a system by constructing an artificial promoter that contained a direct tandem repeat of the VRE1 sequence12, followed by the CaMV 35S minimal promoter13, i.e., a 46-bp fragment containing the TATA box and located immediately upstream of translation initiation codon. This promoter was then used to drive expression of two different reporters, GFP and an intron-containing beta-glucuronidase (GUSintron)14 (Fig. 1A). Unlike previous studies focused on the VRE function in plant protoplasts12, we introduced these expression constructs into plant tissues, either transiently by agroinoculation into tobacco leaves, or stably, in transgenic tobacco plants.
Figure 1. Activation of VRE1-containing promoter in planta.

(A) composition of promoter activation reporter constructs. GFP and GUSintron reporters are driven by the synthetic VRE1-35Smin promoter, composed of two copies of VRE1 (indicated in red), and the 35S minimal promoter (indicated in green), corresponding to 46 bp before the transcription start and containing the TATA box (underlined). (B) activation of VRE1-35Smin-GUSintron reporter. The reporter expression was detected as indigo-blue staining of GUS activity in leaf discs from tobacco transiently cotransformed with the VRE1-35Smin-GUSintron reporter and either an empty pPZP-RCS2 vector (right panel) or pRCS2-VIP1 (right panel). (C) activation of VRE1-35Smin-GFP. GFP expression was detected by confocal microscopy analysis of leaves from the VRE1-35Smin-GFP transgenic tobacco infiltrated with buffer alone (mock inoculation, right panel), transiently transformed an empty pPZP-RCS2 vector (centre panel) or with pRCS2-VIP1 (left panel). GFP is in green, plastid autofluorescence is in red. All images are single confocal sections.
Fig. 1A shows very large areas of GUS histochemical staining on a leaf disk derived from the plant that was transiently co-transformed by agroinoculation with the VRE1-35Smin-GUSintron reporter construct and a VIP1-expressing construct. No GUS expression at all was observed when the reporter construct was co-agroinoculated with the same expression vector, but lacking the VIP1 sequence (empty vector); furthermore, that no GUS staining was observed at the cut edges of the leaf disk indicated that the VRE element was not induced by tissue wounding. Similarly, transient expression of VIP1 in the VRE1-35Smin-GFP transgenic tobacco plants activated expression of the GFP reporter, whereas agroinoculating the empty vector into these transgenic tissues or mock-inoculating them elicited no GFP expression (Fig. 1C). That the endogenous VIP1 did not detectably activate the reporter is consistent with the known naturally low levels of this protein in plant cells1,5. Collectively, the data in Fig. 1B, C, therefore, indicate that the VRE element indeed activates gene expression in the presence of VIP1, and that this control is stringent, i.e., no expression occurs in the absence of VIP1, and specific, i.e., virtually no activation is achieved by wounding or bacterial challenge. These observations also demonstrate that our reporter constructs can be used for simple and specific detection of VIP1-mediated transcriptional activation of the VRE element in plant tissues.
Binding of VIP1 to VRE in vitro
Next, we developed a simple quantitative assay for the VIP1-VRE binding. To this end, we adapted the DNA-Protein-Interaction (DPI)-ELISA technology15. In this method, biotin-labelled DNA probes are bound onto streptavidin-coated 96-well plates; then, binding of VIP1 to DNA probes is detected by anti-VIP1 primary antibody and followed by alkaline phosphatase-conjugated secondary antibody. Based on this rationale, we produced a DNA probe that essentially replicated the VRE1-35Smin-GFP transgene used to detect the VRE response to VIP1 in planta (see Fig. 1A); specifically, it contained the VRE1-35Smin promoter and the full-length GFP coding sequence with a covalently-attached biotin molecule (Fig. 2A). When this probe was incubated with purified recombinant histidine-tagged VIP1 (Fig. 2B), protein-DNA binding was observed (Fig. 2C). Note that our previous studies utilizing histidine-tagged VIP1 demonstrated that this the tagged protein retains its known biological activities, such as specific binding to VirE2 and VirF3. This assay produced only low levels of signal with either no probe or with a non-specific GFP probe, lacking the VRE sequence (Fig. 2C); thus, in all subsequent experiments, the absorbance values obtained with the non-specific GFP probe were subtracted from the total experimental measurements.
Figure 2. VIP1 binding to VRE1 in vitro.

(A) schematic representation of DNA probe, containing the VRE1-35Smin synthetic promoter (indicated in black), GFP coding sequence (indicated in green), and a covalently attached biotin molecule (indicated in yellow). (B) SDS PAGE analysis of purified VIP1. The gel was stained with Coomassie blue. The position of the VIP1 protein band (right lane) is indicated by arrowhead; protein molecular mass standards (left lane) are indicated in kDa. (C) VIP1 binding to VRE1-35Smin-GFP is slightly decreased in the presence of magnesium ions. (D) binding competition by unlabeled VRE1-35Smin-GFP or GFP. Standard deviations are indicated.
The specificity of VIP1-VRE binding was directly demonstrated by competition experiments, using unlabelled competitor DNA6. The results in Fig. 2D show that significant, 80–95%, inhibition of the interaction was achieved in the presence of increasing amounts of the specific VRE1-35Smin-GFP competitor, i.e., 1.0 and 2.5 molar excess, respectively, whereas no such inhibitory effect was observed with 2.5 molar excess of a non-specific GFP competitor.
For some bZIP proteins, such as cAMP responsive element-binding protein (CREB), magnesium ions played a major role in its interaction with the target DNA, enhancing the binding up to 25 fold16. Thus, we tested whether the VIP1-VRE binding also was enhanced by magnesium ions. Fig. 2C shows that this was not the case. In fact, increasing the concentration of the magnesium ions even inhibited binding, potentially via ionic screening. Thus, unlike CREB, VIP1 most likely does not trap magnesium ions in the binding interface with DNA.
Effects of mutations in VRE on VIP1 binding and promoter activation
Having established binding and transcriptional activation assays for the VIP1-VRE interaction, we set out to use this methodology to examine the role of individual VRE nucleotides in VIP1 binding and subsequent function. First, we aimed to assess the importance of nucleotides flanking the core consensus hexamer. Indeed, whereas they are not conserved and not absolutely required for the binding of bZIP proteins to their target sequence, these nucleotides have been shown to affect the specificity of DNA binding of several other bZIP proteins17,18. Using the VRE1 sequence as reference12, we designed a similar construct harbouring modified VRE versions. VRE2, corresponding to the VIP1 response element found in the MYB44 gene promoter region12, differs from VRE1 in the T1G and C10A substitutions, while VRE3 harbours a T1G substitution. Fig. 3B shows that these mutated VRE sequences differed in their ability to be recognized by and bind VIP1. VIP1 binding to VRE2 was reduced by about 25% relative to VRE1 whereas binding to VRE3 was essentially unaffected. That VRE1 and VRE3 displayed similar levels of VIP1 binding suggests that the cytosine nucleotide in position 10 enhances VIP1 binding efficiency (relative to VRE2), whereas the presence of a thymidine or guanosine nucleotides in position 1 produces similar binding levels. Further, we modified the VRE3 sequence by inserting two additional substitutions in the core consensus VRE sequence, A4T and T7A, resulting in VRE4. VRE4 almost completely lost its ability to interact with VIP1. Previous studies indicated that multiple mutations in the VRE core consensus sequence disrupt VIP1 binding and transcriptional activity12. Importantly, however, these mutations also affected the palindromic structure of the VRE sequence, while, in VRE4, the A4T substitution restored the palindromic structure.
Figure 3. Effects of mutations in VRE on VIP1 binding and promoter activation.

(A) sequences of VRE1 and its VRE2-4 mutants. The mutated nucleotides are indicated in red. (B) VIP1 binding to the VRE-35Smin-GFP probe containing VRE1-4 sequences. (C) VIP1 activation of the transiently expressed VRE-35Smin-GFP reporter containing VRE1-4 sequences. GFP is in green, plastid autofluorescence is in red. All images are single confocal sections. (D) quantification of VIP1-induced expression of VRE-35Smin-GFP containing VRE1-4 sequences. GFP signal was calculated as percent of the signal measured with the VRE1-35Smin-GFP reporter, which was defined as 100% signal. All quantified data are shown as mean of three experiments with indicated standard deviations; standard deviation for measurements of the VRE1-35Smin-GFP reporter itself was 9.6%.
These binding data paralleled the biological functionality of the VRE sequence in the promoter activation assay (Fig. 3C, D). Based on the levels of the GFP reporter expression, VRE2 activation was about 30% lower than the VRE1 control, but VRE3 was activated more efficiently, by about 20%. VRE4 activation capacity was very low, about 10% of the VRE1 control.
Effect of coexpression of VIP1 interactors
Besides acting as transcriptional activator of plant defence and stress response genes10,11,12, VIP1 represents one of the major host factors involved in several key steps of plant infection and genetic transformation by Agrobacterium1,4,5,11,19,20. During these activities, VIP1 interacts with several other bacterial and plant proteins, such as VirE21, VirF4, and VIP221, and these interactions are presumed to occur when VIP1 is associated with the chromatin or even bound to DNA. Thus, we set out to assess whether VIP1 retains its transcriptional activation and VRE binding activities in the presence of VirE2, VirF, or VIP2.
In our transcriptional activation assay, we transiently expressed the VRE1-35Smin-GFP reporter construct together with VIP1 alone or VIP1/VirE2, VIP1/VirF, or VIP1/VIP2 pairs; to standardize expression, the tested proteins were coexpressed from the same vector. Fig. 4A shows that the presence of VirE2 did not significantly alter expression of the reporter, whereas coexpression of VirF with VIP1 had a pronounced inhibitory effect. Similarly to VirE2, VIP2 did not affect the reporter expression in a major way when coexpressed with VIP1. Quantification of these data (Fig. 4B) demonstrated that the presence of VirE2 or VIP2 only slightly, i.e., by 10–15%, decreased the activation efficiency of VIP1, but coexpression of VirF reduced this VIP1 activity by more than 50%.
Figure 4. Effect of VIP1 interactors on promoter activation and VRE1 binding by VIP1.

(A) VIP1 activation of the transiently expressed VRE1-35Smin-GFP reporter in the presence of coexpressed VirE2, VirF, and VIP2. GFP is in green, plastid autofluorescence is in red. All images are single confocal sections. (B) quantification of VIP1-induced expression of VRE-35Smin-GFP in the presence of coexpressed VirE2, VirF, and VIP2. GFP signal was calculated as percent of the signal measured with the VRE1-35Smin-GFP reporter coexpressed with VIP1 without interactors, which was defined as 100% signal. All quantified data are shown as mean of three experiments with indicated standard deviations; standard deviation for measurements of the VRE1-35Smin-GFP reporter itself was 11.0%. (C) VIP1 binding to VRE1-35Smin-GFP is not affected by the presence of VirF.
Does VirF interfere with VIP1 binding to VRE to produce this inhibitory effect on transcriptional activation ability of VIP1? To address this question we assayed the VIP1-VRE1 binding in vitro. Comparison between the binding activity of purified recombinant histidine-tagged VIP1 incubated with the VRE1-35Smin-GFP probe alone or in the presence of equimolar amount of purified recombinant histidine-tagged VirF detected only minor, less than 15%, decrease in VRE binding (Fig. 4C). As mentioned above, our earlier studies using similarly tagged and purified VIP1 and VirF demonstrated their protein-protein interaction functionality3. Thus VirF, similarly to VirE2 and VIP2, has no significant effect on VIP1 recognition of VRE. Instead, VirF, which is known to destabilize VIP1 via proteasomal degradation4,8, most likely depletes the amounts of VIP1 in the expressing cells.
Induction of VRE activity by cell division
Besides its functions in defence and stress responses10,11,12, VIP1 is induced during cell dedifferentiation22, which occurs in the course of cell division elicited by growth regulators. Thus, we examined whether induction of cell division by cytokinin and auxin treatments activates the VRE1-35Smin-GUSintron reporter. To this end, leaf explants from the VRE1-35Smin-GUSintron transgenic tobacco plants were grown on a solid medium supplemented with BAP (6-benzylaminopurine) and NAA (naphthalene-1-acetic acid). Fig. 5 shows that, after two weeks of cultivation, the GUS reporter activity was observed in the areas of active callus formation (panels B, C, arrowheads), which indicate the zones where cell division is induced; in control experiments, in which the leaf disks were cultivated in the absence of hormones, no reporter expression or callus formation were observed (panel A). Because these experiments did not involve coexpression of VIP1, the reporter expression, and thus activation of the VRE-containing promoter, was most likely induced by the endogenous VIP1 activated by cell division.
Figure 5. Effect of cell division on activity of VRE1-containing promoter.
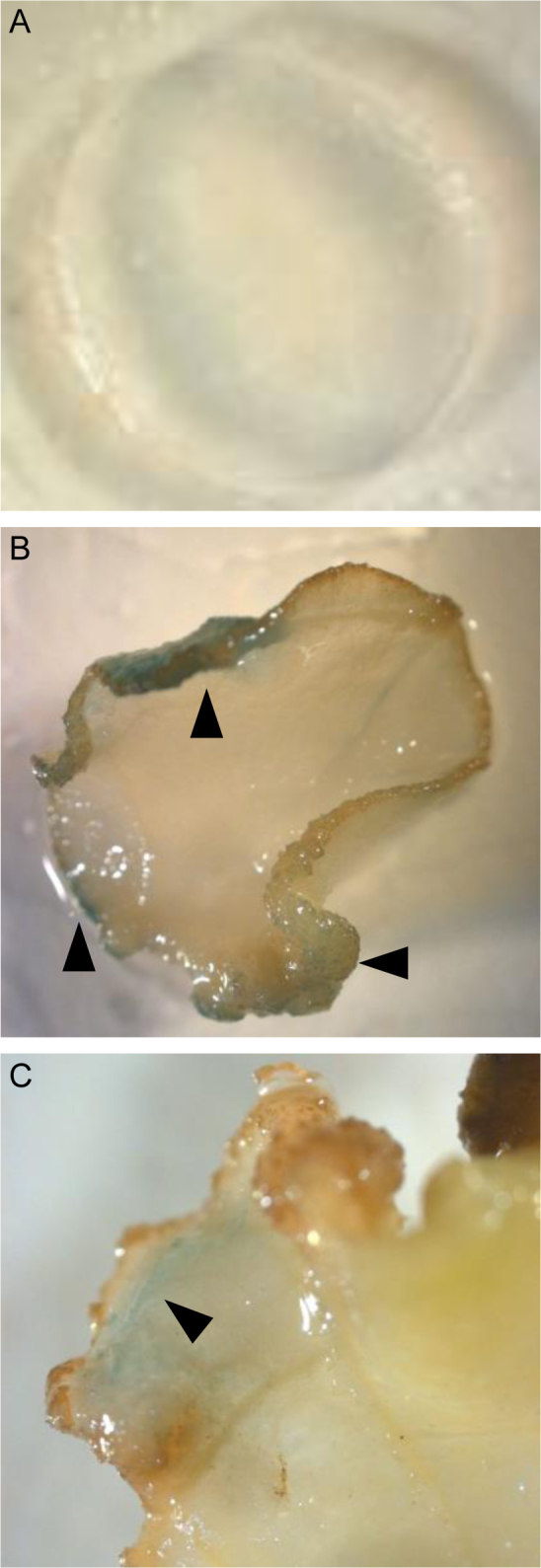
(A) leaf disk from a VRE1-35Smin-GUSintron transgenic tobacco plant cultivated in the absence of hormones. (B) (C) leaf disks from a VRE1-35Smin-GUSintron transgenic tobacco plant cultivated in the in the presence of BAP and NAA. Areas of GUS activity, detected as indigo-blue staining, are indicated by arrowheads.
Discussion
VIP1 is a multifunctional protein involved in several critical aspects of plant interactions with biotic and abiotic environment, such as defence response and osmosensory signalling10,11,12. Evolutionarily, this central role of VIP1 in plant defence makes it a likely target for at least some of the invading pathogens. Indeed, Agrobacterium is thought to subvert some of the VIP1 activities, i.e., import into the nucleus, targeting to the chromatin, and serving as a substrate for the ubiquitin/26S proteasome system (UPS)1,3,4,7, to facilitate its own infection. Thus, it would be useful to understand better the natural VIP1 function, i.e. its binding to the target gene response element (VRE) and subsequent transcriptional activation, and then to examine how they might be affected by interactions with bacterial and cellular factors known to associate with VIP1 during genetic transformation by Agrobacterium. To this end, we have developed methods to assay the VIP1-VRE binding in vitro and VIP1-induced activation of expression of VRE-containing reporter genes in planta. We then used different VRE mutants to demonstrate direct correlation between the protein-DNA binding levels and the resulting transcriptional activity observed in plant tissues. These experiments also revealed the functional significance of the DNA sequences immediately flanking the VRE core consensus. Specifically, the conserved VRE hexamer, ACNGCT, is sufficient for VIP1 binding12, and mutations within this element almost completely disrupt VIP1 binding and transcriptional activation. The nucleotides located outside of and adjacent to the VRE core, on the other hand, had a much milder effect both on the VIP1-VRE binding and on the induction of transcriptional activity. Thus, different target genes of VIP1 may be activated at different levels, depending on the nature of the VRE flanking nucleotides, which therefore might function to fine-tune the VIP1-mediated transcriptional activation. These data are consistent with previous observations that nucleotides at the periphery of the conserved target sequences may modify binding affinity of several other bZIP proteins17.
How are these natural activities of VIP1 influenced by its recruitment by Agrobacterium for molecular reactions of the infection? To address this question, we investigated whether VIP1 is still able to act as a transcriptional activator in presence of its protein interactors involved in Agrobacterium-mediated genetic transformation. These experiments demonstrated that VirE2 and VIP2 interfered with the VIP1 activities only slightly. Whereas the effect of free VirE2 on VIP1 may differ from that of the ssDNA-bound VirE2, as in the bacterial T-complex, our previous study indicates that the VIP1-VirE2 interaction occurs with the free as well as with the ssDNA-bound VirE2 to a comparable degree3. That neither VirE2 nor VIP2 markedly impaired the transcriptional activation activity of VIP1 may be because binding of these proteins to the VIP1 molecule does not significantly affect its domains involved in recognition of VRE and/or activation of transcription. Alternatively, the affinity of VIP1 to VRE might be higher than that toward VirE2 or VIP2, shifting the binding equilibrium toward the formation of VIP1 complexes with DNA rather than its protein interactors. In contrast, the presence of VirF substantially reduced the ability of VIP1 to induce target gene transcription, yet had no major effect on the VIP1-VRE binding. Thus, VirF most likely reduced the overall cellular levels of VIP1 by targeting it to degradation via the SCFVirF pathway, which represents a well-known function of VirF during Agrobacterium infection4,8. In this scenario, VirF would not only act to uncoat the T-complex4,8, but also mitigate the induction of the host defence genes mediated by VIP1.
Our observations also suggest that the activity of VIP1 itself is regulated in a developmentally controlled manner. This is inferred from activation of the VRE element by induction of cell division; potentially, when cells divide, their pool of endogenous VIP1 is increased and/or activated, which in turn activates the VRE-regulated target genes. Potentially, this could be achieved in two ways, both of which have been reported for VIP1: enhancement of nuclear import, and, by implication, transcriptional activity, by phosphorylation11, or simply upregulation of the VIP1 gene transcription22. The latter scenario is consistent with the known increase in VIP1 transcription levels upon cell dedifferentiation22. Collectively, our data shed new light on the function of VIP1 as a transcriptional activator and emphasize how this function fits into participation of VIP1 in the reactions of Agrobacterium-mediated genetic transformation of plant cells.
Methods
Plasmids
For protein expression in E. coli, the full-length VIP1 coding sequence from Arabidopsis thaliana ecotype Col0 (At1G43700) was PCR-amplified from a cDNA library, using the primer pair 5′CCGGAATTCATGGAAGGAGGAGGAAGAGGAC3′/5′CCGCTCGAGTCAGCCTCTCTTGGTGAAATCCATGTAGC3′, and cloned into the EcoRI-XhoI sites of pET-28a(+) (Clontech), resulting in pET28-VIP1. The virF gene from the Agrobacterium strain 15955 was PCR-amplified, using the primer pair 5′CGCGGATTCCGATGAGAAATTCGAGTTTGCGTG3′/5′CGCGTCGACTAGACCGCGCGTTGATCG3′, and cloned into the BamHI-SalI sites of pET28c(+) (Novagen), resulting in pET28-virF.
For VRE-controlled transient expression in plants, a new mini-binary vector was constructed in the following sequential steps. First the pCB302 plasmid23 was PCR-amplified using the primer pair 5′CGCACCGGTAGATTGTCGTTTCCCGCCTTCAG3′/5′GCCGGGCCCCAGTACATCAAAAACGTCCGCAATG3′ and self-ligated, resulting in a plasmid, designated pCB302T, with the pCB302 backbone that includes the T-DNA borders, the restriction sites AgeI and PspOMI between these borders, but lacks the original pCB302 expression cassette. The expression cassette, i.e., the 35Spromoter-MCS-35Sterminator sequence, from the pSAT5A plasmid24 was then inserted as an AgeI-NotI fragment into the AgeI-PspOMI sites of pCB302T, forming pCB302T-MCS. Next, the VRE-35Smin-GFP sequences were produced by PCR amplification, using 35Smin-GFP as template, with forward primers specific for each VRE variant, i.e., 5′CGCACCGGTTACAGCTGTCTACAGCTGTCATGGCAAGACCCTTCCTC3′ (VRE1), 5′CCGACCGGTGACAGCTGTAGACAGCTGTAATGGCAAGACCCTTCCTC3′ (VRE2), 5′CGCACCGGTGACAGCTGTCGACAGCTGTCATGGCAAGACCCTTCCTC3′ (VRE3), 5′CGCACCGGT GACTGCAGTCGACTGCAGTCATGGCAAGACCCTTCCTC3′ (VRE4), and a common reverse primer 5′CCGGAATTCTTACTTGTACAGCTCGTCCATGC3′. Then, each VRE-35Smin-GFP sequence was subcloned into the AgeI-EcoRI sites of pCB302T-MCS, replacing the 35S promoter and resulting in the series of pCB302T-VRE1-4-GFP plasmids. Using the same cloning strategy, the VRE1-35Smin-GUSintron sequence was PCR-amplified with the VRE1-specific forward primer 5′CGCACCGGTTACAGCTGTCTACAGCTGTCATGGCAAGACCCTTCCTC3′, reverse primer 5′CCGGAATTCTCATTGTTTGCCTCCCTGCTGC3′, and 35Smin-GUSintron as template, and inserted into pCB302T-MCS, resulting in pCB302T-VRE1-GUSintron.
For transient expression of VIP1, virE2, virF and VIP2, these genes were first inserted into the pSAT plasmids. Specifically, VIP1 was inserted as PCR-amplified EcoRI-XhoI fragment into the EcoRI-SalI sites of pSAT5A-MCS24, resulting in pSAT5A-VIP1. The nopaline-type virE2 was PCR-amplified from pTiC58 with the primer pair 5′GGAAGATCTATGGATCCGAAGGCCGAAGGCAATG3′/5′CGCGTCGACCTACAGACTGTTTACGGTTGGGC3′ and inserted into the BglII-SalI sites of pSAT1A-MCS24, resulting in pSAT1A-virE2. The octopine-type virF was subcloned as SalI-BamHI fragment from pRTL2-GFP-virF4 into the same sites of pSAT1A-MCS, resulting in pSAT1A-virF. And VIP2 was PCR-amplified from an Arabidopsis cDNA library with the primer pair 5′CGCGGATCCATGTCAAACCTTCATTCATCTCTCAATG3′/5′CGCGTCGACTCAAAGCTGCAGCAAGCTTGGTC3′ and inserted as BamHI-SalI fragment into the BglII-SalI sites of pSAT1A-MCS, resulting in pSAT1A-VIP2. Then, the VIP1 expression cassette was transferred as an ICeuI fragment from pSAT5A-VIP1 into the same site of pPZP-RCS225, forming pRCS2-VIP1, after which, the virE2, virF and VIP2 expression cassettes were transferred as AscI fragments from pSAT1A-virE2, pSAT1A-virF and pSAT1A-VIP2, respectively, into the same site of pRCS2-VIP1, resulting in pRCS2-VIP1-virE2, pRCS2-VIP1-virF, and pRCS2-VIP1-VIP2, respectively. For stable expression of VRE1-GFP and VRE1-GUSintron in plants, the corresponding expression cassettes were PCR-amplified with the primer pair 5′CCCAAGCTTACCGGTTACAGCTGTCTACAGCTGTCATGG3′/5′GGAAGATCTGAGCTCGCGGCCCCGGCGTCACGTGATTTTG3′ using pCB302T-VRE1-GFP and pCB302T-VRE1-GUSintron, respectively, as templates and inserted as HindIII-SacI fragments into the same sites of pBIN1926, resulting in pBIN19-VRE1-GFP and pBIN19-VRE1-GUSintron, respectively. All PCR amplifications were performed using the proofreading DNA polymerase Pfu (Agilent), and all generated clones were verified by DNA sequencing.
VIP1 and VirF purification
Recombinant histidine-tagged VIP1 and VirF proteins were purified as described in3, with modifications. Proteins were expressed in the E. coli strain BL21(DE3) (Novagen) from the pET28-VIP1 or pET28-virF plasmid, respectively, and extracted using a 10 mM phosphate buffer, pH 8, supplemented with 1 M NaCl and 4 M urea, in the presence of 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1 mM β-mercaptoethanol. Protein extract was adsorbed onto a nickel-agarose resin (Qiagen) and partially renatured on the resin by sequential washes with 10 column volumes of 10 mM phosphate buffer, pH 8, containing 1 mM PMSF, 10 mM imidazole, and 1 mM β-mercaptoethanol and supplemented with 1 M NaCl and decreasing concentrations, i.e., 4 M, 3 M, 2 M, and 1 M, of urea. Bound VIP1 or VirF was then eluted with the wash buffer containing 1 M urea and 250 mM imidazole, dialyzed overnight at 4°C against 2,000 volumes of 4 mM HEPES, pH 7.5, 100 mM KCl, 8% (v/v) glycerol, 0.2% bovine serum albumin (BSA), 5 mM dithiothreitol (DTT), and 1 mM PMSF, and aliquoted and stored at −80°C until use. The absence of significant contamination was confirmed by SDS polyacrylamide gel electrophoresis (PAGE) on Coomassie blue-stained 12.5% SDS-polyacrylamide gels as described1. All experiments used the same protein preparation batch, making data comparison more meaningful.
Preparation of biotinylated DNA probes
The EGFP coding sequence was PCR-amplified, using Pfu polymerase (Agilent), with a 5′-biotinylated reverse primer 5′/5Biosg/TTACTTGTACAGCTCGTCCATGC3′ (Integrated DNA Technologies) and forward primers with or without the VRE1-4 sequences, thereby producing probes that corresponded to the full-length GFP with a biotin molecule downstream of GFP and with or without VREs upstream of GFP. Similar probes with a non-biotinylated reverse primer were produced for binding competition experiments. All probes were purified using Zymoclean gel DNA purification kit (Zymoresearch) according to manufacturer's instructions; this method eliminates free biotinylated primer.
In vitro protein-DNA binding assay
The binding of VIP1 to VRE was assayed as described in15, with modifications. A 96-well plate (Nunc, Maxi-Sorp, 442404) was coated with streptavidin (Sigma S4762) by placing 60 μL of a 10-μg.mL−1 streptavidin solution in each well and incubating the plate uncovered at 37°C until the water was completely evaporated, typically, for 6 to 8 h. The wells were then blocked overnight at 4°C with 300 μL/well of biotin-free 5% BSA (Sigma A7906) in TBST (10 mM Tris-HCl, 150 mM NaCl, pH 8.0, 0.05% (v/v) Tween-20). Next, 2 pmoles of biotinylated DNA probe in 60 μL TBST were added to each well and incubated for 1 h at 37°C. After 3 washes with 300 μL TBST per well, a second blocking step was performed with 300 μL/well of 5% non-fat dry milk (BioRad, 170-6404) in TBST, followed by three additional washes with TBST. Purified VIP1, 1 μg protein per well in 60 μL of 4 mM HEPES, pH 7.5, 100 mM KCl, 8% (v/v) glycerol, 0.2% BSA, 5 mM DTT, was added for 1 h at room temperature, followed by three washes with 300 μL TBST per well. When necessary, MgCl2 or unlabeled DNA probe at the concentrations indicated for each specific experiment were preincubated for 20 min at 4°C with the VIP1 solution prior to the binding step.
For the VIP1 binding in presence of VirF, approximately equimolar amounts of VIP1 and VirF, i.e., 1.0 μg of and 1.7 μg, respectively, per well, were preincubated for 20 min at 4°C; the preincubation was performed before the DNA binding step because our objective was to examine whether this protein-protein interaction affects the binding between VIP1 and VRE.
DNA-bound VIP1 was detected using rabbit anti-VIP1 antibody (1:1,000 dilution in TBST) followed by goat anti-rabbit secondary antibody conjugated to alkaline phosphatase (Sigma-Aldrich, 1:500 dilution in TBST). Photometric detection of alkaline phosphatase was performed using p-nitrophenyl phosphate (Sigma, N2770) as substrate according to manufacturer's instructions and measuring the absorbance at 405 nm with a FluoStar Optima plate reader (BMG). For all experiments, three measurements were done for each condition, and each experiment was repeated two or three times. Results are presented as average values with standard deviations.
Plants
Tobacco plants (Nicotiana tabacum, var. Turk) were grown either in soil or on MS medium (10 g.L−1 sucrose, 8 g.L−1 agar) after seed surface sterilization, and maintained in vitro by micro-cuttings on high sucrose MS medium (30 g.L−1 sucrose, 8 g.L−1 agar). All plants were grown in an environment-controlled growth chamber under long day (16 h light/8 h dark) conditions and at 22°C.
Transgenic plants
Transgenic tobacco plants were produced by the classical leaf disc protocol27, using the EHA105 strain of Agrobacterium tumefaciens carrying either pBin19-VRE1-GUSintron or pBin19-VRE1-GFP binary construct. The resulting transformants were selected on MS regeneration medium (30 g.L−1 sucrose, 8 g.L−1 agar, 10 g.L−1 BAP, 1.0 g.L−1 NAA), containing 50 mg.L−1 timentin and 50 mg.L−1 kanamycin, and then transferred to MS rooting medium (30 g.L−1 sucrose, 8 g.L−1 agar).
For induction of calli and, hence cell division, leaf discs from the VRE1-GUSintron transgenic tobacco plants were cultured on MS medium supplemented with 10 g.L−1 BAP and 1.0 g.L−1 NAA for 2 to 3 weeks. The experiment was repeated three times using two leaf discs per experiment.
In planta promoter activation assay
Agrobacterium strain EHA105 was transformed with the binary construct pCB302T-VRE1-GFP or pCB302T-VRE1-GUSintron and one of the constructs pRCS2-VIP1, pRCS2-VIP1-virE2, pRCS2-VIP1-virF, or pRCS2-VIP1-VIP2, grown overnight at 25°C, and agroinfiltrated into intact N. tabacum leaves as described28. The agroinfiltrated tissues were viewed under a Zeiss LSM 5 Pascal confocal laser scanning microscope for detection of GFP expression, or subjected to GUS histochemical assay, as described29. GFP signal was quantified using the LSM Pascal software (Zeiss) by measuring the total GFP fluorescence in one field inside the infiltration area with a low magnification objective (10×); all images used for fluorescence measurement were taken with the same settings. Basal signal measured in area infiltrated with VRE1-35Smin-GFP alone was subtracted from the values measured for each experimental condition. For each vector combination, three agroinfiltrations were performed on three different leaves, and the GFP fluorescence of three microscope fields was measured in each infiltration area; each experiment was repeated twice.
Author Contributions
B.L. designed experiments, performed experiments and wrote manuscript. V.C. designed experiments and wrote manuscript.
Acknowledgments
We thank Dr. Carlos Camacho (University of Pittsburgh) for stimulating discussions and ideas. The work in our laboratory is supported by grants from NIH, NSF, USDA/NIFA, BARD, and BSF to V.C.
References
- Tzfira T., Vaidya M. & Citovsky V. VIP1, an Arabidopsis protein that interacts with Agrobacterium VirE2, is involved in VirE2 nuclear import and Agrobacterium infectivity. Embo J 20, 3596–3607 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citovsky V. et al. Biological systems of the host cell involved in Agrobacterium infection. Cell Microbiol 9, 9–20 (2007). [DOI] [PubMed] [Google Scholar]
- Lacroix B., Loyter A. & Citovsky V. Association of the Agrobacterium T-DNA-protein complex with plant nucleosomes. Proc Natl Acad Sci U S A 105, 15429–15434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzfira T., Vaidya M. & Citovsky V. Involvement of targeted proteolysis in plant genetic transformation by Agrobacterium. Nature 431, 87–92 (2004). [DOI] [PubMed] [Google Scholar]
- Tzfira T., Vaidya M. & Citovsky V. Increasing plant susceptibility to Agrobacterium infection by overexpression of the Arabidopsis nuclear protein VIP1. Proc Natl Acad Sci U S A 99, 10435–10440 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citovsky V. et al. Protein interactions involved in nuclear import of the Agrobacterium VirE2 protein in vivo and in vitro. J Biol Chem 279, 29528–29533 (2004). [DOI] [PubMed] [Google Scholar]
- Loyter A. et al. The plant VirE2 interacting protein 1. a molecular link between the Agrobacterium T-complex and the host cell chromatin? Plant Physiol 138, 1318–1321 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magori S. & Citovsky V. Agrobacterium counteracts host-induced degradation of its effector F-box protein. Sci Signal 4, ra69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaltsman A., Krichevsky A., Loyter A. & Citovsky V. Agrobacterium induces expression of a host F-box protein required for tumorigenicity. Cell Host Microbe 7, 197–209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugama D., Liu S. & Takano T. A bZIP protein, VIP1, is a regulator of osmosensory signaling in Arabidopsis. Plant Physiol 159, 144–155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djamei A., Pitzschke A., Nakagami H., Rajh I. & Hirt H. Trojan horse strategy in Agrobacterium transformation: abusing MAPK defense signaling. Science 318, 453–456 (2007). [DOI] [PubMed] [Google Scholar]
- Pitzschke A., Djamei A., Teige M. & Hirt H. VIP1 response elements mediate mitogen-activated protein kinase 3-induced stress gene expression. Proc Natl Acad Sci U S A 106, 18414–18419 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey P. N. & Chua N. H. The cauliflower mosaic virus 35S promoter: combinatorial regulation of transcription in plants. Science 250, 959–966 (1990). [DOI] [PubMed] [Google Scholar]
- Narasimhulu S. B., Deng X. B., Sarria R. & Gelvin S. B. Early transcription of Agrobacterium T-DNA genes in tobacco and maize. Plant Cell 8, 873–886 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand L. H., Kirchler T., Hummel S., Chaban C. & Wanke D. DPI-ELISA: a fast and versatile method to specify the binding of plant transcription factors to DNA in vitro. Plant Methods 6, 25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M. A., Goodman R. H. & Brennan R. G. The structure of a CREB bZIP.somatostatin CRE complex reveals the basis for selective dimerization and divalent cation-enhanced DNA binding. J Biol Chem 275, 35242–35247 (2000). [DOI] [PubMed] [Google Scholar]
- Izawa T., Foster R. & Chua N. H. Plant bZIP protein DNA binding specificity. J Mol Biol 230, 1131–1144 (1993). [DOI] [PubMed] [Google Scholar]
- Jakoby M. et al. bZIP transcription factors in Arabidopsis. Trends Plant Sci 7, 106–111 (2002). [DOI] [PubMed] [Google Scholar]
- Zaltsman A., Lacroix B., Gafni Y. & Citovsky V. Disassembly of synthetic Agrobacterium T-DNA-protein complexes via the host SCF(VBF) ubiquitin-ligase complex pathway. Proc Natl Acad Sci U S A 110, 169–174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Krichevsky A., Vaidya M., Tzfira T. & Citovsky V. Uncoupling of the functions of the Arabidopsis VIP1 protein in transient and stable plant genetic transformation by Agrobacterium. Proc Natl Acad Sci U S A 102, 5733–5738 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand A. et al. Arabidopsis VIRE2 INTERACTING PROTEIN2 is required for Agrobacterium T-DNA integration in plants. Plant Cell 19, 1695–1708 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avivi Y. et al. Reorganization of specific chromosomal domains and activation of silent genes in plant cells acquiring pluripotentiality. Dev Dyn 230, 12–22 (2004). [DOI] [PubMed] [Google Scholar]
- Xiang C., Han P., Lutziger I., Wang K. & Oliver D. J. A mini binary vector series for plant transformation. Plant Mol Biol 40, 711–717 (1999). [DOI] [PubMed] [Google Scholar]
- Chung S. M., Frankman E. L. & Tzfira T. A versatile vector system for multiple gene expression in plants. Trends Plant Sci 10, 357–361 (2005). [DOI] [PubMed] [Google Scholar]
- Tzfira T. et al. pSAT vectors: a modular series of plasmids for autofluorescent protein tagging and expression of multiple genes in plants. Plant Mol Biol 57, 503–516 (2005). [DOI] [PubMed] [Google Scholar]
- Bevan M. Binary Agrobacterium vectors for plant transformation. Nucleic Acids Res 12, 8711–8721 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsch R. B. et al. Inheritance of functional foreign genes in plants. Science 223, 496–498 (1984). [DOI] [PubMed] [Google Scholar]
- Lacroix B. & Citovsky V. Extracellular VirB5 enhances T-DNA transfer from Agrobacterium to the host plant. PLoS One 6, e25578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J. et al. Identification of T-DNA tagged Arabidopsis mutants that are resistant to transformation by Agrobacterium. Mol Gen Genet 261, 429–438 (1999). [DOI] [PubMed] [Google Scholar]