

Abstract
B12-dependent methionine synthase (MetH) is a large modular enzyme that utilizes the cobalamin cofactor as a methyl donor or acceptor in three separate reactions. Each methyl transfer occurs at a different substrate-binding domain and requires a different arrangement of modules. In the catalytic cycle, the cobalamin-binding domain carries methylcobalamin to the homocysteine (Hcy) domain to form methionine and returns cob(I)alamin to the folate (Fol) domain for remethylation by methyltetrahydrofolate (CH3-H4folate). Here, we describe crystal structures of a fragment of MetH from Thermotoga maritima comprising the domains that bind Hcy and CH3-H4folate. These substrate-binding domains are (βα)8 barrels packed tightly against one another with their barrel axes perpendicular. The properties of the domain interface suggest that the two barrels remain associated during catalysis. The Hcy and CH3-H4folate substrates are bound at the C termini of their respective barrels in orientations that position them for reaction with cobalamin, but the two active sites are separated by ≈50 Å. To complete the catalytic cycle, the cobalamin-binding domain must travel back and forth between these distant active sites.
Cobalamin-dependent methionine synthase (MetH) is a large, modular protein (1) that catalyzes the transfer of a methyl group from methyltetrahydrofolate (CH3-H4folate) to homocysteine (Hcy) to form methionine, using cobalamin as an intermediate methyl carrier (Fig. 1A). MetH enzymes from Escherichia coli (1) and Homo sapiens (2-4) consist of four functional modules arranged in a linear fashion with single interdomain connectors. Each of the three methyl transfer reactions (Fig. 1 A) is catalyzed by a different substrate-binding domain. The N-terminal module (the Hcy domain) utilizes a (Cys)3Zn2+ cluster to bind and activate Hcy (5), the second module (the Fol domain) activates CH3-H4folate for methyl transfer (6), and the third module binds cobalamin (7). The fourth C-terminal module binds S-adenosylmethionine (AdoMet) and is required for reductive reactivation (1).
Fig. 1.

The reactions catalyzed by MetH (A) and the conformational states of MetH (B). (A) During primary turnover (red arrows), a methyl group is transferred from CH3-H4folate to the enzyme-bound cob(I)alamin cofactor, forming H4-folate and methylcobalamin. Methylcobalamin is then demethylated by Hcy, forming methionine (Met) and regenerating cob(I)alamin (1). In reactivation (green arrows), cob(II)alamin is reduced by one-electron transfer from flavodoxin and the resulting cob(I)alamin is methylated by AdoMet (1). (B) Cartoons depicting the arrangements of the modules of MetH. The diagrams designate four states in which the cobalamin-binding domain interacts with different partners: the four-helix bundle or Cap domain, the Fol domain, the Hcy domain, or the AdoMet domain. The Hcy (green) and Fol (gold) barrels are represented by large cylinders, the red oval corresponds to the α/β Rossmann domain (Cob) that binds cobalamin, and the blue oval is the activation (AdoMet) domain. The helical Cap domain is represented as a group of small red cylinders.
A “divide-and-conquer” strategy involving expression or isolation of fragments of the enzyme has provided structures of the individual cobalamin and AdoMet binding modules (7, 8). The AdoMet domain is an unusual α+β helmet-shaped fold that binds AdoMet on its inner surface. The isolated cobalamin-binding module consists of two domains, a four-helix bundle (the Cap domain) lying over the upper (β) face of the cobalamin, and a Rossmann α/β domain (the Cob domain) containing a critical histidine residue that coordinates the cobalt of cobalamin from beneath. In MetH, as in several other B12-dependent enzymes (9, 10), the dimethylbenzimidazole nucleotide is displaced from the lower face of the cobalamin and extends into the core of the Rossmann fold.
Recent studies have demonstrated that the enzyme exists as an ensemble of conformations with equilibria dependent on the oxidation and ligation state of the cobalamin and on the concentrations of substrates or products (11). At a minimum, the ensemble in the intact enzyme comprises four species in which the cobalamin-binding domain is positioned to interact with one or another of its four possible partners (Fig. 1B). Methionine synthesis in the primary catalytic cycle requires switching between a conformation with the Fol and Cob domains positioned for methyl transfer (Fol:Cob) and a conformation with the Hcy and Cob domains positioned for reaction (Hcy:Cob). The rearrangements of domains that allow cobalamin to visit physically separated active sites are reminiscent of the domain rearrangements that occur in signal transduction (12, 13) and may provide a paradigm for some of the domain motions in other complex multimodular enzymes (14-17).
Studies of multiple-domain constructs have furnished direct evidence for conformational rearrangements associated with the reactions of MetH. The structure of a fragment of E. coli MetH that spans both the cobalamin- and AdoMet-binding modules revealed that the four-helix Cap domain moves ≈25 Å to allow formation of an intramolecular complex that is competent for the reactivation reaction in which AdoMet is the methyl donor to cob(I)alamin (18).
Here, we describe crystal structures of the N-terminal substrate-binding modules of MetH from Thermotoga maritima and their complexes with the substrates Hcy and CH3-H4folate. The MetH from this thermophile includes three modules that bind Hcy, CH3-H4folate, and cobalamin; in T. maritima, the activation domain is a separate polypeptide (see annotation for PDB ID 1J6R). The substrate-binding domains (residues 1-566) are intimately associated (βα)8 barrels, with the two active sites distant from one another (Fig. 2). The arrangement of the barrels implies that the cobalamin-binding domain must shuttle back and forth between sites that are separated by ≈50 Å to complete the catalytic cycle.
Fig. 2.

A ribbon drawing of the substrate-binding domains of MetH from T. maritima. The Hcy barrel is shown in green, the linker is shown in gray, and the Fol barrel is shown in gold. Substrates are shown as ball-and-stick models. The view is approximately along the axis of the Hcy barrel and is oriented to show the hairpin component of the domain interface.
Materials and Methods
Protein Expression and Purification. The coding sequence corresponding to residues 1-566 of T. maritima MetH was PCR-amplified from genomic DNA (a gift from James Bardwell, University of Michigan) and inserted into pET29a (Novagen). From alignments (19), this sequence was expected to include all of the structural elements of the substrate-binding modules. The construct was designed as a C-terminal His-6 fusion for ease of purification. Protein expression was induced with 0.4 mM isopropyl β-d-thiogalactoside in BL21(DE3) cells grown in M9 minimal medium supplemented with amino acids and zinc sulfate. Cells were lysed by sonication, and the cell lysate was heated at 70°C for 10 min. The resulting soluble fraction was loaded onto a Hi-Trap Zn(II)-NTA column (Amersham Pharmacia). The protein was eluted with a glycine gradient (50 mM to 1.5 M), dialyzed against 50 mM Tris (pH 8.0)/1 mM Tris(2-carboxyethyl)phosphine, and concentrated to 10-15 mg/ml. Selenomethionine-substituted protein was expressed and purified similarly; incorporation of selenomethionine was validated by amino acid analysis.
Crystallization. Monoclinic crystals of metal-replete protein, space group P21 (a = 59.98 Å, b = 86.07 Å, c = 125.82 Å, β = 100.3°, with two molecules per asymmetric unit) were grown at 4°C and pH 5.7 in 10 μl sitting drops in the presence of 10 mM CdCl2 and 1.25% (wt/vol) 1,2,3-heptanetriol. The reservoir solution, mixed 1:1.5 with the protein and additives, was 25% 1,2-propanediol/10% glycerol/5% poly(ethylene glycol) 3000/100 mM sodium citrate (final pH 5.30). Crystallization in the absence of CdCl2 yielded an oxidized metal-free species with a disulfide cross-link between the metal ligands Cys-207 and Cys-272. The oxidized protein also crystallized in an orthorhombic form at 30°C in 1.4-1.7 M (NH4)2SO4/100 mM Mes/10 mM CoCl2 (final pH, 7.2).
Structure Determination. The structure of the substrate-free form of MetH (1-566), crystallized in space group P21, was initially determined by multiwavelength anomalous diffraction (MAD) using selenomethionine-substituted protein. Structures of complexes with substrates were then obtained by crystallographic refinement (Table 1).
Table 1. Data collection and refinement statistics.
| Structure
|
Wavelength
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Substrate-free | Hcy | Hcy+CH3-H4folate | Oxidized | Oxidized* | λ1 (remote) | λ2 (edge) | λ3 (peak) | |
| Crystal/data | ||||||||
| Resolution, Å | 1.65 | 1.65 | 1.90 | 2.10 | 3.10 | 2.00 | 2.16 | 2.16 |
| Unique reflections | 141,673 | 147,923 | 97,473 | 58,895 | 45,652 | 87,460 | 65,278 | 63,847 |
| Redundancy | 5.1 | 5.8 | 2.2 | 9.2 | 2.6 | 7.5 | 7.1 | 5.3 |
| Completeness, % | 93.1 (82.5) | 99.4 (99.8) | 92.6 (85.2) | 100.0 (100.0) | 89.4 (63.6) | 99.0 (99.3) | 96.7 (74.7) | 93.7 (59.3) |
| I/σ(I) | 15.32 (2.79) | 17.50 (2.96) | 9.86 (3.06) | 25.01/(8.95) | 12.32 (2.54) | 17.0 (8.0) | 13.5 (4.1) | 11.9 (2.2) |
| Rsym | 0.064 (0.424) | 0.064 (0.408) | 0.069 (0.406) | 0.054 (0.222) | 0.074 (0.263) | 0.058 (0.209) | 0.071 (0.308) | 0.077 (0.423) |
| Refinement | ||||||||
| Rwork† | 0.217 | 0.211 | 0.230 | 0.195 | 0.205 | 0.242 | — | — |
| Rfree† | 0.236 | 0.239 | 0.270 | 0.238 | 0.248 | 0.280 | — | — |
| Average B factors, Å2 | ||||||||
| Model atoms | 28.66 | 30.79 | 39.21 | 31.57 | 53.36 | 44.78 | — | — |
| rms deviation | ||||||||
| Bond lengths, Å | 0.0061 | 0.0131 | 0.0154 | 0.0058 | 0.0077 | 0.0060 | — | — |
| Bond angles, ° | 1.319 | 1.687 | 1.691 | 1.346 | 1.360 | 1.288 | — | — |
Space group P212121, a = 92.58 Å, b = 169.02 Å, c = 174.28 Å.
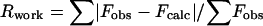 , where Fobs and Fcalc are observed and calculated structure factor amplitudes. Rfree was calculated from a randomly selected 10% of all data.
, where Fobs and Fcalc are observed and calculated structure factor amplitudes. Rfree was calculated from a randomly selected 10% of all data.
Selenomethionine-substituted protein was crystallized as described. Crystals were transferred through Paratone-N, flash-cooled in liquid nitrogen, and shipped to the National Synchrotron Light Source for mail-in data collection by Shai Vaday, who processed and scaled the data using HKL2000 (20). The scaled data were transmitted to us and input to solve (21), which found 16 of the anticipated 18 selenium sites. The initial phases were further refined in sharp (22), which computed a set of experimental phases with a figure of merit of 0.64 (20.0 Å to 2.5 Å). After density modification, performed in resolve (21), the figure of merit was 0.69. Electron density maps calculated with the phases from resolve were readily interpretable, and a model of the structure was built by using xfit (23).
Substrate Complexes. Complexes were prepared by dialyzing substrates into crystals suspended in reservoir solution in 200-μl dialysis buttons. The buttons were placed in anaerobic chambers containing 10 ml of reservoir solution at pH 5.3 that included 1 mM ZnCl2 and 10 mM DTT and had been flushed with argon. This initial dialysis step was intended to replace the Cd2+ found in the active site of the initial structure with Zn2+. Then, 1 mM l-Hcy and 10 mM CH3H4-folate were added individually or in combination. To prepare substrate-free crystals for measurements at higher resolution (Table 1, column 1), the pH was raised to 6.0 in dialyses vs. ZnCl2 and DTT. Except for the selenomethionine MAD data used for experimental phasing, all data were collected at the DuPont-Northwestern-Dow beamline 5-ID at the Advanced Photon Source at Argonne National Laboratory.
Metals. Three metal ions were observed in the initial substrate-free structure (column 1 of Table 1), one at each active site of the asymmetric unit, and a third bridging two symmetry-related chains. The metal ions were assigned as cadmium rather than the natural metal, zinc. The peak electron density for the metal ion was approximately three times the peak densities for the sulfur atoms bound to the metal in the active site clusters, agreeing with expectations for cadmium rather than for zinc. In addition, large positive difference Fourier peaks were centered on the metal positions when the active site metal was modeled as Zn2+. The protocols for addition of substrates did deplete the metal that bridges molecules in the crystal but failed to replace the Cd2+ ion in the active site with Zn2+. Peak densities and difference Fourier maps were also consistent with Cd2+ as the predominant metal ion in the substrate complexes.
Structure Refinement. Structure refinements were carried out in cns (24). The initial model from the Se-MAD experiment performed at National Synchrotron Light Source was refined first with data to 2.5-Å resolution, and then to 2.0 Å. A higher resolution data set collected from native crystals at the DuPont-Northwestern-Dow beamline 5ID at the Advanced Photon Source was then substituted for further refinements. Each step of refinement included two rounds of standard CNS protocols: scaling and overall temperature refinement, minimization, simulated annealing (first at 5,000 K, then at 2,000 K), minimization, and individual temperature factor refinement. Each refinement stage was followed by coordinate adjustment and rebuilding (23) until the R factor converged and no protein density was visible above 3σ in difference Fourier maps (R = 0.266; Rfree = 0.300). Crystallographic waters were then assigned from peaks >3σ in the difference Fourier maps, and were retained based on proper hydrogen bonding and distances from protein atoms or existing solvent atoms. The final models of chain A include residues 1-559 of the protein. Statistics are presented in Table 1.
Structures of the substrate complexes and the oxidized metal-free proteins (Table 1) were determined by using similar refinement protocols. Temperature factors were first reset to a single value (B = 30.0 or 35.0 Å2, depending on resolution), and the model was subjected to group refinement, treating the linker, the Hcy domain, and the Fol domain as rigid units. The remaining steps were the same as those described for the initial structure. Simulated annealing was performed starting at 2,000 K, and was followed by minimization and individual temperature factor refinement. l-Hcy and CH3-H4folate were modeled into difference Fourier density and refined by using parameter and topology files generated with the pro-drg web site (25).
Models of Bound Cobalamin and Analysis of Interfaces. Fitting of the cobalamin cofactor to each of the active sites was explored as the corrin macrocycle was rotated and tilted about the central cobalt. The positions of the substrates and of the metal cluster of the Hcy domain were held fixed as cobalamin orientations were probed, but the protein and corrin side chains were moved manually to relieve close contacts in promising models. Models for methylcobalamin and cob(I)alamin were from the structures of the tryptic fragment (7) and the reactivation complex (18), respectively. Models were assessed by measuring the buried surface areas, the number and kinds of cobalamin-protein interactions, and the gap index, by using the Protein-Protein Interaction Server (www.biochem.ucl.ac.uk/bsm/PP/server) (26) and contacts from Collaborative Computational Project 4 (27). The properties of the domain interface were analyzed similarly.
Coordinates. The coordinates for the structures listed in Table 1 have been deposited in the Protein Data Bank.
Results
The Substrate Barrels and the Interdomain Linker. The substrate-binding fragment of MetH from T. maritima folds into two (βα)8 barrels with distinctive distortions that accommodate the metal sites and substrates (Fig. 2 and Fig. 9, which is published as supporting information on the PNAS web site). In this two-domain structure, the barrels are oriented with their axes perpendicular to one another and are tethered together by an interdomain linker. The active sites of the Hcy and Fol domains, located near the C termini of the barrel strands, are both exposed to solvent but are separated by ≈50 Å. The two barrels of the structure are closely associated, with part of the interdomain linker as well as residues from the outer helices and loops of the barrels forming an extensive interface.
The single linker that connects the Hcy and Fol domains begins at the invariant Pro-292 and ends at His-314 (Fig. 3). The initial extended segment of the linker acts as a spacer that allows the barrels to be joined side-by-side (Fig. 4). Beginning at Phe-301, whose side chain is inserted into the Fol domain, the linker forms a 12-residue β hairpin that is buried between the two domains, with the first strand (Phe-301-Ser-305) completely inaccessible to solvent (Fig. 4). Residues from the hairpin extend on opposite sides of the strands, interacting alternately with residues of the Hcy and Fol domains.
Fig. 3.

The sequence of the substrate-binding modules of MetH from T. maritima. Invariant and conserved residues, framed in dark and light blue, are assigned from a multiple alignment performed in clustalw (19) using 19 sequences from the NCBI database (see Fig. 10). The secondary structure assignments for T. maritima MetH are displayed above the alignments and are labeled H or F to designate Hcy or Fol domains.
Fig. 4.

The interface between the Hcy and Fol domains. Residues 292-300 comprise the extended part of the interdomain linker. The hairpin segment of the linker that packs against both barrels begins at Phe-301 (see text). Residues in a conserved motif (180G-R/K-S/T-L-S/T-G) from the loop β5H-α5H (lavender) contact the conserved Pro-306 of the hairpin.
The properties of the domain interface suggest that this double barrel does not disassemble and reassemble during the catalytic cycle. The barrel-barrel interface is relatively hydrophobic (58% based on atom hydrophobicities) and buries an area of 1,160 Å2 from each of the domain surfaces (20, 22). It incorporates not only the linker hairpin but also nine other structural segments (28) from the two barrels. Approximately 20 hydrogen bonds secure the interfaces in the several high-resolution structures (Table 1). Among these are bonds made by five interdigitating side chains that reach across the interdomain boundary to interact with the backbone of the partner domain. Two Arg-Glu salt bridges link the two domains and entrap two or three waters within the interface.
A useful metric in the interface analysis of Jones et al. is the gap volume index (26, 28). This statistic, the ratio of the interface gap volume to the interface surface area, measures the fit between interacting surfaces, with smaller values indicating closer fits. In an early survey of functional homodimer interfaces, Jones and Thornton (28) found an average gap index of 2.19 ± 0.83 Å. Later studies of domain-domain interfaces in proteins that are single polypeptides reported an average gap volume index of 1.80 ± 0.9 Å (26). Calculations for the interface between the Hcy and Fol domains yield a gap volume index of 2.01 Å, implying excellent complementarity between their interacting surfaces, with interactions comparable to those that maintain functional homodimers in other enzymes.
Analysis of tryptic digests provides supporting evidence that the two domains are firmly associated. The Hcy-Fol linkers of the T. maritima and E. coli (29) enzymes resist proteolysis, even upon treatment with 3% trypsin, and despite the presence of several Arg/Lys residues in these sequences. Tryptic digestion of the T. maritima MetH (1-566) fragment yields two large polypeptides. The N-terminal sequences and the fragment sizes determined by electrospray mass spectrometry indicate cleavages at Arg-327 and Lys-336, downstream of the Hcy-Fol linker in the exposed helix αAF of the Fol domain. Intact E. coli MetH (29) is cleaved at an analogous site within the Fol domain and at sites in the interdomain linkers that precede and follow the cobalamin-binding module.
Further evidence that the two barrels do not readily dissociate comes from the structure of an orthorhombic form of MetH (1-566) (see Table 1), crystallized at 30°C and pH 7.2 in ammonium sulfate rather than in propanediol at 4°C and pH 5.7. There are two copies of the MetH fragment in the asymmetric unit of the orthorhombic crystal form, which diffracts to lower resolution (3.1 Å) and has a higher solvent content (≈70%) than the monoclinic form. Despite these differences in conditions, the interfaces between Fol and Hcy barrels in the structure of the orthorhombic form are identical to those in the monoclinic crystal form.
The Active Site of the Hcy Domain. The Hcy barrel is distorted to form the metal- and substrate-binding sites. To accommodate the substrate, strands 1 and 2 of the barrel are loosely joined by nonclassic hydrogen bonds; to accommodate the metal, strands 6 and 8 are drawn together and strand 7 is extruded from the end of the barrel. These distortions were first observed in the related enzyme, betaine-Hcy methyltransferase (BHMT) (30). Like MetH, BHMT binds Hcy at an essential (Cys)3Zn2+ cluster. In the Hcy complex of MetH, the substrate sulfur is also attached to the metal ion, with the remainder of the amino acid protruding into the barrel (Figs. 2 and 5). As in BHMT, the amino group of Hcy is bound by a conserved glutamate from strand β4H (Glu-146 in MetH) and the carboxylate group forms hydrogen bonds to the backbone just beyond the fingerprint sequence 19D-G-A/G at the end of strand β1H (Fig. 5).
Fig. 5.

Substrate interactions in the Hcy domain. The sulfur of Hcy coordinates the metal, the amino group is hydrogen-bonded to Glu-146, and the carboxyl group interacts with backbone amides following the fingerprint sequence, 19D-G-A. Phe-66 stacks against the substrate, and Thr-147 is hydrogen-bonded to the sulfur of Hcy.
Binding of Hcy forms a metal-ligand cluster with approximately tetrahedral geometry. This result agrees with extended x-ray absorption fine structure measurements showing four sulfur ligands to zinc in Hcy complexes of E. coli MetH (5). However, the electron density at the metal site and the metal-sulfur geometries suggested that the Cd2+ used in crystallization had replaced the native zinc (Materials and Methods). The most satisfactory fits to the electron densities of the Hcy complexes were obtained with Cd2+ and Cd-S bond distances of ≈2.53 Å. These Me2+-S bond lengths are typical for Cd2+ and significantly longer than Zn2+-S bonds (2.35 Å). Cd2+ is also the predominant metal ion in the substrate-free structure.
The Hcy domain of MetH is a homolog of BHMT, with >80% of the α carbons in equivalent positions, but in BHMT the methyl donor is glycine betaine rather than methylcobalamin. Important differences between the Hcy domains of BHMT and T. maritima MetH reflect their very different methyl donors. In BHMT, a ring of hydrophobic residues encircles the binding site for betaine and sequesters the substrates from solvent. In the Hcy domain of MetH, the δ-thiol of Hcy is actually solvent accessible, and except for Tyr-247, the ring of hydrophobic residues has been deleted, leaving room for the much larger second substrate of MetH, methylcobalamin. Models of bound cobalamin (see below) suggest that the cofactor itself closes the active site, providing a relatively hydrophobic environment. It has been proposed that a low dielectric environment is important for methyl transfer to Hcy (31).
The Active Site of the Fol Barrel. In the Fol domain, the final barrel strand, β8, is shorter than the other strands, leaving an opening like a tent flap from which the folate side chain emerges (Figs. 2 and 9). Methyltetrahydrofolate is bound in an extended mode that resembles the proposed solution conformation (32). This binding mode contrasts with the conformations adopted by folates productively bound to dihydrofolate reductase (33) and thymidylate synthase (34), where the pterin and p-aminobenzoic acid (PABA) moieties are partly folded into L- or V-shapes.
The pterin ring is positioned by hydrogen bonds with four invariant residues: Asp-390, Asn-411, Asp-473, and Asn-508 (Fig. 6). Contributions of the two aspartates to binding and activity have already been explored by Asp → Asn mutations in E. coli MetH (35) of the residues that correspond to Asp-390 and Asp-473. These residues form hydrogen bonds to NH8 and NH3, respectively. The mutants were inactive in methyl transfer from CH3-H4folate to cob(I)alamin (35). Binding of the CH3-H4folate substrate was only slightly impaired in the mutant corresponding to Asp-390-Asn but could not be measured in the mutant corresponding to Asp-473-Asn, where the interaction with N3 is lost. The complex of MetH with CH3-H4folate confirms several features of the model proposed for the binding of CH3-H4-folate to the homologous corrinoid/iron-sulfur protein methyltransferase from Moorella thermoacetica (AcsE, a subunit of acetyl-CoA synthase) (36).
Fig. 6.

CH3-H4folate and its interactions with conserved residues from the Fol barrel. The extended arrangement of the folate is determined by the interactions of the p-aminobenzoic acid (PABA) side chain: the ring stacks with Glu-321, N10 makes a through-water interaction with the conserved Asn-323, and Arg-516 binds the PABA carbonyl oxygen. In the saturated pyrazine ring of the pterin, the C6 side chain substituent is axial and the N5-methyl group is equatorial, pointing toward the reader. Hydrogen bonds are indicated for donor-acceptor pairs that are closer than 3.2 Å. The side chain amide of Asn-508 may reorient in the ternary complex to allow the oxygen to interact with a protonated pterin N5 (see text). The viewpoint is approximately along the expected approach of the corrin ring.
For an SN2 displacement reaction, the N5-methyl bond is expected to be almost perpendicular to the corrin ring of cob(I)alamin (37). The observed tilt of the pterin ring would help to achieve this geometry (Fig. 6). The short axis of the pterin, defined by the bond between C5a and C8a, is inclined to the barrel axis at an angle of ≈50°. In addition, the methyl group that is transferred to cobalamin is displaced from the pterin plane in a direction that brings the N5-methyl bond closer to alignment with the barrel axis and points it toward the postulated binding site for cobalamin.
Protonation of the N5 leaving group is an essential step in methyl transfer from CH3-H4folate to cob(I)alamin (35), but the mechanism of protonation remains to be determined. Data for E. coli MetH show that protonation does not accompany formation of the binary complex with the folate substrate (35), but must occur in the ternary E·CH3-H4folate·cob(I)alamin complex. Curiously, there is no general acid in the neighborhood of N5, either in MetH or in related enzymes where N5 is protonated during the reaction, such as the homologous corrinoid/iron-sulfur protein methyltransferase (AcsE) (38). The invariant Asn-508, which can interact with both O4 and N5 of the pterin ring (Fig. 6), may be important for the methyl transfer reaction. We do not expect Asn-508 to be the direct donor of a proton to N5, as there is no precedent for proton donation by amides. The amide oxygen might stabilize a protonated N5 in the transition state, a role analogous to that proposed for an asparagine in purine nucleotide phosphorylase (39).
Modeling the Molecular Complexes with Cobalamin. The bound substrates and the residues surrounding the Hcy and CH3-H4folate binding sites are expected to interact with the cobalamin species that serve as methyl donor and methyl acceptor during primary turnover. Models of cobalamins bound to each of the substrate barrels were generated manually (Materials and Methods). The reactants were positioned so that the methyl carbons of methylcobalamin and CH3-H4folate were in van der Waals contact with the attacking sulfur and cobalt, respectively.
Criteria for the choice of the orientation of the corrin ring were derived in part from the known structures of B12 proteins where adenosylcobalamin is complexed with (βα)8 barrels. There are four helpful examples: glutamate mutase (PDB ID code 1I9C) from Clostridium cochlearium (10), methylmalonyCoA mutase (MMCoA mutase) from P. shermanii (9) (PDB ID code 1REQ), diol dehydratase (40) (PDB ID codes 1DIO and 1EEX), and glycerol dehydratase (PDB ID code 1IWP) from Klebsiella pneumoniae (41). Interactions of cobalamin with these proteins all follow a pattern in which the methyl groups of rings C and D, at the hydrophobic rim of the corrin macrocycle (Fig. 7), are surrounded by hydrophobic groups, whereas the A, B, and D ring acetamides are hydrogen bonded to protein backbone or side chains.
Fig. 7.

Models of cobalamin binding to each of the substrate domains. (A) (Left) The surface of the cobalamin binding site of the Hcy barrel, with Hcy represented as van der Waals spheres; the Hcy sulfur is yellow. Hydrophobic patches are yellow, and hydrophilic regions are cyan. (Right) A schematic diagram of the modeled orientation of the corrin macrocycle, showing the possible interactions between cobalamin and residues of the Hcy domain. Cys-207 and -273 of the (Cys)3 Hcy-metal cluster are positioned behind the B and C rings of the corrin. Invariant residues that interact with cobalamin side chains are labeled, and hydrophobic contacts are shaded. Preliminary characterization of the Tyr247Phe mutant shows an 8-fold decrease in the rate constant for the reaction of Hcy with methylcobalamin (D. Touw, L. Paul, and R.G.M., unpublished data). (B) (Left) The surface of the cobalamin binding site of the Fol barrel, with the atoms of CH3-H4folate represented as van der Waals spheres; the 5N-methyl group is pale yellow. Hydrophobic patches are yellow, and hydrophilic regions are cyan. (Right) A schematic diagram showing the orientation and interactions of cobalamin in the model complex with the Fol domain. Invariant residues that interact with cobalamin side chains are labeled; Asp-390 and Asn-411 also interact with the pterin ring of CH3-H4folate (Fig. 6).
The α/β domain that binds cobalamin was added by superimposing the known structures of cobalamin-binding domains from MetH (7, 18) onto the modeled cobalamin complexes. The resulting model structures resemble glutamate mutase and MMCoA mutase, which are also (βα)8 barrels positioned atop a related Rossmann domain that binds cobalamin. Fig. 8 displays the models with the Cob domain (residues 745-896 from E. coli MetH), attached in turn to the Hcy and Fol barrels of the structure of the T. maritima fragment. It vividly illustrates the large movement of the Cob domain that is essential to carry the methyl group from one active site to the other.
Fig. 8.

Models of the complexes of the cobalamin-binding domain with the Hcy and Fol barrels. The α/β (Cob) domain, represented in surface mode (red), interacts with the Hcy domain (Left) or with the Fol domain (Right). The Hcy:Cob and Fol:Cob conformations are shown side by side to illustrate the large displacement of the Cob domain that occurs during the reaction cycle; the centers of the Cob domains are separated by ≈70 Å. During turnover, exchange of product for substrate is assumed to occur only in “open” accessible substrate barrels, as proposed earlier (46).
Discussion
The Conformational Landscape. Intact MetH appears to sample a limited number of domain arrangements that are not vastly different in energy (11) (Fig. 1B). The factors that govern the distribution of these species are now partly understood. The oxidation and ligation states of cobalamin strongly influence the population of the accessible domain arrangements (11, 42). Recent studies of MetH from E. coli have demonstrated that substrates and products can also alter the relative stabilities of the different conformations (11).
The structures reported here suggest that the effects of substrates on the distributions of conformations are not mediated by allosteric mechanisms. Comparison of the substrate-free structure with the substrate complexes reveals only local rearrangements at each substrate site. For example, upon binding of Hcy, the helix αBH that interacts with the substrate carboxylate (Fig. 5) shifts toward the metal cluster of the Hcy domain, but this movement is not propagated to the neighboring Fol domain. Although interactions between the active sites and more distant residues might be revealed by further studies (43), current observations from the structure fit the earlier view that changes in the distribution of conformations (Fig. 1B) are largely governed by local interactions (11). The modeling studies (Fig. 7) also suggest that the local changes elicited by binding of Hcy or CH3-H4folate increase the affinity for cobalamin so that E·S·cobalamin complexes are formed by a path in which E·S is the preferred intermediate. Thermodynamic interactions between the binding of substrates and the binding of cobalamin would minimize the populations of “empty” complexes between the substrate-binding barrels and the cobalamin-binding domain.
The tight association of the substrate-binding barrels that is inferred from the structure of the N-terminal domains restricts the conformational space available to the polypeptide chain of MetH. Rather than serving as a flexible linker that would allow the Hcy and Fol domains to adopt a variety of relative orientations, the connector helps to cement the interdomain interface and limits the number of accessible conformations.
Acrobatics in the Catalytic Cycle. The chemical reactions are not rate-limiting in MetH (44); instead, the domain rearrangements are likely to be the slow steps in turnover (V. Bandarian and R.G.M., unpublished data). The observed kcat sets limits on the time scale of the domain motions: two transitions between the Hcy:Cob and Fol:Cob states of Fig. 1B must occur within the 1/27 sec required to complete each catalytic cycle at 25°C (42). The distance between the sites where the cobalamin cofactor is alternately demethylated and remethylated is ≈50 Å,§ but from the structures it is evident that the cobalamin-binding domain cannot take a straight path between the active sites of Hcy and Fol barrels. It must travel a more circuitous route, perhaps swinging away from the substrate domains, using residues following the Fol barrel as a flexible string (15). In fact, kinetic studies of reactions in trans between the two halves of MetH indicate that the cobalamin domain dissociates from the substrate domains after each half-reaction rather than sliding from one site to the other (45). During these rearrangements, the substrate domains presumably move as a discrete structural unit, maintaining the relative orientation of the two active sites. Concerted motions of paired domains may occur in other modular proteins that are believed to undergo large conformational changes, such as fatty acid synthases (15) and polyketide synthases (17).
The sequences that connect the Fol barrel with the α/β cobalamin-binding domain (Fig. 10, which is published as supporting information on the PNAS web site) are expected to facilitate the domain motions that occur in the catalytic cycle. Capturing structures of the cobalamin domain in productive complexes with the substrate barrels should provide descriptions of the Fol-Cob connector and the nature of the rearrangements that occur in the transition between the Fol:Cob and Hcy:Cob states.
Supplementary Material
Acknowledgments
We thank Dr. Shai Vaday (National Synchrotron Light Source, Brookhaven National Laboratory) for the initial MAD data collection and the staff at DuPont-Northwestern-Dow Collaborative Access Team at the Advanced Proton Source for assistance with additional x-ray measurements. Sizes of the tryptic fragments of Thermotoga maritima MetH were determined by Elise Manning. Diffraction data collection at the National Synchrotron Light Source beamline X12C is supported by the National Institutes of Health National Center for Research Resources and the U.S. Department of Energy. Use of the APS is supported by the U.S. Department of Energy. This research was supported by National Institutes of Health Grants GM16429 (to M.L.L.) and GM24908 (to R.G.M.) and by the Michigan Molecular Biophysics Training Program, GM08270 (J.C.E.).
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 29, 2003.
Abbreviations: MetH, cobalamin-dependent methionine synthase; Hcy, homocysteine; AdoMet, S-adenosylmethionine; BHMT, betaine-homocysteine methyltransferase; MAD, multiwavelength anomalous diffraction.
Data deposition: The structure coordinates reported in this study have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1Q7Z for the substrate-free structure; 1Q85 for the selenomethionine substrate-free structure (MAD data in Table 1); 1Q8A for the complex with Hcy; 1Q8J for the complex with Hcy and CH3-H4folate; 1Q7Q for the oxidized structure (orthorhombic); and 1Q7M for the oxidized structure (monoclinic)].
See accompanying Biography on page 3727.
Footnotes
The possibility that methyl transfers occur between subunits in a dimer can be eliminated. Assays show no significant change in specific activity over a 104 range of enzyme concentration that spans concentrations where the enzyme is known to be a monomer.
References
- 1.Ludwig, M. L. & Matthews, R. G. (1997) Annu. Rev. Biochem. 66, 269-313. [DOI] [PubMed] [Google Scholar]
- 2.Chen, L. H., Liu, M.-L., Hwang, H.-Y., Chen, L.-S., Korenberg, J. & Shane, B. (1997) J. Biol. Chem. 272, 3628-3634. [PubMed] [Google Scholar]
- 3.van der Put, N. M., van der Molen, E. F., Kluijtmans, L. A., Heil, S. G., Trijbels, J. M., Eskes, T. K., van Oppenraaij-Emmerzaal, D., Banerjee, R. & Blom, H. J. (1997) Q. J. Med. 90, 511-517. [Google Scholar]
- 4.LeClerc, D., Campeau, E., Goyette, P., Adjalla, C. E., Christensen, B., Ross, M., Eydoux, P., Rosenblatt, D. S., Rozen, R. & Gravel, R. A. (1996) Hum. Mol. Genet. 5, 1867-1874. [DOI] [PubMed] [Google Scholar]
- 5.Peariso, K., Goulding, C. W., Huang, S., Matthews, R. G. & Penner-Hahn, J. E. (1998) J. Am. Chem. Soc. 120, 8410-8416. [Google Scholar]
- 6.Goulding, C. W., Postigo, D. & Matthews, R. G. (1997) Biochemistry 36, 8082-8091. [DOI] [PubMed] [Google Scholar]
- 7.Drennan, C. L., Huang, S., Drummond, J. T., Matthews, R. G. & Ludwig, M. L. (1994) Science 266, 1669-1674. [DOI] [PubMed] [Google Scholar]
- 8.Dixon, M., Huang, S., Matthews, R. G. & Ludwig, M. L. (1996) Structure (London) 4, 1263-1275. [DOI] [PubMed] [Google Scholar]
- 9.Mancia, F., Keep, N. H., Nakagawa, A., Leadlay, P. F., McSweeney, S., Rasmussen, B., Bosecke, P., Diat, O. & Evans, P. R. (1996) Structure (London) 4, 339-350. [DOI] [PubMed] [Google Scholar]
- 10.Reitzer, R., Gruber, K., Jogl, G., Wagner, U. G., Bothe, H., Buckel, W. & Kratky, C. (1999) Structure (London) 7, 891-902. [DOI] [PubMed] [Google Scholar]
- 11.Bandarian, V., Ludwig, M. L. & Matthews, R. G. (2003) Proc. Natl. Acad. Sci. USA 100, 8156-8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison, S. C. (2003) Cell 112, 737-740. [DOI] [PubMed] [Google Scholar]
- 13.Lim, W. A. (2002) Curr. Opin. Struct. Biol. 12, 61-68. [DOI] [PubMed] [Google Scholar]
- 14.Milne, J. L., Shi, D., Rosenthal, P. B., Sunshine, J. S., Domingo, G. J., Wu, X., Brooks, B. R., Perham, R. N., Henderson, R. & Subramaniam, S. (2002) EMBO J. 21, 5587-5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith, S., Witkowski, A. & Joshi, A. K. (2003) Prog. Lipid Res. 42, 289-317. [DOI] [PubMed] [Google Scholar]
- 16.Gokhale, R. S. & Khosla, C. (2000) Curr. Opin. Chem. Biol. 4, 22-27. [DOI] [PubMed] [Google Scholar]
- 17.Tsuji, S. Y., Wu, N. & Khosla, C. (2001) Biochemistry 40, 2317-2325. [DOI] [PubMed] [Google Scholar]
- 18.Bandarian, V., Pattridge, K. A., Lennon, B. W., Huddler, D. P., Matthews, R. G. & Ludwig, M. L. (2002) Nat. Struct. Biol. 9, 53-56. [DOI] [PubMed] [Google Scholar]
- 19.Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994) Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-325. [DOI] [PubMed] [Google Scholar]
- 21.Terwilliger, T. C. (2002) Acta Crystallogr. D 58, 1937-1940. [DOI] [PubMed] [Google Scholar]
- 22.de La Fortelle, E. & Bricogne, G. (1997) Methods Enzymol. 276, 471-493. [DOI] [PubMed] [Google Scholar]
- 23.McRee, D. (1999) Practical Protein Crystallography (Academic, San Diego).
- 24.Brünger, A. T., Adams, P. A., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. Q., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 25.vanAalten, D. M. F., Bywater, R., Findlay, J. B. C., Hendlich, M., Hooft, R. W. W. & Vriend, G. (1996) J. Comput. Aided Mol. Des. 10, 255-262. [DOI] [PubMed] [Google Scholar]
- 26.Jones, S., Marin, A. & Thornton, J. M. (2000) Protein Eng. 13, 77-82. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 28.Jones, S. & Thornton, J. M. (1995) Prog. Biophys. Mol. Biol. 63, 31-65. [DOI] [PubMed] [Google Scholar]
- 29.Jarrett, J. T., Huang, S. & Matthews, R. G. (1998) Biochemistry 37, 5372-5382. [DOI] [PubMed] [Google Scholar]
- 30.Evans, J. C., Huddler, D. P., Jiracek, J., Castro, C., Millian, N. S., Garrow, T. A. & Ludwig, M. L. (2002) Structure (London) 10, 1159-1171. [DOI] [PubMed] [Google Scholar]
- 31.Jensen, K. P. & Ryde, U. (2003) J. Am. Chem. Soc. 125, 13970-13971. [DOI] [PubMed] [Google Scholar]
- 32.Poe, M., Hensons, O. D. & Hoogsteen, K. (1979) J. Biol. Chem. 254, 10881-10884. [PubMed] [Google Scholar]
- 33.Sawaya, M. R. & Kraut, J. (1997) Biochemistry 36, 586-603. [DOI] [PubMed] [Google Scholar]
- 34.Stroud, R. M. & Finer-Moore, J. S. (2003) Biochemistry 42, 239-247. [DOI] [PubMed] [Google Scholar]
- 35.Smith, A. E. & Matthews, R. G. (2000) Biochemistry 39, 13880-13890. [DOI] [PubMed] [Google Scholar]
- 36.Doukov, T., Seravelli, J., Stezowski, J. J. & Ragsdale, S. W. (2000) Structure (London) 8, 817-830. [DOI] [PubMed] [Google Scholar]
- 37.van Tenud, L., Farooq, S., Seibl, J. & Eschenmoser, A. (1970) Helv. Chim. Acta 53, 2059-2069. [Google Scholar]
- 38.Zhao, S., Roberts, D. L. & Ragsdale, S. W. (1995) Biochemistry 34, 15075-15083. [DOI] [PubMed] [Google Scholar]
- 39.Kicska, G. A., Tyler, P. C., Evans, G. B., Furneaux, R. H., Shi, W., Fedorov, A., Lewandowicz, A., Cahill, S. M., Almo, S. C. & Schramm, V. L. (2002) Biochemistry 41, 14489-14498. [DOI] [PubMed] [Google Scholar]
- 40.Shibata, N., Masuda, J., Tobimatsu, T., Toraya, T., Suto, K., Morimoto, Y. & Yasuoka, N. (1999) Structure (London) 7, 997-1008. [DOI] [PubMed] [Google Scholar]
- 41.Yamanishi, M., Yunoki, M., Tobimatsu, T., Sato, H., Matsui, J., Dokiya, A., Iuchi, Y., Oe, K., Suto, K., Shibata, N., et al. (2002) Eur. J. Biochem. 269, 4484-4494. [DOI] [PubMed] [Google Scholar]
- 42.Jarrett, J. T., Amaratunga, M., Drennan, C. L., Scholten, J. D., Sands, R. H., Ludwig, M. L. & Matthews, R. G. (1996) Biochemistry 35, 2464-2475. [DOI] [PubMed] [Google Scholar]
- 43.Benkovic, S. J. & Hammes-Schiffer, S. (2003) Science 301, 1196-1202. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee, R. V., Frasca, V., Ballou, D. P. & Matthews, R. G. (1990) Biochemistry 29, 11101-11109. [DOI] [PubMed] [Google Scholar]
- 45.Bandarian, V. & Matthews, R. G. (2001) Biochemistry 40, 5056-5064. [DOI] [PubMed] [Google Scholar]
- 46.Matthews, R. G. (1999) in Enzymatic Mechanisms, eds. Frey, P. A. & Northrup, D. B. (IOS, Amsterdam), pp. 155-161.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}