

Abstract
Type II dihydrofolate reductase (DHFR) is a plasmid-encoded enzyme that confers resistance to bacterial DHFR-targeted antifolate drugs. It forms a symmetric homotetramer with a central pore which functions as the active site. Its unusual structure, which results in a promiscuous binding surface that accommodates either the Dihydrofolate (DHF) substrate or the NADPH cofactor, has constituted a significant limitation to efforts to understand its substrate specificity and reaction mechanism. We describe here the first structure of a ternary R67 DHFR•DHF•NADP+ catalytic complex, resolved to1.26 Å. This structure provides the first clear picture of how this enzyme, which lacks the active site carboxyl residue that is ubiquitous in Type I DHFRs, is able to function. In the catalytic complex, the polar backbone atoms of two symmetry-related I68 residues provide recognition motifs that interact with the carboxamide on the nicotinamide ring, and the N3-O4 amide function on the pteridine. This set of interactions orients the aromatic rings of substrate and cofactor in a relative endo geometry in which the reactive centers are held in close proximity. Additionally, a central, hydrogen-bonded network consisting of two pairs of Y69-Q67-Q67′-Y69′ residues provides an unusually tight interface, which appears to serve as a “molecular clamp” holding the substrates in place in an orientation conducive to hydride transfer. In addition to providing the first clear insight regarding how this extremely unusual enzyme is able to function, the structure of the ternary complex provides general insights into how a mutationally-challenged enzyme, i.e., an enzyme whose evolution is restricted to four-residues-at-a-time active site mutations, overcomes this fundamental limitation.
Keywords: R67 DHFR, Dihydrofolate Reductase, X-ray crystallography, ternary complex, Type II DHFR
Antifolate drug therapy plays a critical role in the treatment of pathogenic and neoplastic diseases. The evolution of a plasmid-encoded, Type II dihydrofolate reductase (DHFR) provides one mechanism for bacterial evasion of drugs such as trimethoprim that target the bacterial dihydrofolate reductase enzyme (1–4). Type II DHFR is an extremely unusual enzyme that exhibits no apparent structural or evolutionary relationship with the type I (chromosomal) enzyme. It is one of the smallest enzymes known to self-assemble into an active quaternary structure, forming a homotetramer consisting of four 78-residue peptides organized into a toroidal structure with a central pore (5). Structural studies indicate that both the substrate (5) and cofactor (6–8) bind in this central pore, which functions as the active site. While a shared active site between protomers is not surprising, the presence of only one active site per multimer is quite novel. This single active site pore is characterized by 222 symmetry, which presents fundamental problems for binding different ligands, as well as for enzyme catalysis. In particular, R67 DHFR lacks separately defined substrate and cofactor binding sites, and thus can form DHFR•(DHF)2 and DHFR•(NADPH)2 complexes as well as the active DHFR•DHF•NADPH ternary complex (9). Cooperative binding leads to a strong preference for the productive, catalytic complex. It has been unclear how an enzyme without specific substrate and cofactor binding sites is nevertheless able to catalyze a stereospecific hydride transfer reaction. A recent study by Alonso et al. (10) used a docking approach (AutoDock and FlexX) to model the ternary complex. Because of the large pore volume, numerous conformers were found. To sort through these alternate possibilities, they performed a comparative scoring analysis as well as molecular dynamics simulations. Ultimately, multiple binding modes for the ligands in R67 DHFR were proposed.
This type of active site structure also creates substantial evolutionary and mutational challenges to the enzyme. Since each mutation will alter four active site residues at a time, most of the evolutionary pressure that would normally optimize enzyme function is compromised by the need to balance the effects of substitutions at all four symmetry-related sites. For example, a residue substitution on one monomer, which might promote folate N5 protonation, may also interfere with NADPH binding when it is present on a symmetry-related chain. For the vast majority of oligomeric enzymes, this difficulty is simply avoided by allowing each monomeric unit to possess its own catalytic site. A second example of an oligomer that possesses a single active site is the dimeric AIDS protease (11).
The catalytic mechanism of R67 DHFR must also must differ significantly from that of the extensively studied chromosomal DHFRs, since the type II enzyme lacks an active site acidic residue which appears to facilitate hydride transfer from NADPH by elevating the pKa of bound DHF (12–15) or by promoting enolization of the pteridine N3-O4 in order to facilitate N5 protonation (16).
In addition to presenting fundamental problems in enzyme biochemistry, structural analysis of the Type II DHFR ternary complex poses some unique problems. The four-fold symmetric central pore contains asymmetrically bound ligands, which are presented as a four-fold superposition of partially occupied atom positions. The symmetry-related ligands overlap extensively with each other, which makes deconvolution of the single asymmetric structure extremely problematic. To our knowledge, this is the first example of deconvolution of overlapping densities describing two different ligands bound to a promiscuous binding surface.
The present study presents the first crystallographic data for a ternary Type II DHFR•DHF•NADP+ complex
In addition to confirming the relative endo orientation of the nicotinamide and pteridine ring systems deduced on the basis of interligand Overhauser effect (ILOE) studies (7), the results explain how the enzyme is able to use structurally similar binding sites to accommodate both NADP+ and DHF, exploiting subtle similarities between the two different molecules. The structure of the ternary complex demonstrates the critical significance of backbone interactions as a means for resolving the mutational challenge noted above. The structure also demonstrates the importance of a central, hydrogen bonded network formed from two pairs of Y69-Q67-Q67’-Y69’ residues that sandwich the substrates tightly in place, facilitating catalysis.
Methods
Recombinant R67 DHFR was purified as previously described (17). The 16 N-terminal residues were removed using chymotrypsin, yielding a fully active protein composed of 62-residue peptides (17). DHF and NADP+ were obtained from Sigma and used without additional purification.
Crystallization and data collection
Crystals were obtained using the hanging drop vapor diffusion technique at 4 °C by mixing 2 μl of protein solution at a concentration of about 15 mg/ml in 50 mM phosphate buffer, 1 mM EDTA, pH 8.0, with 2 μl of the reservoir solution consisting of 50% 2-methyl-2,4-pentanediol (MPD), 100 mM Tris at pH 8.0. Of the conditions screened, MPD appeared to be an absolute requirement for crystallization. Crystals grew to about 0.1 × 0.8 mm in 3–4 days. The crystals belong to the space group I4122, with a=b≈67.8 Å and c≈52.2 Å with one 62 amino-acid subunit per asymmetric unit (Vm=1.97 Å3 A.M.U.−1 and 40% solvent content).
The ternary DHFR•DHF•NADP+ complex was prepared by soaking a crystal in saturated (~1mM) DHF, 10 mM NADP+, 55% MPD, 10% ethylene glycol, 100 mM Tris at pH 8.0, degassed with nitrogen. The binary complex was prepared by soaking a crystal in 20 mM NADP+, 60% MPD, 100 mM Tris at pH 8.0. For data collection, crystals were frozen by submersion in liquid nitrogen and placed on the goniometer in a stream of nitrogen gas at −180 °C. Data were collected using a Rigaku 007HF X-ray generator equipped with Osmic VariMax HF mirrors and a Saturn 92 area detector.
Structure determination and refinement
The starting model for the apo-enzyme model was derived from the previously determined isomorphous structure, PDB code 1VIE (5), with the D2 center of symmetry shifted to the origin. The model was initially refined including all non-solvent protons with CNS 1.1 (18) and force-field restraints derived from AMBER98 (19). Ligand restraint parameters were derived from similar ligands from CNS, with additional parameters derived from small-molecule statistics of the Cambridge structure database (20). Final refinement was done with REFMAC5 using anisotropic temperature factors (21). The quality of the final structures was assessed using the programs Procheck (22) and Molprobity (23). Structural data has been deposited with the protein data bank (Table 1).
Table 1.
Data collection and refinement statistics
| Ternary | Binary | Apo | |
|---|---|---|---|
|
| |||
| PDB Accession code | 2RK1 | 2RK2 | 2RH2 |
| Data collection1 | |||
| Space group | I4122 | I4122 | I4122 |
| Cell dimensions | |||
| a, b, c (Å) | 67.8, 67.8, 52.2 | 67.5, 67.5, 52.1 | 67.81, 67.81, 51.97 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 1.26 (1.31–1.26)2 | 1.90 (1.97–1.90) | 0.96 (0.99–0.96) |
| Rmerge3 (%) | 7.2 (49.9) | 11.5 (21.9) | 10.0(65.7) |
| I / σI | 14.1 (3.0) | 13.0 (4.5) | 27.1 (3.6) |
| Completeness (%) | 99.6 (96.5) | 97.8 (83.7) | 96.8 (91.1) |
| Redundancy | 12.4 (5.4) | 6.4 (4.0) | 16.3 (6.7) |
| No. reflections | 15838 (1096) | 4883 (396) | 32858 (3331) |
| Refinement4 | |||
| Resolution (Å) | 1.26(1.29–1.26) | 1.90 (1.933–1.90) | 0.96 (0.99–0.96) |
| Rwork3 (%) | 12.4 (30.6) | 14.1 (18.7) | 10.4 (23.3) |
| Rfree3 (%) | 15.2 (37.6) | 20.0 (34.2) | 11.7 (24.8) |
| Number of atoms | |||
| Protein | 1034 | 530 | 1082 |
| Ligand | 61 | 48 | 0 |
| Solvent5 | 146 | 114 | 161 |
| Average B-factors | |||
| Protein | 12.4 | 13.2 | 8.6 |
| Ligand | 13.7 | 11.3 | - |
| Solvent5 | 32.0 | 34.3 | 23.6 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.016 | 0.023 | 0.016 |
| Bond angles (°) | 2.12 | 2.30 | 1.65 |
Each data set was obtained from a single crystal.
Values in parentheses represent statistics from the highest resolution shell.
R-factor values according to standard equations.
All statistics include hydrogens, where present (no hydrogens are present in the binary complex). Counts include all partial-occupancy sites. B-factor averages are weighted by occupancy.
Solvent includes MPD molecules.
RESULTS AND DISCUSSION
X-ray crystallography
Initial studies focused on the R67 DHFR•folate•NADP+ ternary complex, in order to avoid potential redox chemistry (see Figure 1 for atom labels). However, occupancy of folate in the active site was extremely low, as judged on the basis of electron density as well as the absence of yellow coloring. This likely results from the poor solubility of folate in high concentrations of 2-methyl-2,4-pentanediol (MPD). Crystals were successfully transferred to different stabilizing solutions, but these encountered similar folate solubility problems. Precipitants also appear to interfere with binding. Indeed, the structure determined in the presence of folate was found to be nearly identical to that subsequently determined using only NADP+. For this reason, ternary complex formation was then evaluated using various folate analogs. Of the folate and related analogs tested, dihydrofolate proved to be the most useful, and it was immediately apparent from the yellow color of the crystal that a higher level of occupancy of the DHF binding site was obtained. This probably results from the fact that DHF is the preferred substrate, and is also more soluble than folate in the MPD-containing medium.
Figure 1.

Structure and numbering of the dihydrofolate and NADP+ ligands present in the R67 DHFR•DHF•NADP+ ternary crystal structure.
The inherent D2 symmetry of the type II dihydrofolate reductase, which simplifies crystallographic analysis of the uncomplexed enzyme, results in a significant analytical problem for the ternary complex due to the presence of four-fold overlapping, symmetry-related ligand binding sites (Figure 2). The ternary DHFR•DHF•NADP+ complex is derived from four symmetry-related substrate arrangements, each having 25 % occupancy. Interpretation of the density requires a four-fold deconvolution, in which the individual ligand conformations initially are unknown. Although we anticipated the need for exhaustive ligand conformational searches in order to determine a reasonable model for the ligand positions, we were able to obtain interpretable electron density corresponding to the nicotinamide and pteridine portions of the molecules due to high resolution diffraction data, and parallel modes of ring moiety binding that results in additive electron density, simplifying the identification of the ring structures. The positions of bound NADP+ and the pteridine ring of DHF were ultimately determined with a high level of confidence (Figure 3), however, as in the initially reported folate complex (5) we were not able to identify electron density for the p-aminobenzoyl glutamate tail.
Figure 2.

The crystallographic problem posed by the DHFR•DHF•NADP+ ternary complex. A schematic, cross sectional view of R67 DHFR is shown in blue, with the pore shown in gray. The three perpendicular two-fold axes are indicated in red. On the left, a single pair of ligands is shown, while on the right, the four symmetry related pairs are indicated. In the crystal, each relative orientation is expected to have 25 % occupancy.
Figure 3.



Fits of electron density. a) Fit of the electron density at the center of the pore for the binary complex, revealing the nicotinamide rings for two of the four symmetry-related NADP+ positions. Electron density represents 2Fobs-Fcalc, contoured at 0.5σ in order to illustrate density for ligands at 25% occupancy. Each color (red, blue, green, and magenta) identifies a unique set of NADP+ or water ligands among the four symmetry-related groups which are averaged in the crystal. Two symmetry-related nicotinamide rings are shown. Since only one NADP+ binds per tetramer (9), the ligand is present at 25% occupancy at each position. Symmetry-related water molecules bind to each of the sites not occupied by NADP+, resulting in 75% occupancy. For example, the green, magenta and blue water molecules describe water seen in three symmetry related sites when the red colored cofactor is bound. The protein side chains at the top and bottom of the figure correspond to the NH and O backbone atoms from symmetry related Ile68 residues, which interact with the amide of the nicotinamide. b) Electron density for one side of the pore in the ternary complex (contoured the same as a), showing overlapping nicotinamide and pteridine groups. Color-coding is as in panel a, however in this case, the two ligands exclude several water molecules from two of the four sites. Electron density for the pyridine ring of the nicotinamide overlaps closely with that arising from the pyrazine ring of DHF. Note that these figures illustrate electron density for symmetry-related molecules, rather than for an interacting substrate-cofactor pair, which is discussed below. WAT114, positioned near the pteridine O4, has only 25 % occupancy and appears to be associated with the pteridine ring. c) Electron density corresponding to the phospho-adenosine pyrophosphate group of NADP+. The bracketed PN5′ and PA5′ from a symmetry-related NADP+ molecule, shown in lighter shades of magenta and red, are also included. The density was taken from data for the ternary complex. Symmetry-related water molecules are also indicated by faded coloring.
The structures of the apo enzyme and the binary DHFR•NADP+ complex have been included for comparison with the ternary complex. The statistics for the data collection and results from refinement of the three structures are reported in Table I, and the structures have been deposited in the protein data bank with accession codes 2RH2 (apo), 2RK2 (DHFR•NADP+), and 2RK1 (DHFR•DHF•NADP+). The structure of the apo enzyme includes three MPD molecules, positioned near Gly25, Leu50, and Tyr55. The MPD molecule closest to Tyr55 fits into a hydrophobic pocket defined primarily by Phe24, Met26, Ile42, Trp45, and Tyr55. This pocket is located on the outer surface of the molecule, so that binding interactions would not be predicted to interfere with substrate binding. Also of interest, no MPD molecules were observed in the enzyme pore. Alternative conformations are observed for Trp45 and Gln67. These residues are paired at monomer interfaces, resulting in conformations that are correlated. Both pairs of conformations differ from those reported in the previous structure 1VIE (5).
In principle, other complexes, such as the DHFR•(DHF)2 can also be present. Further, the use of DHF introduces the possibility of additional chemical conversions, such as the reduction of the NADP+ by the DHF which will result in the production of NADPH, which in turn can reduce DHF to tetrahydrofolate (THF) as well. Due to the poor solubility and presumed low concentration of folate in the 50% MPD solution, folate-containing complexes are very unlikely to be significant. In addition, the other possible ternary complexes have substantially lower affinity for the enzyme active site, compared with the ternary DHFR•DHF•NADP+ complex (9). Substantial occupancy of the active site by NADPH and THF, formed from the back reaction, is ruled out by the absence of the significant ring puckering expected for these species. Finally, refinement of the model with 25% occupancy, as expected for a ternary complex, fits the electron density sufficiently well so that all other reasonable complexes that might form are not significantly populated.
Structural features of the DHFR•DHF•NADP+ ternary complex
The structure of the ternary DHFR•DHF•NADP+ complex is characterized by a well organized arrangement of the nicotinamide and pteridine ring systems (Figure 4a). The enzyme achieves a multiple layered stacking arrangement that includes the indole rings of the four Trp38 residues, and a hydrogen bonded network formed from pairs of Gln67 and Tyr69 residues (Figures 4a, b) which sandwich the nicotinamide and pteridine ring systems. We characterize this multiple-tiered stacking arrangement as a “club sandwich motif”. In addition to the stacking interaction with Gln67, Trp38 is stacked against His62 from an adjacent chain, explaining its significant contribution to the stability of the R67 DHFR tetramer (24, 25).
Figure 4.


The “club sandwich motif” of R67 DHFR. Panel a shows a crystallographic tetramer with a single (asymmetric) pair of ligands. Each monomer is color-coded and labeled A-D. The active site pore faces the viewer. The four Trp38 residues, which contribute to the multi-tiered stack, occur on the outer layers of the “sandwich”. Moving inwards, the next layers are the symmetry-related Y69-Q67-Q67′-Y69′ residues, which “clamp” the pteridine and nicotinamide rings at the center. These residues sandwich the ligands, DHF (yellow) and NADP+ (magenta; only the ribonicotinamide ring is shown for simplicity). A red line shows the proximity of the C4 (nicotinamide) and C6 (pteridine) atoms, which are involved in hydride transfer. Panels b and c show details of the R67 DHFR “clamp”. In panel b, the structure of one of the two hydrogen bonding networks formed from Q67 and Y69 residues on chains A and B is shown. This panel corresponds to the top of the pore in panel a, rotated 90° about the horizontal axis. In panel c, the structure of the Y69-Q67-Q67′-Y69′ group in the apo enzyme is shown. In this case, one of the Gln67 residues tends to adopt an alternate conformation, which relieves the congestion and allows the sidechain to form an alternate hydrogen bond with Ile68 NH instead of with Tyr69 OH.
The arrangement of the central Gln67 residues differs from that originally reported for uncomplexed R67 DHFR (5), but appears to be similar to a more recently reported structure (26). Our own data for the uncomplexed enzyme show that the tight packing at the center of the pore results in conformational heterogeneity of the Gln67 sidechains, which occasionally are displaced from the framework shown in Figure 4b to adopt the structure pictured in Figure 4c. In this structure, one of the Gln67 sidechains bends into the pore, where it forms an apparent hydrogen bond with the amide proton of Ile68. This interaction substitutes for the hydrogen bond with the tyrosine, which may then hydrogen bond with water molecules in the pore. Movement of the Gln67 sidechain into the pore also loosens the packing near Trp38, which then also tilts towards the center of the pore. Thus, the main conformational response of the enzyme to the presence of substrates is the displacement of some of the Gln67 sidechains from the pore, resulting in the H-bonded network shown in Figure 4b and in tight packing of the Gln67 residues against the pteridine and nicotinamide rings (Figure 4a). This packing, along with expulsion of much of the pore water, stabilizes the hydrogen-bonded network shown in Figure 4b, which in turn packs more tightly against the Trp38 residues (Figure 3b). The extended planes formed by the two pairs of Y69-Q67-Q67′-Y69′ residues tightly clamp the substrate and cofactor together, promoting the reaction. This arrangement is characterized by a fundamental asymmetry in the structure of the pore, with one tyrosine acting as an H-bond donor, and the paired tyrosine acting as an H-bond acceptor (Figure 4b).
Substrate/Cofactor Recognition
One of the central questions posed by the symmetric structure of Type II DHFR concerns how the enzyme is able to affect a stereo-specific hydride transfer in the absence of specific DHF and NADPH binding sites. This puzzle is directly addressed by the present structure, which demonstrates that the enzyme recognizes a subtle structural analogy between the nicotinamide and pteridine ring systems. Specifically, the enzyme interacts with the nicotinamide ring and with a substructure of the pteridine ring system in an identical fashion, treating the pteridine N-3 and O-4 atoms as an analog of the nicotinamide carboxamide group (Figure 3a, b and Figure 5a). In each case, a pair of hydrogen bonds is formed with the backbone amide and carbonyl groups of a single Ile68 residue. An analogous interaction of the nicotinamide ring is present in Type I DHFR, utilizing an alanine residue rather than an isoleucine residue (27–29). This parallel mode of binding anchors the substrate and cofactor in an ideal orientation for the hydride transfer reaction. Thus, the interaction of the N3-O4 amide of the pteridine ring with Ile68 makes a critical contribution to DHF recognition and to reactivity. Interestingly, the pteridine ring of DHF is laterally displaced relative to its position in the previously reported folate binary complex (5). This leads to hydrogen bond distances of 2.88 Å and 2.98 Å between N3-Ile68 O and O4-Ile68 N in the present structure, compared with corresponding distances of 4.75 and 3.07 Å in the DHFR•(folate)2 structure (5). Apparently, the corresponding N3-Ile68 O hydrogen bonding interaction is not present in the earlier structure. Most significantly, this mode of DHF recognition has important implications for both the selectivity of the enzyme and for the catalytic mechanism.
Figure 5.


Recognition of NADP+. Panel a shows the interactions of the ribonicotinamide-ribose moiety of bound NADP+ with the protein. The extensive hydrogen bond network to the ribose helps to position the reactive centers and also tilts the rings into a more reactive geometry. Panel b illustrates many of the remaining interactions of the R67 DHFR with the cofactor. Ionic interactions between symmetry related Lys32 residues and two of the phosphate groups as well as several H-bonds are involved in binding. The ionic interactions have been experimentally monitored by ionic strength effects on binding (33). The terminal subscript indicates the chain identity of the residue.
Interestingly, isothermal titration calorimetry studies indicate that aminopterin (APTER) forms a DHFR•(APTER)2 complex, but does not bind to R67 DHFR•NADPH (30), suggesting that folate analogs in the binary and ternary complexes can adopt different conformations, consistent with the above differences noted for folate versus DHF in the binary and ternary complexes.
As indicated in Figure 4a, the nicotinamide ring is sandwiched between the Gln67 sidechain of monomer A and the pteridine ring of DHF. The ribonicotinamide is also positioned by a network of H-bonds that involves residues from chains A and B (Figure 5a). These interactions not only hold the nicotinamide ring in position for hydride transfer for the DHF, but also tilt the ring so that the reactive centers on the DHF and nicotinamide ring adopt a geometry that more closely approximates that of an endo transition state as modeled by Andres et al. (31). Additionally, two Lys32 residues (chains A and D) interact with the NADP+ cofactor (Figure 5b). The Lys32 residue on chain A is positioned closest to the nicotinamide phosphate, while Lys32 on chain D can form a salt bridge with the adenosyl-2′-phosphate group. In addition to these electrostatic interactions, one of the adenosyl-2′-phosphate oxygen atoms is positioned 3.0 Å from the amide nitrogen of Ala36 (chain D). Specific interactions with this phosphate are required to explain the 20-fold preference of the enzyme for NADPH over NADH (30, 32, 33). The adenine base is positioned so that one side is solvent exposed, while the other side lies directly over Ala72-Ala73 (chain D), consistent with the large amide chemical shift previously observed in NMR studies of the R67 DHFR•NADP+ complex (8). The imidazole ring of the adenine base is positioned near Pro70 (chain D). This structure differs from the recently reported binary complex structure for a Q67H mutant of R67 DHFR bound to NADP+ (34), perhaps due to the presence of the mutation.
As in the previously reported structure of the binary R67 DHFR•(folate)2 complex (5), there was insufficient electron density to allow positioning of the 4′-aminobenzoylglutamate group of DHF, despite the fact that it contributes significantly to binding (35). Modeling this portion of the molecule indicates that the binding site can provide favorable electrostatic interactions for both the α- and γ-carboxyl groups of the glutamate involving two of the Lys32 residues, but that the mouth of the pore is too wide to be consistent with a tight fit. This suggests that there may be considerable disorder of this portion of the molecule, minimizing the entropic penalty of binding while providing a favorable enthalpic interaction. This result is consistent with earlier NMR data indicating disorder of the glutamyl tail of the bound folate (7). The observation of significant conformational disorder and simultaneously a significant contribution to binding affinity is somewhat reminiscent of recent NMR observations on the mobility of the arginine sidechains in the HIV REV-RRE complex (36). In this case, electrostatic interactions between the arginine sidechains of the HIV REV protein make an important contribution to the binding affinity for the RNA target within the Rev Response element (RRE), while the arginine sidechains remain highly mobile as judged by NMR relaxation data. Other examples exist where mobile guest molecules bind tightly to their hosts (37, 38).
Relative Substrate/Cofactor Orientation
The structure of a ternary R67 DHFR•folate•ribonicotinamide complex was first predicted by the addition of a modeled ribonicotinamide to the crystallographically determined R67 DHFR•(folate)2 complex (5). In this model, the relative orientation of the nicotinamide and folate ring systems is similar to that found in chromosomal DHFR. Subsequently, interligand Overhauser effect (ILOE) studies were found to be inconsistent with this model, and suggested a relative endo orientation of the reactive ring systems (7), a result consistent with the structure of the ternary complex determined here (Figure 6a). The nicotinamide ring is stacked over the pyrazine ring of DHF, indicating an endo hydride transfer. The relative orientations of the pteridine and nicotinamide rings are consistent with the transfer of the pro-R hydride ion (A-side hydride transfer) previously demonstrated for a Type II DHFR (39). The closest approach of heavy atoms for the two ring systems corresponds to the nicotinamide C-4 - pteridine C-6 distance of 3.4 Å. The ring systems tilt away from each other, so that the distance between the nicotinamide N-1 and the pteridine C-4a is 3.9 Å. This ring stacking geometry is not observed in the Type I dihydrofolate reductase (Figure 6b), but has recently been observed in pteridine reductase (40) (Figure 6c), although in that case, the nicotinamide ring is flipped, leading to a B-side hydride transfer. Theoretical studies have suggested that this relative orientation may be optimally efficient for hydride transfer (31, 41–43).
Figure 6.


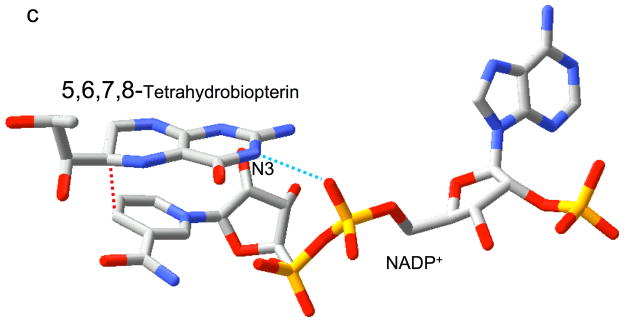
Relative substrate orientation. a) Relative orientation of NADP+ and the pteridine ring of DHF determined here for the R67 DHFR•DHF•NADP+ ternary complex. The backbone of Ile68 on chain D, which interacts with the N3-O4 amide of DHF, is also shown. The nicotinamide and pteridine ring systems adopt an endo conformation in which the closest approach corresponds to the reactive nicotinamide C4 and pteridine C6 carbons. The NADP+ is wrapped around the pteridine ring, so that the phosphates are positioned near the 2-amino group: distances: Ad-5′-P---N = 5.4 Å; Nic-5′-P---N = 6.1 Å; Ad-2′-P---N = 6.8 Å. b) Relative substrate orientation in a ternary DHFR•folate•NADP+ complex corresponding to the E. coli (Type I) enzyme (pdb entry 1RX2; (27)). The Asp-27 sidechain from the E. coli enzyme, which binds to folate N2 and N3, is also shown. The relative exo orientation contrasts with that observed for the Type II enzyme. c) Relative substrate orientation in a ternary PTR•tetrahydrobiopterin•NADP+ complex corresponding to the leishmania pteridine reductase (pdb entry 2BFP (67)). The relative endo orientation is analogous to that observed for R67 DHFR, however the enzyme catalyzes a B-side hydride transfer, so that the orientaton of the nicotinamide ring is flipped. Hydrogen bonding/salt bridge interactions with N3 are shown as blue dotted lines, and the reactive centers on the substrates are connected with red dotted lines. In order to facilitate comparison, the orientation of the pteridine ring system is similarly oriented in each frame.
In type I DHFR, a carboxylate sidechain from a glutamate (mammalian) or an aspartate (bacterial) sidechain is positioned to hydrogen bond with the pteridine N-3 and with the exo amino group of the pteridine ring (Figure 6b) (27, 44). In pteridine reductase, the adenosyl-5′-phosphate group is analogously positioned near the pteridine N3 (Figure 6c). The proximity of the negatively charged phosphate group could facilitate protonation of the pteridine N-5, consistent with a catalytic role (40). In the R67 DHFR ternary complex, the NADP+ molecule wraps around the pteridine so that several of the phosphate groups are positioned near the pteridine ring (Figure 6a). Although this binding mode may to some extent substitute for the absence of the carboxylate group in the Type I enzyme, none of the NADP+ phosphate groups makes close contact with the pteridine ring, or with the pteridine N3, which is instead hydrogen bonded to an I68 carbonyl (Figure 6a). Consistent with this structural observation, Raman studies of the R67 DHFR•DHF•NADP+ complex (15) have demonstrated that, in contrast with the Type I enzyme, the pKa of DHF is not significantly elevated in the complex. This observation is consistent with the lower kcat of the Type II enzyme (17).
Relationship to NMR Data
As noted above, the relative endo orientation of the folyl pteridine ring and the nicotinamide ring of the cofactor observed in the crystal structure (Figure 6a) are consistent with the ILOE observed between the two enzyme substrates (7). Despite some differences in the complexes studied and in the conditions of the measurements, distances determined from transferred NOE and ILOE data are generally in excellent agreement with the corresponding distances in the R67 DHFR•DHF•NADP+ complex ternary complex determined here (Supplementary Material; Supplementary Table S1).
The crystal structure also is generally consistent with previous chemical shift mapping studies of the interaction of NADP+ with the enzyme. In addition to the expected large shifts for residues 64–69 located in the pore of the enzyme, additional large shifts for resonances corresponding to residues located near the mouth of the pore were observed. The position of the adenosine ring of NADP+ is consistent with the large amide shifts observed for Ala73 and Leu74, as well as for the somewhat smaller shifts of Lys33 and Ser34 (8), which are also positioned near the adenosyl binding site. Analysis of the signs of these shifts indicates that they are not consistent with a ring current contribution arising from the proximity to the adenine ring, and hence the perturbation of the hydrogen bonding interactions for these amides may be the dominant factor in causing the observed shift perturbations.
Catalytic Mechanism
The NADPH-dependent reduction of DHF by DHFR takes place in two steps – a hydride transfer reaction in which a hydride is donated by the NADPH, and a protonation step. For Type I DHFR, it is generally thought that the protonation step precedes hydride transfer, thus creating a cationic intermediate which will more readily accept the hydride ion (45–47). Consistent with this view, vibrational spectroscopic studies have demonstrated that the N5 pKa of DHF is increased from 2.6 to 6.5 upon complex formation with E. coli DHFR (12–15). In the type I enzyme, this protonation step is thought to involve an active site aspartyl (bacterial) or glutamyl (mammalian) residue. Nevertheless, this residue is not positioned to bind directly to the N5-protonated pteridine, but rather is positioned near N3 and the C-2 exo amino group (Figure 6b) (27). The interaction can nonetheless stabilize a protonated form of the pteridine ring, and it has been suggested that the arrangement promotes a tautomeric equilibrium that facilitates transfer of a water proton to N5, followed by hydride transfer (16). The recently determined structure of pteridine reductase is perhaps even more relevant to the mechanism of the Type II enzyme, since the NADPH cofactor wraps around the pteridine ring in a manner that is reminiscent of the relative orientation determined here for a Type II DHFR (40). Furthermore, in pteridine reductase, the bridging adenosyl-5′-phosphate is positioned close to the pteridine N3 (Figure 6c), suggesting that it can play a role analogous to the aspartyl/glutamyl sidechain in the Type I enzyme.
In contrast with the functionally analogous enzymes discussed above, interaction of the DHF amide with the Ile68 backbone in the structure determined here effectively eliminates its role in catalysis. Therefore, the catalytic mechanism of Type II DHFR must differ significantly from that of the chromosomal DHFR (12, 13, 16, 48, 49) and also from the mechanism postulated for pteridine reductase (40). More specifically, there would appear to be no possibility of a keto-enol tautomeric equilibrium to facilitate N5 protonation, as has been proposed for the chromosomal DHFR (16). The importance of the N3-H – Ile68 hydrogen bond is supported by isothermal titration calorimetry studies showing that folate binds to R67 DHFR with a neutral pteridine ring (30). In the anionic form, the N3-O4 group deprotonates (50, 51), so that no proton is available for hydrogen bond formation with the Ile68 CO group.
The simplest catalytic model consistent with the structural data presented here is shown in Figure 7. This model uses water 114 (WAT114) as the proton donor to DHF N5. Based on the electron density, this water position shows 25 % occupancy, consistent with its association with the DHF, and appears to be H-bonded to DHF O4. The involvement of DHF N3-O4 in interactions with the Ile68 backbone should restrict the possibility of keto/enol tautomerism which has been proposed to facilitate N5 protonation in the Type I enzyme (16). The primary remaining questions are: 1) to what extent is a WAT114 proton transferred to the DHF N5, i.e., does the pKa of N5 become elevated in the bound state? 2) Does WAT114 function as the final water in a charge transfer complex so that some other functional group on either the enzyme or the substrate provides the main source for N5 protonation? 3) To what extent do other mechanisms contribute to the observed enzymatic activity?
Figure 7.

Enzyme-substrate interactions and catalytic mechanism. The enzyme exploits a subtle symmetry between the NADP+ cofactor and the DHF substrate by interacting similarly with the nicotinamide amide group in the first case, and the N3-O4 amide group of the pteridine. In both cases, a pair of hydrogen bonds is formed with the backbone carbonyl and amide groups of the Ile68 residues on chains A and D. As a result of these and other interactions described in the text, the relative positions of two ring systems are optimized for hydride transfer to C6. This transfer follows or is concerted with N5 protonation, presumably from WAT114. WAT114 is within hydrogen bonding distance of DHF O4, but is solvent accessible and does not otherwise appear to be specifically activated.
The first question noted above has been largely answered by Raman spectroscopic studies performed on the same R67 DHFR•NADP+•DHF complex studied here (15). These studies have shown that the pKa of N5 in the complex is < 4 (15), compared to the free pKa of 2.60 (35, 52). Both these values compare with an N5 pKa value of 6.5 for DHF bound to the Type I enzyme from E. coli (12, 13). These results demonstrate that elevation of the N5 pKa represents an important strategy for optimization of the catalytic rate constant for the chromosomal enzyme, while this perturbation apparently is not achieved for the Type II enzyme. The absence of a structural motif that would promote DHF N5 protonation is also consistent with kinetic studies demonstrating that the Type II DHFR is considerably less efficient than the type I enzyme, exhibiting an activity that is more similar to mutated forms of Type I DHFR in which the active site aspartyl residue has been replaced by an asparagine or serine residue (49, 50). In summary, the structural, kinetic and Raman data obtained for this enzyme all support a consistent conclusion that the Type II DHFR lacks an effective mechanism for stabilizing the N5-protonated intermediate or for charge relay to this position, resulting in lowered catalytic efficiency. Based on the above discussion, N5 protonation may be concerted with hydride transfer, as suggested in Figure 7.
Conversely, we may consider the factors that do appear to be significant for Type II DHFR: (1) Positioning of substrate/cofactor. As determined here, interactions of the nicotinamide and pteridine rings with the backbone amide and carbonyl groups of Ile68 position the reactive portions of the molecules in close proximity. Further, hydrogen bonding interactions of the nicotinamide ribose with backbone groups of Val66 and Gly64 also contribute to the positioning of the NADP+ and tilt the nicotinamide ring toward the pteridine ring of the DHF (Figure 6a). This tilted ring structure is similar to that calculated for transition state models (e.g., (31)). Interligand interactions, which clearly contribute to binding affinity (9, 53, 54) also contribute to the relative positioning of the substrates. Finally, two K32 residues interact with the phosphate moieties of NADPH and two others presumably bind to the glutamate tail of DHF, contributing to positioning and substrate affinity. (2)Steric compression. As originally noted by Narayana et al. (5), the diameter of the pore decreases at the center. The H-bonded Y69-Q67-Q67′-Y69′ network forms a substantial steric barrier that clamps the two substrates in place for optimal reactivity. The degree of compression appears to be significant, as judged by the observation that in the absence of substrate, the central Gln67 residues tend to adopt alternate conformations in which one extends into the center of the pore. Compression, as a physical chemical mechanism, could provide independence from a specific protein sequence and could be a useful device for R67 DHFR, which is limited in its catalytic choices by its 222 symmetry. Theoretical calculations have also indicated that compression of substrate and cofactor into a conformation close to a transition state contributes to the activity of the Type I DHFR (41). Further, the puckered ring structure of NADPH would be expected to result in even greater compression. (3) Substrate-assisted catalysis. Another catalytic strategy that may be operative for R67 DHFR is substrate assisted catalysis (11, 55, 56). As noted above, the adenosyl-2′-phosphate group of the NADP+ could provide a useful proton source for N5 protonation, although no enzyme-facilitated pathway has been identified. Alternatively, proton donation by the p-aminobenzoylglutamate tail of the DHF could serve as a proton source if either of the glutamate residues is protonated. In this context, we note recent data indicating that the kcat value for dihydropteroate (DHP – a DHF analog that lacks the glutamyl residue) reduction is decreased 1600 fold compared to the value for DHF (35). This observation thus lends some support to the possible significance of the glutamate residues as proton sources. According to this model, the enhanced catalytic rate at lower pH could result from the increased fraction of protonated glutamate carboxyl groups. A second type of substrate-assisted catalysis may result from internal mobility of the p-aminobenzoylglutamate moiety of the DHF. By allowing movement of the large benzoyl group while constraining the position of the pteridine ring, the C6 position of the pteridine could be subject to forces which may strain the ring structure and move the system toward a more reactive transition state structure (57, 58). In this view of catalysis, dynamics of the bound substrate complex could ultimately lead to transition state formation (59, 60). Potentially, molecular dynamic simulations can provide further insight into the feasibility of this potential catalytic mechanism.
An additional constraint on the possible catalytic mechanism is derived from the recent combinatorial studies reported by Schmitzer et al. (61) In particular, they demonstrated that co-evolution of residues 66–69 in the active site could produce variants with kinetic behavior that are generally similar to that observed for the native enzyme. The wt sequence is V66-Q67-I68-Y69 while the active mutant sequences are S66-K67-I68-H69 or I66-N67-R68-Y69 or G66-E67-L68-Y69. Since no residue remains conserved, specific side chains must not be essential for catalysis. This surprising behavior is nevertheless consistent with many aspects of the structure and the catalytic mechanism outlined above. In particular, substrate/cofactor positions are constrained by interactions with backbone atoms and so would be relatively independent of the identities of the sidechains. Similarly, the steric compression at the center of the pore might also be achieved with different sidechain combinations. This result is additionally consistent with the conclusion that none of the sidechains plays a critical role in N5 protonation. These minimalist catalytic strategies are consistent with the proposal that R67 DHFR is a primitive enzyme (62).
Insensitivity to Trimethoprim
Presumably, the primary advantage conferred by the plasmid-encoded DHFR is the resistance to diaminopyrimidine anti-folate drugs such as trimethoprim (1–4). The main basis for this insensitivity is immediately suggested by the structure of the R67 DHFR•DHF•NADP+ complex in which the enzyme recognizes the N3-O4 amide group of the DHF. Substitution of the pteridine O4 with an amino group in folate analogs such as aminopterin or methotrexate blocks this critical binding interaction, and hence is consistent with substantially weaker binding (30). Similarly, the diaminopyrimidine analogs such as trimethoprim also lack the amide functionality required to interact with Ile68, explaining their poor affinity for the enzyme (63). Additionally, substitution of the C-4 oxygen with an amino group as in aminopterin or methotrexate substantially elevates the pKa values for N1 protonation. This results in salt bridge formation with the Type I enzyme, in which the N1-protonated inhibitor is salt bridged with the active site carboxyl group (64, 65). A similar effect occurs for trimethoprim complexes with bacterial DHFR (66). However, as discussed above, the Type II DHFR lacks an active site carboxyl group and does not appear to utilize a mechanism that strongly favors N5-protonated DHF, and hence will have lower affinity for high-pK analogs such as trimethoprim or aminopterin. This behavior is also indicated by Raman studies demonstrating the failure of the Type II enzyme to significantly increase the N5 pKa value for DHF (15).
The reduced affinity for trimethoprim may also result from steric crowding of the trimethoxybenzyl group, which does not fit well in the restricted volume at the center of the pore. In addition, trimethoprim lacks carboxyl groups to contribute to the binding interaction by electrostatic interactions with the Lys32 residues in the pore. These observations are consistent with the mM Ki associated with TMP binding to R67 DHFR (63).
Conclusions
This study reports the first structure of a ternary enzyme-substrate-cofactor complex involving a Type II dihydrofolate reductase (R67 DHFR). The structure resolves a number of fundamental questions related to the substrate recognition and catalytic activity of Type II DHFR. The primary novel conclusions are: 1) The backbones of two I68 residues on symmetry-related chains serve as amide-recognition motifs that position the nicotinamide and pteridine rings so that their reactive centers are able to interact. 2) Additional important interactions include the hydrogen bonded Y69-Q67-Q67′-Y69′ surfaces which sandwich the two ring systems in an endo orientation, several additional hydrogen bonds, particularly with the nicotinamide ribose, which help to tilt the ring systems, electrostatic interactions with the K32 residues in the pore, and finally stacking of the nicotinamide and pteridine rings with each other, which also helps to explain the cooperativity of binding (9). The tilted, endo ring orientation closely approaches the theoretical transition state modeled by Andres et al. (31). The reliance on critical backbone interactions for optimal positioning provides a mechanism that relieves the evolutionary problem caused by the inability of the enzyme to mutate individual active site residues in order to optimize binding and activity. 4) The use of the pteridine N3-O4 amide function as a recognition motif appears to largely preclude its involvement in the catalytic mechanism. This function is in sharp contrast with the proposed catalytic role for N3-O4 in both chromosomal DHFR (16) and in pteridine reductase (40). In these systems, amide group interactions have been proposed to play an important role in N5 protonation. The absence of analogous interactions in the Type II DHFR ternary complex is probably an important reason for its relatively low catalytic activity (5). In addition to the limited role that the pteridine amide can play in N5 protonation, there is no obvious protonation mechanism/pathway that can be identified in the ternary complex studied here. Thus, protonation of N5 must rely on a chain of water molecules connecting N5 to the ultimate proton donor. This result is consistent with previous kinetic characterization of the enzyme indicating that activity is strongly enhanced as the pH is lowered. 6) The structure of the ternary complex provides significant insight into the ability of the enzyme to retain activity in the face of a massive level of mutation of the active site residues (61). Thus, the major structural features outlined above can be maintained in the presence of multiple mutations, at least from a qualitative standpoint. Finally, the present structure offers significant insights for the future development of inhibitors that can target Type II dihydrofolate reductase.
Acknowledgments
This research was supported by the intramural program of NIEHS, and by NSF grant MCB-0445728 (to E.E.H.).
Abbreviations
- APTER
aminopterin
- DHF
dihydrofolate
- DHFR
dihydrofolate reductase
- DMDDF - 2-desamino-2-methyl-5
8-dideazafolate
- MPD
2-methyl-2,4-pentanediol
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- NADP+
oxidized nicotinamide adenine dinucleotide phosphate
- NMR
nuclear magnetic resonance
- TMP
trimethoprim
- DHP
dihydropteroate
- ITC
isothermal titration calorimetry
- ILOE
interligand overhauser effect
References
- 1.Fleming MP, Datta N, Gruneberg RN. Trimethoprim resistance determined by R factors. Br Med J. 1972;1:726–8. doi: 10.1136/bmj.1.5802.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pattishall KH, Acar J, Burchall JJ, Goldstein FW, Harvey RJ. Two distinct types of trimethoprim-resistant dihydrofolate reductase specified by R-plasmids of different compatibility groups. J Biol Chem. 1977;252:2319–23. [PubMed] [Google Scholar]
- 3.Stone D, Smith SL. The amino acid sequence of the trimethoprim- resistant dihydrofolate reductase specified in Escherichia coli by R-plasmid R67. J Biol Chem. 1979;254:10857–61. [PubMed] [Google Scholar]
- 4.Smith SL, Stone D, Novak P, Baccanari DP, Burchall JJ. R plasmid dihydrofolate reductase with subunit structure. J Biol Chem. 1979;254:6222–5. [PubMed] [Google Scholar]
- 5.Narayana N, Matthews DA, Howell EE, Nguyen-huu X. A plasmid-encoded dihydrofolate reductase from trimethoprim-resistant bacteria has a novel D2-symmetric active site. Nat Struct Biol. 1995;2:1018–1025. doi: 10.1038/nsb1195-1018. [DOI] [PubMed] [Google Scholar]
- 6.Brito RM, Rudolph FB, Rosevear PR. Conformation of NADP+ bound to a type II dihydrofolate reductase. Biochemistry. 1991;30:1461–9. doi: 10.1021/bi00220a003. [DOI] [PubMed] [Google Scholar]
- 7.Li D, Levy LA, Gabel SA, Lebetkin MS, DeRose EF, Wall MJ, Howell EE, London RE. Interligand Overhauser effects in type II dihydrofolate reductase. Biochemistry. 2001;40:4242–52. doi: 10.1021/bi0026425. [DOI] [PubMed] [Google Scholar]
- 8.Pitcher WH, 3rd, DeRose EF, Mueller GA, Howell EE, London RE. NMR studies of the interaction of a type II dihydrofolate reductase with pyridine nucleotides reveal unexpected phosphatase and reductase activity. Biochemistry. 2003;42:11150–60. doi: 10.1021/bi0349874. [DOI] [PubMed] [Google Scholar]
- 9.Bradrick TD, Beechem JM, Howell EE. Unusual binding stoichiometries and cooperativity are observed during binary and ternary complex formation in the single active pore of R67 dihydrofolate reductase, a D2 symmetric protein. Biochemistry. 1996;35:11414–24. doi: 10.1021/bi960205d. [DOI] [PubMed] [Google Scholar]
- 10.Alonso H, Gillies MB, Cummins PL, Bliznyuk AA, Gready JE. Multiple ligand-binding modes in bacterial R67 dihydrofolate reductase. J Comput Aided Mol Des. 2005;19:165–87. doi: 10.1007/s10822-005-3693-6. [DOI] [PubMed] [Google Scholar]
- 11.Xu Q, Guo H, Wlodawer A, Guo H. The importance of dynamics in substrate-assisted catalysis and specificity. J Am Chem Soc. 2006;128:5994–5. doi: 10.1021/ja058831y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen YQ, Kraut J, Blakley RL, Callender R. Determination by Raman spectroscopy of the pKa of N5 of dihydrofolate bound to dihydrofolate reductase: mechanistic implications. Biochemistry. 1994;33:7021–6. doi: 10.1021/bi00189a001. [DOI] [PubMed] [Google Scholar]
- 13.Deng H, Callender R. Structure of dihydrofolate when bound to DHFR. J Am Chem Soc. 1998;120:7730–7737. [Google Scholar]
- 14.Rod TH, Brooks CL., 3rd How dihydrofolate reductase facilitates protonation of dihydrofolate. J Am Chem Soc. 2003;125:8718–9. doi: 10.1021/ja035272r. [DOI] [PubMed] [Google Scholar]
- 15.Deng H, Callender R, Howell E. Vibrational structure of dihydrofolate bound to R67 dihydrofolate reductase. J Biol Chem. 2001;276:48956–60. doi: 10.1074/jbc.M105107200. [DOI] [PubMed] [Google Scholar]
- 16.Cannon W, Garrison B, Benkovic S. Electrostatic Characterization of Enzyme Complexes: Evaluation of the Mechanism of Catalysis of Dihydrofolate Reductase. J Am Chem Soc. 1997;119:2386–2395. [Google Scholar]
- 17.Reece LJ, Nichols R, Ogden RC, Howell EE. Construction of a synthetic gene for an R-plasmid-encoded dihydrofolate reductase and studies on the role of the N-terminus in the protein. Biochemistry. 1991;30:10895–904. doi: 10.1021/bi00109a013. [DOI] [PubMed] [Google Scholar]
- 18.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 19.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 20.Bruno IJ, Cole JC, Kessler M, Luo J, Motherwell WDS, Purkis LH, Smith BR, Taylor R, Cooper RI, Harris SE, Orpen AG. Retrieval of Crystallographically-Derived Molecular Geometry Information. J Chem Inf Comput Sci. 2004;44:2133–2144. doi: 10.1021/ci049780b. [DOI] [PubMed] [Google Scholar]
- 21.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. Procheck: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291a. [Google Scholar]
- 23.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 24.West FW, Seo HS, Bradrick TD, Howell EE. Effects of single-tryptophan mutations on R67 dihydrofolate reductase. Biochemistry. 2000;39:3678–89. doi: 10.1021/bi992195x. [DOI] [PubMed] [Google Scholar]
- 25.Nichols R, Weaver CD, Eisenstein E, Blakley RL, Appleman J, Huang TH, Huang FY, Howell EE. Titration of histidine 62 in R67 dihydrofolate reductase is linked to a tetramer to two-dimers equilibrium. Biochemistry. 1993;32:1695–706. doi: 10.1021/bi00058a002. [DOI] [PubMed] [Google Scholar]
- 26.Narayana N. High-resolution structure of a plasmid-encoded dihydrofolate reductase: pentagonal network of water molecules in the D2-symmetric active site. Acta Crystallogr D Biol Crystallogr. 2006;62:695–706. doi: 10.1107/S0907444906014764. [DOI] [PubMed] [Google Scholar]
- 27.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 28.Bystroff C, Oatley SJ, Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase: the NADP+ holoenzyme and the folate. NADP+ ternary complex Substrate binding and a model for the transition state. Biochemistry. 1990;29:3263–77. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- 29.Filman DJ, Bolin JT, Matthews DA, Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. II Environment of bound NADPH and implications for catalysis. J Biol Chem. 1982;257:13663–72. [PubMed] [Google Scholar]
- 30.Jackson M, Chopra S, Smiley RD, Maynord PO, Rosowsky A, London RE, Levy L, Kalman TI, Howell EE. Calorimetric studies of ligand binding in R67 dihydrofolate reductase. Biochemistry. 2005;44:12420–33. doi: 10.1021/bi050881s. [DOI] [PubMed] [Google Scholar]
- 31.Andres J, Moliner V, Safont BS, Domingo LR, Picher MT, Krechl J. On Transition Structures for Hydride Transfer Step: A Theoretical Study of the Reaction Catalyzed by Dihydrofolate Reductase Enzyme. Bioorganic Chem. 1996;24:10–18. [Google Scholar]
- 32.Smith SL, Burchall JJ. Alpha-pyridine nucleotides as substrates for a plasmid-specified dihydrofolate reductase. Proc Natl Acad Sci USA. 1983;80:4619–23. doi: 10.1073/pnas.80.15.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hicks SN, Smiley RD, Hamilton JB, Howell EE. Role of ionic interactions in ligand binding and catalysis of R67 dihydrofolate reductase. Biochemistry. 2003;42:10569–78. doi: 10.1021/bi034643d. [DOI] [PubMed] [Google Scholar]
- 34.Divya N, Grifith E, Narayana N. Structure of the Q67H mutant of R67 dihydrofolate reductase-NADP+ complex reveals a novel cofactor binding mode. Protein Sci. 2007;16:1063–8. doi: 10.1110/ps.062740907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chopra S, Lynch R, Kim SH, Jackson M, Howell EE. Effects of temperature and viscosity on R67 dihydrofolate reductase catalysis. Biochemistry. 2006;45:6596–605. doi: 10.1021/bi052504l. [DOI] [PubMed] [Google Scholar]
- 36.Wilkinson TA, Botuyan MV, Kaplan BE, Rossi JJ, Chen Y. Arginine side-chain dynamics in the HIV-1 rev-RRE complex. J Mol Biol. 2000;303:515–29. doi: 10.1006/jmbi.2000.4143. [DOI] [PubMed] [Google Scholar]
- 37.Schneider HJ, Blatter T, Simova S. Host guest chemistry.26 NMR and fluorescence studies of cyclodextrin complexes with guest molecules containing both phenyl and naphthyl units. J Am Chem Soc. 1991;113:1996–2000. [Google Scholar]
- 38.Garel L, Lozach B, Dutasta JP, Collet A. Remarkable effect of the receptor size in the binding of acetylcholine and related ammonium ions to water-soluble cryptophanes. J Am Chem Soc. 1993;115:11652–11653. [Google Scholar]
- 39.Brito RM, Reddick R, Bennett GN, Rudolph FB, Rosevear PR. Characterization and stereochemistry of cofactor oxidation by a type II dihydrofolate reductase. Biochemistry. 1990;29:9825–31. doi: 10.1021/bi00494a011. [DOI] [PubMed] [Google Scholar]
- 40.Gourley DG, Schuttelkopf AW, Leonard GA, Luba J, Hardy LW, Beverley SM, Hunter WN. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat Struct Biol. 2001;8:521–5. doi: 10.1038/88584. [DOI] [PubMed] [Google Scholar]
- 41.Castillo R, Andres J, Moliner V. Catalytic Mechanism of Dihydrofolate Reductase Enzyme. A Combined Quantum-Mechanical/Molecular-Mechanical Characterizaton of Transition State Structure for the Hydride Transfer Step. J Am Chem Soc. 1999;121:12140–12147. [Google Scholar]
- 42.Wu YD, Houk KN. Transition structures for hydride transfers. J Am Chem Soc. 1987;109:906–8. [Google Scholar]
- 43.Wu YD, Houk KN. Theoretical transition structures for hydride transfer to methyleniminium ion from methylamine and dihydropyridine. On the nonlinearity of hydride transfers. J Am Chem Soc. 1987;109:226–227. [Google Scholar]
- 44.Davies JF, 2nd, Delcamp TJ, Prendergast NJ, Ashford VA, Freisheim JH, Kraut J. Crystal structures of recombinant human dihydrofolate reductase complexed with folate and 5-deazafolate. Biochemistry. 1990;29:9467–79. doi: 10.1021/bi00492a021. [DOI] [PubMed] [Google Scholar]
- 45.Morrison J, Stone S. Mechanism of the reaction catalyzed by DHFR from E. coli: pH and deuterium isotope effects with NADPH as the variable substrate. Biochemistry. 1988;27:5499–5506. doi: 10.1021/bi00415a017. [DOI] [PubMed] [Google Scholar]
- 46.Pu J, Ma S, Garcia-Viloca M, Gao J, Truhlar DG, Kohen A. Nonperfect synchronization of reaction center rehybridization in the transition state of the hydride transfer catalyzed by dihydrofolate reductase. J Am Chem Soc. 2005;127:14879–86. doi: 10.1021/ja054170t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Viloca M, Truhlar DG, Gao J. Reaction-path energetics and kinetics of the hydride transfer reaction catalyzed by dihydrofolate reductase. Biochemistry. 2003;42:13558–75. doi: 10.1021/bi034824f. [DOI] [PubMed] [Google Scholar]
- 48.Casarotto MG, Basran J, Badii R, Sze KH, Roberts GC. Direct measurement of the pKa of aspartic acid 26 in Lactobacillus casei dihydrofolate reductase: implications for the catalytic mechanism. Biochemistry. 1999;38:8038–44. doi: 10.1021/bi990301p. [DOI] [PubMed] [Google Scholar]
- 49.Cummins PL, Gready JE. Energetically most likely substrate and active-site protonation sites and pathways in the catalytic mechanism of dihydrofolate reductase. J Am Chem Soc. 2001;123:3418–28. doi: 10.1021/ja0038474. [DOI] [PubMed] [Google Scholar]
- 50.Poe M. Acidic dissociation constants of folic acid, dihydrofolic acid, and methotrexate. J Biol Chem. 1977;252:3724–8. [PubMed] [Google Scholar]
- 51.Kallen RG, Jencks WP. The dissociation constants of tetrahydrofolic acid. J Biol Chem. 1966;241:5845–50. [PubMed] [Google Scholar]
- 52.Maharaj G, Selinsky BS, Appleman JR, Perlman M, London RE, Blakley RL. Dissociation constants for dihydrofolic acid and dihydrobiopterin and implications for mechanistic models for dihydrofolate reductase. Biochemistry. 1990;29:4554–60. doi: 10.1021/bi00471a008. [DOI] [PubMed] [Google Scholar]
- 53.Strader MB, Chopra S, Jackson M, Smiley RD, Stinnett L, Wu J, Howell EE. Defining the binding site of homotetrameric R67 dihydrofolate reductase and correlating binding enthalpy with catalysis. Biochemistry. 2004;43:7403–12. doi: 10.1021/bi049646k. [DOI] [PubMed] [Google Scholar]
- 54.Strader MB, Smiley RD, Stinnett LG, VerBerkmoes NC, Howell EE. Role of S65, Q67, I68, and Y69 residues in homotetrameric R67 dihydrofolate reductase. Biochemistry. 2001;40:11344–52. doi: 10.1021/bi0110544. [DOI] [PubMed] [Google Scholar]
- 55.Kosloff M, Selinger Z. Substrate assisted catalysis -- application to G proteins. Trends Biochem Sci. 2001;26:161–6. doi: 10.1016/s0968-0004(00)01748-5. [DOI] [PubMed] [Google Scholar]
- 56.Dall’Acqua W, Carter P. Substrate-assisted catalysis: molecular basis and biological significance. Protein Sci. 2000;9:1–9. doi: 10.1110/ps.9.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fersht A. Structure and Mechanism in Protein Science. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 58.Ranaghan KE, Ridder L, Szefczyk B, Sokalski WA, Hermann JC, Mulholland AJ. Transition state stabilization and substrate strain in enzyme catalysis: ab initio QM/MM modelling of the chorismate mutase reaction. Org Biomol Chem. 2004;2:968–80. doi: 10.1039/b313759g. [DOI] [PubMed] [Google Scholar]
- 59.Bruice TC, Benkovic SJ. Chemical basis for enzyme catalysis. Biochemistry. 2000;39:6267–74. doi: 10.1021/bi0003689. [DOI] [PubMed] [Google Scholar]
- 60.Schramm VL. Transition state variation in enzymatic reactions. Curr Opin Chem Biol. 2001;5:556–63. doi: 10.1016/s1367-5931(00)00246-5. [DOI] [PubMed] [Google Scholar]
- 61.Schmitzer AR, Lepine F, Pelletier JN. Combinatorial exploration of the catalytic site of a drug-resistant dihydrofolate reductase: creating alternative functional configurations. Protein Eng Des Sel. 2004;17:809–19. doi: 10.1093/protein/gzh090. [DOI] [PubMed] [Google Scholar]
- 62.Howell EE. Searching sequence space: two different approaches to dihydrofolate reductase catalysis. Chem Bio Chem. 2005;6:590–600. doi: 10.1002/cbic.200400237. [DOI] [PubMed] [Google Scholar]
- 63.Amyes SG, Smith JT. The purification and properties of the trimethoprim-resistant dihydrofolate reductase mediated by the R-factor, R388. Eur J Biochem. 1976;61:597–603. doi: 10.1111/j.1432-1033.1976.tb10055.x. [DOI] [PubMed] [Google Scholar]
- 64.Coco L, Groff J, Temple C, Jr, Montgomery J, London R, Blakley R. Carbon-13 nuclear magnetic resonance study of protonation of methotrexate and aminopterin bound to dihydrofolate reductase. Biochemistry. 1981;20:3972–3978. doi: 10.1021/bi00517a005. [DOI] [PubMed] [Google Scholar]
- 65.Coco L, Temple C, Jr, Montgomery J, London R, Blakley R. Protonation of methotrexate bound to the catalytic site of dihydrofolate fromLactobacillus casei. Biochem Biophys Res Commun. 1981;100:413–419. doi: 10.1016/s0006-291x(81)80112-x. [DOI] [PubMed] [Google Scholar]
- 66.Coco L, Roth B, Temple C, Jr, Montgomery J, London R, Blakley R. Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch Biochem Biophys. 1983;226:567–577. doi: 10.1016/0003-9861(83)90326-0. [DOI] [PubMed] [Google Scholar]
- 67.Schuttelkopf AW, Hardy LW, Beverley SM, Hunter WN. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J Mol Biol. 2005;352:105–16. doi: 10.1016/j.jmb.2005.06.076. [DOI] [PubMed] [Google Scholar]