

Abstract
Proper development of the skeleton in utero and during growth requires mechanical stimulation. Loading results in adaptive changes in bone that strengthen bone structure. Bone’s adaptive response is regulated by the ability of resident bone cells to perceive and translate mechanical energy into a cascade of structural and biochemical changes within the cells — a process known as mechanotransduction. Mechanotransduction pathways are among the most anabolic in bone, and consequently, there is great interest in elucidating how mechanical loading produces its observed effects, including increased bone formation, reduced bone loss, changes in bone cell differentiation and lifespan, among others. A molecular understanding of these processes is developing, and with it comes a profound new insight into the biology of bone. In this article, we review the nature of the physical stimulus to which bone cells mount an adaptive response, including the identity of the sensor cells, their attributes and physical environment, and putative mechanoreceptors they express. Particular attention is allotted to the focal adhesion and Wnt signaling, in light of their emerging role in bone mechanotransduction. The cellular mechanisms for increased bone loss during disuse, and reduced bone loss during loading are considered. Finally, we summarize the published data on bone cell accommodation, whereby bone cells stop responding to mechanical signaling events. Collectively, these data highlight the complex yet finely orchestrated process of mechanically regulated bone homeostasis.
Keywords: mechanical loading, bone mechanotransduction, bone strength, adaptation, osteoblasts, osteocytes, Wnt signaling, focal adhesion, bone turnover, bone modeling, nitric oxide, prostaglandin, desensitization
I. INTRODUCTION
Bones serve as shields for vital organs and levers for muscles to contract against to facilitate locomotion. Like muscles, bones add mass when we exercise and atrophy if we do not. The adaptation of bone architecture to mechanical loads has been a subject of great interest for many years. The most well-known treatise on the subject was written by Julius Wolff in 1892.1 Wolff proposed that the patterns trabeculae form near joints are dictated by mechanical stress patterns and can be explained by mathematical laws. Wolff was inspired by the observation that trabecular trajectories within the proximal femur were similar to the pattern of stresses in a curved crane calculated by Culmann2,3 (Fig. 1). Wolff’s ideas are often summarized by the simple rule that bone adjusts its structure to adapt to the loads placed on it. Indeed mechanical loading is essential during growth for the development of robust weight-bearing bones. In the absence of mechanical usage, limbs develop with only 30–50% of normal bone mass. A child growing without skeletal loading will develop thin, fragile long bones with diminished periosteal circumference (Fig. 2A).4 In addition, a long bone in a paralyzed limb does not achieve its characteristic cross-sectional shape (Fig. 2B).5
FIGURE 1.
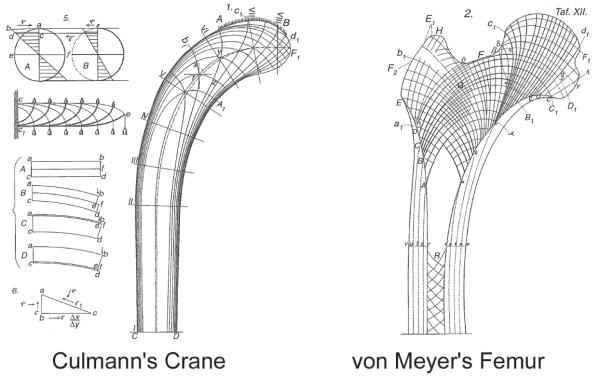
The stress directions in a curved crane (Left) estimated by the Swiss engineer Culmann compared with the drawings of trabecular trajectories in the proximal femur by Swiss anatomist von Meyer (Right).
FIGURE 2.
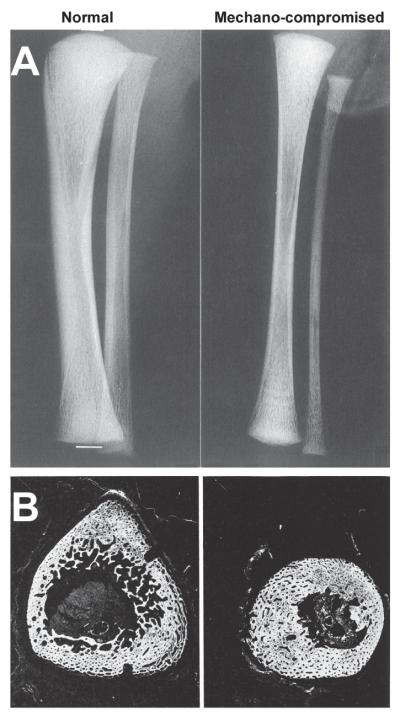
Radiographs of tibiae from two newborns with reduced loading of the tibia due to (A) congenital neuromuscular disease and (B) spina-bifida. The reduced mechanical environment of the tibia results in a narrow, underdeveloped diaphysis that remains circular and small, rather than triangular and large, in cross section. (A) Reprinted from Rodríguez et al.4 with permission from Springer-Verlag New York Inc. (B) Reprinted from Ralis et al.5 with permission from Cambridge University Press.
On the other hand, exercise strengthens bones. A tennis player will develop larger bones in his or her playing arm compared to the nonplaying arm (Fig. 3).6 We found that applying loading to the forelimbs of rats improved bone strength by 64%. Interestingly the improvement in bone mineral content (BMC) in these rats was only a modest 7%. The greater increase in bone strength, compared to BMC, occurred because new bone formed where the stresses were highest but not where the stresses were low producing a mechanically efficient structure. Consequently, mechanical loading adds mass but also causes the cross-sectional shape of long bones to become more structurally efficient. The reshaping of bones is most effective during skeletal growth. Once growth is completed, the effects of exercise shift from bone building to prevention of bone loss. In the adult skeleton, exercise reduces bone turnover by reducing bone resorption, which results in better preservation of bone mass and a more youthful skeleton.
FIGURE 3.
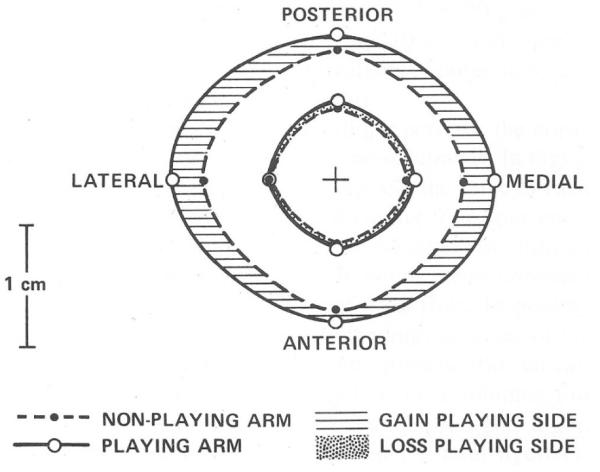
Comparison between the humerus cross-section in the playing arm and nonplaying arm of a professional tennis player. The periosteal circumference of the humerus in the playing arm has expanded substantially and there is also a slight expansion of the marrow cavity. Reprinted from Jones et al.6 with permission from the publisher (Journal of Bone and Joint Surgery).
II. WHAT CELLS DETECT MECHANICAL LOADS?
Osteocytes within the bone and lining cells on the bone surface are thought to be the primary mechanosensors in bone tissue. Their abundance and connectivity make them an antenna for detecting mechanical strains. These cells are the most abundant in bone — they outnumber osteoblasts and osteoclasts by over 20 to 1. Osteocytes have long dendritic processes that contact surrounding cells at gap junctions. Deformation of the bone matrix caused by mechanical loads is detected by osteocytes, which in turn send paracrine signals to osteoblasts and osteoclasts. Osteocytes form a functional network that passes intracellular calcium signals from cell to cell, either intracellularly through gap junctions or extracellularly by paracrines like adenosine triphosphate (ATP).7 Lining cells cover over 90% of the surfaces of adult bone and they contain gap junction connections to osteocytes and osteoblasts. Expression of several genes has been observed in osteocytes and bone lining cells in vivo after a mechanical stimulus (Fig. 4). These include the proto-oncogene c-fos (30 min after loading) and insulin-like growth factor I (IGF-I) (6 h after loading).8 Moreover, osteocytes appear to provide key chemotactic signals that target osteoclasts to repair focal damage and microcracks.9
FIGURE 4.
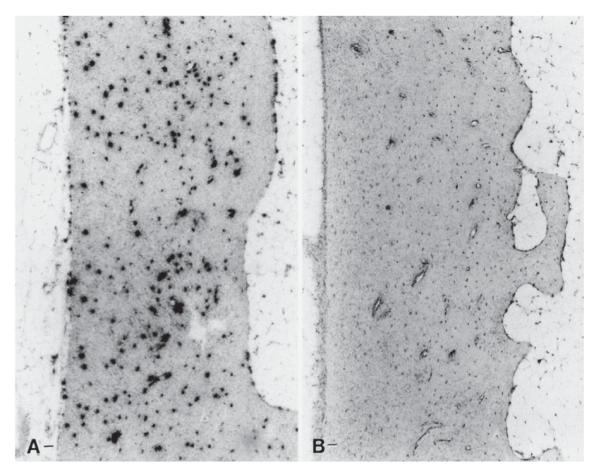
In situ hybridization for c-fos mRNA in rat vertebral bone. (A) Vertebral cortex hybridized for c-fos mRNA 1 h after applied mechanical loading. (B) Similar region of the vertebral cortex from a nonloaded control vertebra hybridized for c-fos mRNA. Reprinted from Lean et al.8 with permission from The American Physiological Society.
Osteocytes occupy cavities within the mineralized matrix called lacunae and their processes travel though channels called canaliculae. They are surrounded by fluid-filled space and connected to the bone extracellular matrix at focal adhesions. As bone is loaded, extracellular fluid is pushed back and forth through the extracellular space surrounding the osteocytes and their cell processes. The fluid has viscosity that creates shear stress on the osteocyte cell membrane and drag forces on the extracellular tethering proteins. Therefore, it is said that osteocytes are viscously coupled to the bone tissue. The magnitude of the fluid forces is proportional to loading rate and this explains why bone is more sensitive to dynamic rather than static loading (Fig. 5).10 The microstructure of the osteocyte processes has been modeled by Han and colleagues.11 Their model predicts that fluid drag forces bow the extracellular matrix proteins and create tension in the cell membrane (Fig. 6). Other studies suggest that stretch opens ion channels within the membrane resulting in intracellular calcium signaling and activation of calcium-dependent pathways within the osteocyte.12 The model predicts membrane stretch increases as loading frequency increases, up to around 10 cycles/sec (Hz) above which the response plateaus, consistent with experimental findings.13,14 Our studies show that bone becomes more sensitive to loading as the loading frequency is increased from 1 to 10 Hz. We subjected rats to forelimb loading and measured the bone formation response at different frequencies.13 As the frequency of loading was raised from 1 cycle/sec (Hz) to 5 Hz to 10 Hz, the slope each dose-response curve significantly increases (Fig. 7). It takes much less bone strain to stimulate bone formation when loading at 10 Hz compared to 5 or 1 Hz. The minimum strain needed to initiate bone formation is 1820 microstrain at 1 Hz but drops to 1180 microstrain at 5 Hz and to only 650 microstrain at 10 Hz.
FIGURE 5.
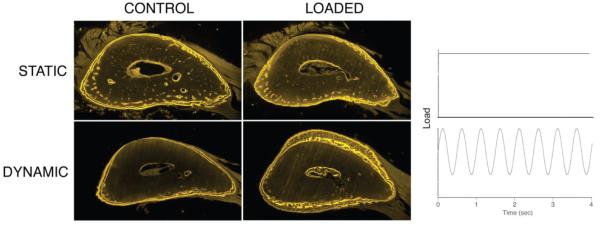
Dynamic, cyclical loading produces new bone formation whereas static, stationary loading does not.10 Here we see ulna sections that were loaded similarly, except the bone on top received stationary loading and the bone below received cyclic loading at 2 cycles/sec. Fluorochrome (yellow) labels indicate the bone boundary at the beginning and end of the experiment. It is clear that a significant amount of new bone was formed under cyclic loading. The new bone can be seen on the medial (Top) and lateral (Bottom) bone surfaces where the mechanical strain within the bone tissue is highest. Reprinted from Robling et al.10 with permission from Elsevier.
FIGURE 6.
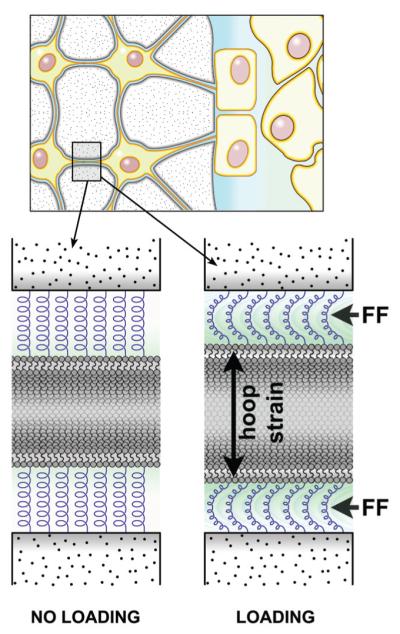
Physical deformations of bone tissue that occur during functional use (e.g., muscle contractions) create hydrostatic pressure gradients within bone’s lacunar-canalicular network. As those pressure gradients are equilibrated via movement of extracellular fluid from regions of high pressure to regions of low pressure, the glycocalyx (a structure that suspends/tethers the osteocyte cell membrane to the canalicular wall) is exposed to drag forces from the fluid, which create a “hoop strain” on the cell process. This hoop strain is one mechanism by which smaller strains in the tissue can be amplified to larger strains at the cell surface by fluid movement.11
FIGURE 7.

Bone is more sensitive to loading at higher loading frequencies. (Left) The graph on the left shows dose-response curves for the bones’ response to mechanical loading at 1 cycle/sec (Hz), 5 Hz, and 10 Hz.13 The y-axis is average bone formation rate and the x-axis is the peak strain engendered in the bone tissue. (Right) The graph on the right shows the minimum strain needed to initiate bone formation. This value is 1820 microstrain at 1 Hz but drops to 1180 microstrain at 5 Hz and to only 650 microstrain at 10 Hz. Reprinted from Hsieh and Turner13 with permission from the publisher (American Society for Bone and Mineral Research).
III. BIOMECHANICAL REGULATION OF BONE REMODELING
Mechanical loading has profound influences on bone remodeling. Disuse or lack of loading causes an acceleration of bone turnover with bone resorption overwhelming bone formation resulting in a rapid loss of bone mass. This type of bone loss is observed in astronauts who spend extended periods of time in the weightless environment of a space station or shuttle. Overuse of bone can cause small cracks or focal damage within the tissue, which in turn stimulates bone remodeling as a reparative process. One of the important roles of bone turnover is to continuously replace and repair damaged bone tissue. Osteoclasts target microcracks within bone tissue preferentially and remove compromised bone tissue.15 The damaged tissue is then replaced by new bone tissue. If damage accumulates faster than the tissue can be repaired, larger microcracks develop and propagate to form a stress fracture.
The increased number of bone remodeling sites with disuse or overuse is preceded by programmed cell death in osteocytes.16 The mechanisms that initiate osteocyte death (apoptosis) are not well understood but may include direct damage to osteocytes via microcracks in the bone matrix or lack of convective fluid flow during disuse. Prolinerich tyrosine kinase 2 (Pyk2) must be activated before osteocyte apoptosis will occur.17
The effects of loading on bone remodeling follows a U-shaped curve — remodeling is increased in disuse (insufficient loading) or overuse (overloading causing damage) (Fig. 8). There is a range of loading within which bone remodeling is minimized. This is often called the physiological range.18 Periosteal bone formation, on the other hand, steadily increases with increased loading, indicating that the mechanisms controlling the growth and reshaping of bones differ from those controlling of bone remodeling.
FIGURE 8.
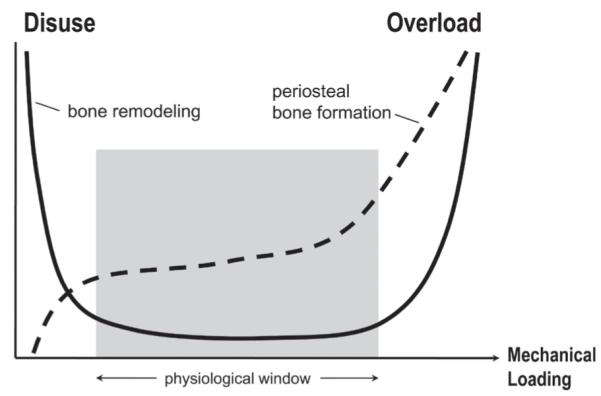
Bone turnover (remodeling) increases in states of disuse (Left) and overuse (Right). A physiological window falls between the extreme loading conditions, in which bone turnover is low. Bone formation at the periosteal bone surfaces is also affected by mechanical loading, but the relationship is different. Periosteal bone formation is very low in states of disuse and increases with increasing mechanical stimulus. Reprinted from Robling et al.,18 Annual Review of Biomedical Engineering ©2006 by Annual Reviews.
IV. MECHANICAL LOADING AND BONE FORMATION
Our studies employ a machine that applies axial loads to the forelimbs of rats or mice.19 The ulna is curved so when it is loaded it bows. This bowing causes a predictable pattern of deformation (strain) within the bone tissue. The highest strain is compressive and located on the medial surface. The second highest strain value is tensile, on the lateral surface. This strain distribution results in a reproducible pattern of strain energy (Fig. 9). Mechanical loading stimulates bone formation following a pattern consistent with the distribution of strain energy. This process of growth directed at regions of high strain energy is similar to strategies employed by structural engineers to achieve optimum structure. For instance, one engineering algorithm allows parts to “grow” in areas of high stress,21 thus reducing the stress concentrations. Parts that have been redesigned using this algorithm have sustained over 40-fold more loading cycles before failing. In the rat ulna, where bone growth is directed to regions of high strain energy, improvement in function is dramatically improved. After 16 weeks of loading, the rat ulna will sustain 100-fold more loading cycles before failure.22 These experimental results suggest that bone tissue contains sensors of strain energy. These sensors are mostly contained in the osteocytes embedded within the bone matrix. Since the osteocytes themselves cannot form new bone, we may assume that they send a signal to osteoblasts that enhances their bone forming activity. While several factors might fill the role of the osteocyte signal to osteoblasts, the most likely candidate is a protein called sclerostin, an osteocyte-specific secreted protein that is a potent inhibitor of Wnt signaling.
FIGURE 9.

The forelimb loading system causes the ulna to bow, which produces a distinct distribution of strain energy within the bone tissue. The highest strain energy is shown in red and the lowest in white. The amount of new bone formed in response to loading is proportional to the strain energy magnitude. A cross-section from a rat ulna is shown in the right panel. This rat was subjected to forelimb loading 3 times/wk for 16 weeks. The duration of each loading session was only 3 min. This short loading program substantially changed the shape of the bone section. The red line is a label showing the outline of the bone at the beginning of the experiment. One can clearly see where the bone was formed and these locations correspond with the red and orange regions in the middle panel.20
V. MECHANICAL SIGNALING IN BONE CELLS – THE ROLE OF THE WNT/LRP5 PATHWAY
The search for pathways involved in load-induced bone formation took a giant leap forward around the turn of this century, when it was discovered that the low-density lipoprotein receptor-related protein 5 (LRP5) had important functions in the mammalian skeleton. It was reported that the autosomal recessive human disease Osteoporosis-Pseudoglioma syndrome (OPPG) is caused by loss-of-function mutations in LRP5.23 Patients with OPPG have BMDs more than 5 standard deviations below the mean and are prone to skeletal fracture and deformity. Cultured fibroblasts and iliac crest biopsies from OPPG patients revealed no obvious abnormalities in collagen or other bone matrix proteins, nor did they exhibit abnormal morphologic changes in mineralized bone, osteoid, osteoblasts, or osteoclasts. Instead, patients with OPPG simply made too little bone.
Importantly, mice engineered with a loss-of-function mutation in Lrp5 exhibited low bone mass, but the weight-bearing portions of the skeleton bore a greater deficit in bone mass than the non-weight-bearing portions. Those observations led us to test the hypothesis that Lrp5 is important in mechanical signaling. When subjected to in vivo ulnar loading, we found a an 88–99% reduction in load-induced bone formation among Lrp5−/− mice, compared to wild-type controls.24 In a large human sample, Kiel et al. confirmed the role of LRP5 in mechanotransduction by reporting that several single nucleotide polymorphisms (SNPs) in LRP5, located in exons 10 and 18, significantly affected the relation between physical activity and bone mass.25 Taken together, these data indicate that LRP5 has an important role in translating mechanical signaling into the proper skeletal response.
Most data support an integral role for LRP5 in the “canonical” Wnt signaling pathway. This cascade begins by binding of secreted Wnt proteins to Lrp5 and a seven pass transmembrane co-receptor called Frizzled. Activation of the receptor complex at the cell surface by Wnt ultimately leads to the nuclear accumulation of β-catenin (β-cat), which complexes with members of the TCF/LEF transcription factor family to regulate gene transcription. Between the cell membrane (where Wnt, LRP5, and Frizzled interact) and the nucleus (where β-catenin accumulates) is a complex regulatory process that involves a large number of cytosolic proteins including disheveled (Dsh), Axin, adenomatous polyposis coli gene product (Apc), glycogen synthase kinase 3β, (Gsk3β), and casein kinase 1 (Ck1), among others. Normally, when Wnt signaling is not active, the Axin protein complex is intact and serves to bring Ck1 and Gsk3β into close proximity to β-cat, where they can (sequentially) phosphorylate β-cat.26 Phosphorylated (inactive) β-cat is ubiquitinated and consequently targeted for proteosomal degradation by the 26S proteosome. Wnt signaling causes the Axin complex to dismantle, which allows β-cat to avoid phosphorylation by Ck1 and Gsk3β. Activated (nonphosphorylated) β-cat can then accumulate in the cytoplasm, and subsequently translocate to the nucleus.
Several experiments have demonstrated the involvement of canonical Wnt signaling in mechanotransduction. Osteoblasts cultured from transgenic mice expressing a TCF/β-catenin transcription reporter (TopGal mice) exhibited activation of canonical Wnt signaling (β-galactosidase expression) when subjected to mechanical strain.27 In addition, fluid shear stress and mechanical strain both stimulate the translocation of β-catenin to the nucleus in MC3T3-E1, UMR-106, ROS 17/2.8, and primary rodent calvarial osteoblasts.28,29 Moreover, this translocation event — the hallmark of Wnt signaling — was accompanied by glycogen synthase kinase Gsk3β phosphorylation, further implicating the activation of canonical Wnt signaling. A host of Wnt/β-cat target genes (e.g., Wnt10b, sFrp1, Dkk3, Ccnd1, Wisp2) are upregulated after loading in vivo, and a more dramatic upregulation of those targets is seen after loading in the high-bone mass Col3.6::G171V transgenic mice.30 Similar results have been found in mechanically strained calvarial osteoblasts cultured from these mice. Furthermore, ulnar loading in TopGal mice (β-catenin/TCF transcriptional reporter) results in a rapid induction of β-gal activity in osteocytes, which precedes activation in surface osteoblasts by at least 20 h.31 Those studies indicate that Wnt signaling is initially activated by the resident osteocyte network, which supports the role of osteocytes in strain reception and translation.
While it is clear that Wnt/Lrp5 signaling is important for properly transmitting mechanical signaling in bone, an important consideration in this process is to define how mechanical stimulation enhances Lrp5 signaling. Activation of the receptor could be the result of enhanced agonism (e.g., enhanced Wnt secretion), reduced antagonism, or both. We investigated an Lrp5 antagonist — the protein sclerostin (product of the Sost gene) — as a potential modulator of Lrp5 activity in response to loading. Sclerostin is an osteocyte-specific, cysteine-knot secreted glycoprotein that is a potent inhibitor of bone formation. Mutations in the SOST gene are associated with very high bone mass.32,33 Sclerostin was originally characterized as a bone morphogenic protein (Bmp) antagonist because of its sequence homology to other members of the DAN family of cysteine-knot proteins, but more recently it has been shown to bind Lrp5/6 with high affinity.34-36 We looked at the mechanoregulation of Sost and sclerostin by subjecting adult mice and rats to ulnar loading in vivo and probing the tissue for protein and mRNA levels. We and others have found a dramatic reduction in sclerostin protein levels and in Sost transcripts (Fig. 10).31,37,38 Moreover, the regions of the bone that exhibited the greatest reductions in Sost and sclerostin were those that endured the greatest strains and underwent the greatest degree of osteogenesis. It should also be mentioned that we found a significant reduction in another Wnt signaling antagonist Dkk1 in the rat ulna as a result of loading. In addition, we found that tail suspension, a rodent model for disuse-induced bone loss, resulted in an increase in Sost mRNA levels in the mouse tibia (Fig. 11). Thus, modulation of sclerostin levels appears to be a finely tuned mechanism by which osteocytes coordinate regional and local osteogenesis in response to increased mechanical stimulation, perhaps via releasing the local inhibition of Wnt/Lrp5 signaling. It remains to be determined whether simply releasing the Lrp5 receptor from inhibition by downregulating sclerostin is sufficient to transduce mechanical signaling through the pathway, or if enhanced Wnt secretion is also required. Further studies involving genetic models within the Wnt pathway are underway to address this issue.
FIGURE 10.

Sclerostin mediation of Lrp5 signaling during mechanical loading. (Bottom) In the unstimulated state, osteocytes produce and secrete an ample supply of sclerostin, which serves to keep Wnt signaling low in the nearby osteoblast populations by antagonizing the LRP5 receptor. This process keeps β-catenin levels low (high level of Gsk-mediated β-catenin degradation). (Top) After mechanical stimulation, sclerostin levels drop and LRP5 becomes more available for Wnt binding, which triggers the accumulation of β-catenin in the cytosol, and eventually, the nucleus.
FIGURE 11.

Mechanical stimulation of the rodent forearm induces a decrease in Sost and Dkk1 mRNA after 2 days, whereas hinblimb unloading (disuse) induces an increase Sost but not Dkk1 transcripts after 3 days. sFrp1, another secreted Wnt pathway antagonist, remains unchanged by either stimulus. Reprinted from Robling et al.38 with permission from the publisher (American Society for Biochemistry and Molecular Biology).
VI. TEMPORAL SEQUENCE OF EVENTS IN MECHANOTRANSDUCTION
Once a mechanical stimulus is perceived by the receptor cells — most likely the osteocytes — a cascade of signaling events is activated that promotes local bone gain and inhibits local bone loss. Significant effort has been spent trying to delineate the cellular events — including ion channel activity, release of paracrine/autocrine factors, activation of enzymes, and movement of proteins from one subcellular domain to another — that occur in the seconds, minutes, and hours that follow reception of a mechanical signal in bone. There is some debate as to what serves as a mechanosensor in bone cells. Three candidates are mechanically sensitive ion channels, a G protein-coupled mechanoreceptor or focal adhesion signaling molecules like PYK2 and FAK (Fig. 12). As noted above, fluid forces are thought to activate mechanically sensitive ion channels (MSCs) in osteocytes.12 Some have proposed that activation of MSCs causes the cell membrane to depolarize and subsequently activate voltage sensitive calcium channels (VSCs).39 Indeed blocking VSCs has been shown to suppress bone mechanotransduction in rats.40 Others have shown that mechanosensing involves both MSCs and a G protein-coupled receptor.41 G protein-coupled mechanosensors have been shown to be primary mechanosensors in endothelial cells42 and they may play a similar role in osteocytes.
FIGURE 12.
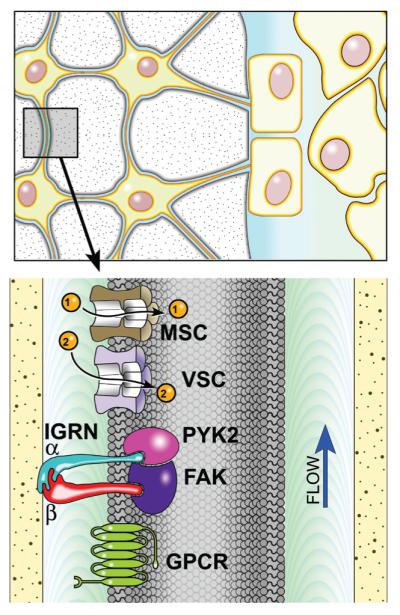
Mechanical stimuli may be sensed by mechanically sensitive ion channels (MSC), voltage sensitive ion channels (VSC), G protein-coupled mechanoreceptors (GPCR), or the focal adhesion complex made up of trans membrane integrins (IGRN) and several signaling molecules including focal adhesion kinase (FAK) and proline-rich tyrosine kinase 2 (PYK2).
Signaling through focal adhesions may provide another mechanism for mechanotransduction. Focal adhesion kinase (FAK) appears to play a role in bone mechanotransduction. For instance, bone regeneration around a mechanically loaded implant is suppressed in FAK-deficient mice.43 Another focal adhesion signaling molecule important in bone biology is Proline-rich tyrosine kinase 2 (PYK2). Pyk2 knockout mice have high bone mass resulting from increased bone formation44 and decreased bone resorption.45 PYK2 also mediates osteocyte apoptosis17 and may be important in mechanotransduction since it is known that mechanical loading suppresses osteocyte apoptosis and unloading induces osteocyte apoptosis.46,47 PYK2 is activated by phosphorylation of the Y402F site prior to osteocyte apoptosis. Furthermore, osteocyte apoptosis is blocked by calcium chelating agents BAPTA and EGTA and by gadolinium, a blocker of stretch activated channels demonstrating that PYK2 calcium binding is necessary to activate PY2 in osteocytes.
Two of the earliest events in mechanotransduction signaling, occurring within one minute of stimulus application, are an increase in intracellular Ca2+ concentrations and the release of adenosine triphosphate (ATP) from the cell (Fig. 13). Significant increases in intracellular Ca2+ can be detected in fluid-sheared osteoblasts within 20 sec of flow application.48 ATP release from fluid-sheared MC3T3 osteoblasts can be blocked by inhibitors of voltage sensitive calcium channels,49 indicating that the increase in intracellular Ca2+ is required for, and consequently precedes, ATP signaling. We confirmed the requirement of Ca2+ signaling for mechanotransduction in vivo by treating rats with two different L-type VSC antagonists (nifedipine or verapamil) 30 min before a loading bout. Both compounds reduced load-induced bone formation by 50–60%, highlighting the importance of Ca2+ signaling in that process.40 Furthermore, we have validated the role of ATP signaling in bone mechanotransduction in vivo by applying ulnar loading to mice harboring loss-of-function mutations in P2X7, an ionotropic ATP receptor. Those mice exhibited a 50–70% reduction in load-induced bone formation.50
FIGURE 13.
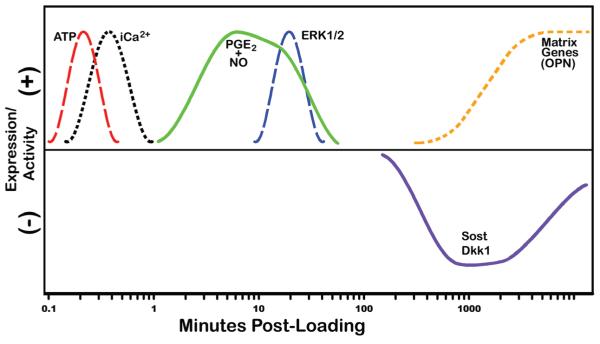
Timeline of major mechanotransduction signaling events. Within 1 minute of loading, bone cells activate Ca2+ signaling and release a bolus of ATP. A few minutes later, prostaglandin E2 (PGE2) and nitric oxide (NO) are released, followed by MAP-kinase signaling (ERK1/2 activation), Sost/Dkk1 downregulation, and ultimately, the expression of bone matrix genes, including osteopontin and collagen.
Soon after the Ca2+ and ATP signaling events take place, two other important second messengers participate in the response — prostaglandin and nitric oxide. Histological sections from mechanically loaded rat tibiae show strong immunostaining for Cox2 — a key enzyme in the synthesis of prostaglandins — in osteocytes immediately after application of load.51 In vital trabecular bone cores explanted from dog femora, mechanical loading at physiological strains stimulated a significant increase in PGE2 levels within minutes; no change in PGE2 was observed in nonloaded explants from contralateral femora.52 In human subjects, PGE2 levels in the proximal tibia were elevated 3.5-fold immediately after vigorous mechanical loading (originating from voluntary heel-drop exercises).53 The crucial role of prostaglandins in mechanotransduction has been demonstrated in several animal models. Animals administered compounds that block the enzymatic synthesis of PGE2 (indomethacin or NS-398) prior to loading showed no osteogenic response to an otherwise potent mechanical stimulus.54-56 Mice lacking the gene encoding one of the four prostaglandin receptors (EP-2) exhibit impaired response to mechanical loading of the tibia.57 Similarly, mice administered an EP-1 antagonist prior to loading show a reduced response to mechanical stimulation.58
Nitric oxide (NO) is another key early mediator of mechanotransduction. Osteocytes are a rich source of NO, more so that the other populations of bone cells,59 and they actively release NO upon mechanical stimulation. Ulnar explants from growing rats loaded in vitro release 50% more NO into the culture media than nonloaded controls after only 8 min of loading.60 Watanuke et al.61 tested the role of NO in mechanotransduction by subjecting wild-type mice and inducible nitric oxide synthase (iNOS) knockout mice to tail suspension for 1 week, then allowed the mice to reambulate normally to stimulate load-induced bone formation. Both genotypes lost bone during the tail suspension period, but reambulation restored bone mass in wild-type but not iNOS−/− mice. While mechanically-induced NO appears to have both anti-catabolic effects (via suppression of RANKL) and anti-apototic effects (via Bcl-2 and caspase-3), the target of NO in the effector cells remains unclear.62-64
Further downstream, elements of the Wnt pathway are activated/inactivated as a result of mechanical loading. By 12 h, there is a strong upregulation of β-catenin–induced transcriptional activity among osteocytes in loaded rodent bone.31 This activity occurs concomitantly with a reduction in Sost/sclerostin and Dkk1 levels.31,37,38 These alterations in the Wnt pathway are important for generating a matrix-formation response, based on the observation that Lrp5−/− mice are able to recruit osteoblasts to strained surfaces, but matrix formation is inhibited.24
VII. MECHANICAL CONTROL OF BONE RESORPTION
Unlike cells of the osteoblastic lineage, osteoclasts and their precursors are not likely to be sensors of the mechanical environment, but rater, rely on pro- or anti-resorptive signals originating from mechanically stimulated cells of the stromal lineage (e.g., osteocytes, osteoblasts, bone lining cells, and undifferentiated mesenchymal stem cells) residing nearby. In light of this control sequence, studying the direct effects of mechanical stimulation on osteoclasts probably would not yield meaningful information. Rather, the mechano-molecular regulation of osteoclastogenesis can be best ascertained by studying the signal output of stromal-derived cells. Consequently, the mechanical regulation of resorption can be, in theory, studied in the absence of osteoclasts, simply by mechanically manipulating osteoblast-lineage cells and probing for generation/expression of the relevant osteoclastogenic signaling molecules. In practice, however, it is obviously more attractive to maintain the effector cells (osteoclasts) in the experimental model to ascertain whether a particular mechanical stimulus is indeed associated with an increase or decrease of bone resorption. Those considerations make in vivo models of mechanotransduction more attractive, although significant progress in our understanding of mechanically controlled resorption has come from a number of sophisticated in vitro models (e.g., coculture).
The control of osteoclastogenesis by stromal-derived cells occurs via the M-CSF/RANKL/OPG signaling axis. This axis is responsible for controlling osteoclastogenesis and viability in response to a wide variety of stimuli (e.g., parathyroid hormone, prostaglandins, interleukins, vitamin D3, corticosteroids), including mechanical stimulation. Osteoclastogenesis begins when a hematopoietic stem cell residing in the marrow or spleen is stimulated to generate mononuclear cells, which then become committed osteoclast precursors and are introduced into the blood stream. Circulating precursors exit the peripheral circulation at or near the site to be resorbed, and in the presence of macrophage colony stimulating factor (M-CSF), fuse with one another to form a multinucleated immature osteoclast (polykaryon). Local expression of M-CSF (originating from osteoblast-lineage cells) is required for both fusion of mononuclear cells and maturation of the polykaryon into a mature osteoclast (Fig. 14A). At this stage, the receptor activator of nuclear factor κB (RANK-L), a tumor-necrosis factor family member is also required to achieve differentiation into a mature osteoclast and to maintain viability. RANKL, like M-CSF, is a locally produced osteoblast-lineage cell product that binds to receptors expressed on the cell surface of osteoclasts. Another relevant molecule produced by the osteoblast-lineage cells is osteo-protegerin (OPG), a secreted decoy receptor that binds to the available RANK-L in an autocrine fashion, and consequently competitively inhibits RANK signaling in the osteoclasts (Fig. 14B). By adjusting the relative levels of OPG and RANK-L, the stromal-lineage cells can control the local resorptive activity of osteoclasts in response to mechanical loading. Because these molecules, and the signaling pathways in which they participate, have such a profound effect on osteoclastogenesis and viability, they represent a sufficient and physiologically relevant set of endpoints to assess the mechanical effects on resorption in stromal-derived cells.
FIGURE 14.

Molecular control of bone resorption by the stroma-derived cell population. (Left) The stromal cells can be thought of as a rheostat in the resorptive process. M-CSF is required to activate the system, and once activated, the relative levels of RANK-L and OPG control osteoclastogenesis and resorptive activity. (Right) Both RANK receptors on the osteoclast (and pre-osteoclast) surface, and the soluble decoy OPG receptors in the extracellular fluid, bind RANK-L. The level of RANK-L and OPG therefore control the resorptive signal.
Studying the effect of mechanical stimulation on resorption in animal models has been much more difficult than studying the effects on formation, and consequently, our understanding of the mechanisms involved is less developed. Part of the problem is that most of the animal models developed for studying the effects of increased loading on the skeleton are designed for addressing cortical bone in rodents. Because rodent cortical bone does not undergo Haversian remodeling, resorption data are scarce. Rodent trabecular bone exhibits osteoclast-initiated turnover and therefore would be amenable to measurements of resorption (or osteoclast prevalence), but most of the load application models exclude the bone ends, where the trabecular bone is found.65-67 Only larger mammals exhibit significant intracortical turnover and it is to these models we must turn to look at actual resorptive activity (rather than pro-resorption molecular signaling) under enhanced loading conditions. The majority of larger animal studies, using a variety of models to induce enhanced loading conditions (e.g., squeezing of surgically implanted transcortical pins, administering a hard diet to mechanically stimulate mandibular bone, osteotomy to induce overloading of intact bones), indicate that loading increases Haversian remodeling rates, that is, stimulates resorption.65,66,68-71 However, it is unclear how much of the recruitment of osteoclasts, in the form of remodeling teams, was driven by the tissue strains and how much was driven by microdamage created in the tissue, a stimulus known to recruit osteoclasts.72 One clever method to circumvent the issue of intracortical damage was developed by Hillam and Skerry.73 They studied the periosteal osteoclasts that are involved in a coordinated modeling drift in the growing rat ulna, which help move the ulnar cortex through tissue space in a dorsomedial direction. These cells are not part of a remodeling team, but rather, continuously resorb bone on one surface of the bone in order to maintain proper cross-sectional size and shape as the bone lengthens. They applied axial loads to the growing ulnae and found not only a total disappearance of osteoclasts from the loaded cortex, but also a transformation of the formerly resorbing surface into a forming (osteoblast-lined) surface. Thus, in this model, loading had a clear suppressive effect on resorption.
The anti-catabolic effects of mechanical loading are modulated at least in part by nitric oxide (NO). Mechanical stimulation stimulates the release of NO from bone cells,74,75 and NO induces a decrease in the RANKL/OPG ratio, at both the mRNA and protein levels, in stromal cells.76 Inhibition of NO synthases during mechanical stimulation results in reduced anabolism77 and abrogates the anti-osteoclastogenic effects.64,78,79 The mechanism of action that translates the NO signal (i.e., the NO target) into the appropriate RANKL/OPG output is unclear, but it appears to involve the MapK pathway given that the Erk1/2 inhibitor PD98059 abolishes the mechanically induced RANKL expression.79
At the tissue and cellular level, mechanical stimulation is a strong modulator of osteocyte apoptosis, a process known to induce resorption. Too little mechanical stimulation induces osteocyte apoptosis via hypoxia mechanisms.46,80-82 Too great a mechanical stimulus generates physical damage to the bone matrix, and consequently, induces apoptosis and triggers osteoclast invasion via unknown mechanisms.9,16,83 Enhanced loading within the physiologic range is anti-apoptotic for osteocytes.16,84 The anti-apoptotic effects of physiologic mechanical stimulation are mediated by an integrin–MapK pathway that involves the β1, α2 and α5 integrin subunits, activation of several integrin-associated kinases (Fak, Src), and activation/translocation of Erk to the nucleus.47 Interestingly, mechanically induced Erk activation is modulated by the estrogen receptor, in a nonligand (i.e., even in the absence of estrogen analogs) dependent manner.46 Furthermore, mechanical stimulation recruits Erα to the membrane, where it might interact with the Fak/Src/Erk complex.85 NO signaling might also be involved in the anti-apoptotic effects of mechanical stimulation on osteocytes. Fluid flow normally upregulates Bcl-2 and downregulates caspase-3 expression in osteocytes, promoting survival, but those genes fail to change expression in the presence of the NOS inhibitor L-NAME.63
A number of in vitro mechanotransduction models (e.g., pressure, stretch, fluid flow) have been applied to study osteoclastogenic signaling, with mixed results. Mechanically stimulated stromal-lineage bone cell cultures have been reported to downregulate62,86 or upregulate87-89 RANKL transcripts. Likewise, OPG transcripts in cultured bone cells have been reported to decrease90 or increase89 in response to mechanical stimulation. Thus, the published data on the effects of mechanical loading on the RANKL/OPG signaling axis are inconsistent. This might be due, in part, to the wide variety of in vitro loading models employed, and the variation in the types of responses that they generate.
While the effects of increased loading on osteoclastogenesis markers and bone resorption per se can appear complicated, data on the effects of underloading, or disuse, are more consistent. Tail suspension is a widely used model to reduce ground reaction forces in the hindlimbs of rodents, and it results in rapid osteoclast-mediated bone loss.91,92 Not surprisingly, tail suspension induces an upregulation of RANKL and a downregulation of OPG mRNA in the tibiae, compared to ground control mice.92,93 Moreover, administration of recombinant OPG to tail suspended rats and mice protects the hindlimbs from bone loss, resulting in similar bone mass as ground-control rats.94,95
VIII. DESENSITIZATION (ACCOMMODATION) TO MECHANICAL SIGNALS
Bone cells are responsive to mechanical stimuli in the short term, but their sensitivity to the stimulus wanes quickly after its initiation. Consequently, loading cycles that occur toward the end of a loading bout are not as osteogenic as those that occurred toward the beginning of the bout. This phenomenon has been described as “diminishing returns”; that is, as the duration of the loading bout increases without interruption, the osteogenic response tends to saturate. The principle of mechanosensory saturation has been demonstrated in vivo in several different animal models (Fig. 15). Those experiments highlight two key points about bone mechanosensitivity: (1) that mechanical loading sessions need not be long to maximize bone formation, and (2) that extending the loading session beyond a few minutes does not contribute any additional osteogenic effect. Implicit in the mechanosensory saturation phenomenon is the existence of a recovery period. Saturated cells do become responsive again, as indicated by the observation that rat tibiae loaded once per day are capable of mounting as robust a response on loading day 2 as they did on loading day 1.98 Therefore, there must be a biological process involved in mechanosensory saturation, that prevents cells from responding further to mechanical stimuli, and in mechanosensory recovery, that restores sensitivity to the cells. Currently, the cellular and molecular mechanisms involved in these processes in bone cells are unclear, although several potential “off switches” in mechanotransduction have been identified (see Section V.).
FIGURE 15.

Bone mass and formation rates saturate relatively soon after a mechanical stimulus is initiated, as demonstrated by (A) the transcortically pinned turkey ulna,66 (B) the jumping rat model,96 and (C) in our lab using the rat tibia 4-point bending model.97
One of the notable features of the mechanosensory saturation/recovery phenomenon is that it occurs on several different time scales, ranging from seconds to weeks. Moreover, the existence and time constraints of these phenomena can be easily defined by manipulating the recovery periods introduced between mechanical stimulations — be they individual cycles, whole loading bouts, or week-long blocks of loading — and measuring the magnitude of the osteogenic response. Proof of this concept was published by Srinivasan et al.99 using two different animal models. They applied 100 load cycles per day to the turkey ulna (via the transcortical pin model) and to the mouse tibia (via the cantilever tibia bending model). Within each species, half of the animals were given all 100 cycles back to back, that is, with no rest between cycles, whereas the remaining animals were administered the 100 cycles with a 10-sec “no load” rest period between cycles. Among the turkeys receiving the back-to-back loading protocol, the loaded ulna exhibited a ~2-fold increase in labeled surface compared to the nonleaded ulna, whereas the rest-inserted protocol yielded a ~13-fold increase in labeled surface.99 We have followed up on this result by probing further into the duration of the short-term rest period required to increase osteogenesis. We administered 36 loading cycles per day to the tibiae of adult female rats in a single bout, 5 days a week for 2 weeks. Some of the rats were given the 36 daily cycles consecutively, with no time between each cycle (back-to-back cycles), while other rats were given different durations of rest between each of their 36 daily cycles, ranging from 3.5 to 14 sec. Rats given back-to-back cycles (0 sec rest), 3.5 sec of rest, or 7 sec of rest all exhibited statistically equivalent load-induced osteogenesis, but rats given 14 seconds between each load cycle formed 67% more bone than the remaining groups.100 Thus, the minimum recovery period required to enhance osteogenesis appears to be between 7 and 14 sec.
We have also observed a recovery effect on mechanically induced bone formation that appears to operate on the order of hours. In another rat tibial bending experiment, we subjected the right tibiae of rats to 4 separate bouts of loading per day for three nonconsecutive days. Each bout comprised 90 load cycles of an identical loading waveform (2 Hz, 54 N). The only difference among the 6 groups of rats was the amount of time allotted between each of the 4 daily loading bouts. The rats received 8, 4, 2, 1, 0.5, or 0 (equivalent to 360 back-to-back cycles) h between loading bouts, and their bone formation response was assessed 2 weeks later. The 8-h recovery group exhibited ~100% more bone formation that the 0-h or 0.5-h recovery group, suggesting that the recovery periods restored a significant amount of mechanosensitivity to the bone. In a subsequent experiment, we selected two groups—back-to-back cycles (360 × 1) and recovery group given 3 h between the 4 bouts of 90 cycles (90 × 4)—and conducted a longer-term experiment, lasting 16 weeks. We sought to learn whether the improvements in osteogenesis associated with the recovery protocol, observed after a week of loading, would be maintained in the long run (after 16 weeks of the protocol), or if the two protocols would yield similar bone mass and strength if the bone tissue was allowed sufficient time to fully adapt. After the 16 weeks of loading, DEXA measurements revealed significantly greater bone mineral content and areal bone mineral density in the ulnae of rats in the 90 × 4 group when compared to rats in the 360 × 1 group.20,101 Second moments of area at the midshaft were calculated from pQCT scans and revealed significantly greater resistance to bending in the direction of bending (which occurs in the IMIN plane), a result that was later confirmed in mechanical testing of the ulnae: bones from the 90 × 4 group exhibited a 30% greater ultimate force and a 45% greater energy to failure than the 360 × 1 group when tested in axial compression (Fig. 16). Our long-term data confirm the conclusions drawn from the short-term experiments highlighting the importance of recovery periods in restoring mechanosensitivity. Further, the ability of scheduling differences to maintain such dramatic differences in bone formation, strength, mass, and density, even after an extended period of loading, underscores the therapeutic potential of understanding the mechanisms controlling sensitization/desensitization in bone cells.
FIGURE 16.

Recovery periods on several time scales restore sensitivity to mechanical loading in bone cells. Mechanical stimulation is represented by blue wedges; recovery periods are represented by white wedges. Columns A-C represent the experimental designs (pie charts) and results (bar charts) of three separate mechanical loading experiments designed to reveal the effects of allowing bone cells to recover from individual mechanical loading cycles, bouts, and blocks. (A) Rat tibiae were administered 36 load cycles/day in a single daily bout for 2 weeks. Some of the rats were given 36 back-to-back cycles (lower pie chart), while others were given 14 seconds of recovery between each of their 36 daily cycles (upper pie chart). The relative bone formation rate was significantly greater in the recovery group compared to the back-to-back-cycles group, despite the fact that both groups received identical mechanical inputs.100 (B) Rat ulnae were subjected to 4 four bouts of loading per day (90 load cycles per bout), 3 days/wk for 16 weeks. Some of the rats were given all 4 bouts back to back (lower pie chart), while others were allotted 3 h between each of the bouts (upper pie chart). The increase in ulnar work to failure, a measure of whole bone toughness, was significantly greater in the recovery group compared to the back-to-back-bouts group, despite the fact that both groups received identical mechanical inputs.20 (C) Rat ulnae were exposed to two (upper pie chart) or three (lower pie chart) 5-week blocks of loading. The group exposed to only 2 blocks of loading was given a 5-week recovery period in the middle of the experiment. The recovery period group exhibited significantly greater ulnar work to failure, despite receiving less mechanical stimulation than the 15-week continuous loading group.102
Further experiments aimed at expanding the temporal spectrum of recovery period effects have revealed an effect of several weeks’ recovery on bone formation. We designated a 15-week experimental period comprising three time blocks: weeks 1–5, weeks 6–10, and weeks 11–15.102 One group was loaded during the first and third period (a 5-week recovery was introduced during the second period), while a second group was loaded during all three periods. Loading was administered once per day, 3 days/wk. Despite receiving less mechanical stimulation, the recovery group exhibited 73% greater work to failure than the group loaded for all three periods. Another benefit of the recovery protocol included a more advantageous geometric change in the ulnar diaphysis during the final stages of the experiment. Desensitization to mechanical loading appears to be an unavoidable consequence of mechanical stimulation, and it occurs on several time scales in bone.
IX. CONCLUDING REMARKS
Mechanical loading is a simple yet effective way to increase bone mass, decrease bone loss, and improve bone strength. As the cellular and molecular mechanisms involved in bone cell mechanotransduction become better defined, new targets for therapeutic intervention will be identified. Eventually, as these pathways become more thoroughly characterized, it will be possible to trigger the signaling cascades activated by mechanical loading without applying any force to the bone. Currently, parathyroid hormone (PTH) is the only FDA-approved compound for osteoporosis that has anabolic properties. As discussed above, a number of mechanotransduction pathways are highly anabolic in bone, so learning how to activate those pathways without inducing unwanted side effects presents itself as the next great challenge.
ACKNOWLEDGMENTS
The authors are grateful to the National Institutes of Health for supporting this work (Grants AR053237 to Alexander G. Robling and AR046530 to Charles H. Turner).
REFERENCES
- 1.Wolff J. The law of bone transformation. A. Hirschwald; Berlin: 1892. [Google Scholar]
- 2.Murray PDF . Bones – A study of the development and structure of the vertebrate skeleton. Cambridge University Press; Cambridge: 1936. [Google Scholar]
- 3.von Meyer C. Die architektur der spongiosa. Arch Anat Physiol Wiss Med Reichert DuBois-Reymonds Arch. 1867;34:615–28. [Google Scholar]
- 4.Rodriguez JI, Palacios J, Garcia-Alix A, Pastor I, Paniagua R. Effects of immobilization on fetal bone development. A morphometric study in newborns with congenital neuromuscular diseases with intrauterine onset. Calcif Tissue Int. 1988;43:335–9. doi: 10.1007/BF02553275. [DOI] [PubMed] [Google Scholar]
- 5.Ralis ZA, Ralis HM, Randall M, Watkins G, Blake PD. Changes in shape, ossification and quality of bones in children with spina bifida. Dev Med Child Neurol Suppl. 1976:29–41. doi: 10.1111/j.1469-8749.1976.tb04278.x. [DOI] [PubMed] [Google Scholar]
- 6.Jones HH, Priest JD, Hayes WC, Tichenor CC, Nagel DA. Humeral hypertrophy in response to exercise. J Bone Joint Surg Am. 1977;59:204–8. [PubMed] [Google Scholar]
- 7.Jorgensen NR, Geist ST, Civitelli R, Steinberg TH. ATP- and gap junction-dependent intercellular calcium signaling in osteoblastic cells. J Cell Biol. 1997;139:497–506. doi: 10.1083/jcb.139.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lean JM, Mackay AG, Chow JW, Chambers TJ. Osteocytic expression of mRNA for c-fos and IGF-I: an immediate early gene response to an osteogenic stimulus. Am J Physiol. 1996;270:E937–45. doi: 10.1152/ajpendo.1996.270.6.E937. [DOI] [PubMed] [Google Scholar]
- 9.Verborgt O, Gibson GJ, Schaffler MB. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000;15:60–7. doi: 10.1359/jbmr.2000.15.1.60. [DOI] [PubMed] [Google Scholar]
- 10.Robling AG, Duijvelaar KM, Geevers JV, Ohashi N, Turner CH. Modulation of appositional and longitudinal bone growth in the rat ulna by applied static and dynamic force. Bone. 2001;29:105–13. doi: 10.1016/s8756-3282(01)00488-4. [DOI] [PubMed] [Google Scholar]
- 11.Han Y, Cowin SC, Schaffler MB, Weinbaum S. Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci U S A. 2004;101:16689–94. doi: 10.1073/pnas.0407429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, McNamara LM, Schaffler MB, Weinbaum S. A model for the role of integrins in flow induced mechanotransduction in osteocytes. Proc Natl Acad Sci U S A. 2007;104:15941–6. doi: 10.1073/pnas.0707246104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh YF, Turner CH. Effects of loading frequency on mechanically induced bone formation. J Bone Miner Res. 2001;16:918–24. doi: 10.1359/jbmr.2001.16.5.918. [DOI] [PubMed] [Google Scholar]
- 14.Warden SJ, Turner CH. Mechanotransduction in the cortical bone is most efficient at loading frequencies of 5–10 Hz. Bone. 2004;34:261–70. doi: 10.1016/j.bone.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Burr DB, Martin RB, Schaffler MB, Radin EL. Bone remodeling in response to in vivo fatigue microdamage. J Biomech. 1985;18:189–200. doi: 10.1016/0021-9290(85)90204-0. [DOI] [PubMed] [Google Scholar]
- 16.Noble BS, Peet N, Stevens HY, Brabbs A, Mosley JR, Reilly GC, Reeve J, Skerry TM, Lanyon LE. Mechanical loading: biphasic osteocyte survival and targeting of osteoclasts for bone destruction in rat cortical bone. Am J Physiol Cell Physiol. 2003;284:C934–43. doi: 10.1152/ajpcell.00234.2002. [DOI] [PubMed] [Google Scholar]
- 17.Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival. Evidence for inside-out signaling leading to anoikis. J Biol Chem. 2007;282:24120–30. doi: 10.1074/jbc.M611435200. [DOI] [PubMed] [Google Scholar]
- 18.Robling AG, Castillo AB, Turner CH. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng. 2006;8:455–98. doi: 10.1146/annurev.bioeng.8.061505.095721. [DOI] [PubMed] [Google Scholar]
- 19.Torrance AG, Mosley JR, Suswillo RF, Lanyon LE. Noninvasive loading of the rat ulna in vivo induces a strain-related modeling response uncomplicated by trauma or periostal pressure. Calcif Tissue Int. 1994;54:241–7. doi: 10.1007/BF00301686. [DOI] [PubMed] [Google Scholar]
- 20.Robling AG, Hinant FM, Burr DB, Turner CH. Improved bone structure and strength after long-term mechanical loading is greatest if loading is separated into short bouts. J Bone Miner Res. 2002;17:1545–54. doi: 10.1359/jbmr.2002.17.8.1545. [DOI] [PubMed] [Google Scholar]
- 21.Mattheck C. Design in nature: learning from trees. Springer; Berlin: 1997. [Google Scholar]
- 22.Warden SJ, Hurst JA, Sanders MS, Turner CH, Burr DB, Li J. Bone adaptation to a mechanical loading program significantly increases skeletal fatigue resistance. J Bone Miner Res. 2005;20:809–16. doi: 10.1359/JBMR.041222. [DOI] [PubMed] [Google Scholar]
- 23.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 24.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 25.Kiel DP, Ferrari SL, Cupples LA, Karasik D, Manen D, Imamovic A, Herbert AG, Dupuis J. Genetic variation at the low-density lipoprotein receptor-related protein 5 (LRP5) locus modulates Wnt signaling and the relationship of physical activity with bone mineral density in men. Bone. 2007;40:587–96. doi: 10.1016/j.bone.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–47. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 27.Hens JR, Wilson KM, Dann P, Chen X, Horowitz MC, Wysolmerski JJ. TOPGAL mice show that the canonical Wnt signaling pathway is active during bone development and growth and is activated by mechanical loading in vitro. J Bone Miner Res. 2005;20:1103–13. doi: 10.1359/JBMR.050210. [DOI] [PubMed] [Google Scholar]
- 28.Norvell SM, Alvarez M, Bidwell JP, Pavalko FM. Fluid shear stress induces beta-catenin signaling in osteoblasts. Calcif Tissue Int. 2004;75:396–404. doi: 10.1007/s00223-004-0213-y. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE. Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J Biol Chem. 2007;282:20715–27. doi: 10.1074/jbc.M703224200. [DOI] [PubMed] [Google Scholar]
- 30.Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, Cullen DM, Zhao W, Li C, Kharode Y, Sauter L, Babij P, Brown EL, Hill AA, Akhter MP, Johnson ML, Recker RR, Komm BS, Bex FJ. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–8. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- 31.Kim-Weroha NA, Ferris A, Holladay B, Kotha SP, Kamel MA, Johnson ML. In vivo load activated propagation of b-catenin signaling in osteocytes through coordinated downregulation of inhibitors of Lrp5. J Bone Miner Res. 2008;23:S13. [Google Scholar]
- 32.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 33.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–7. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–5. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 35.Semenov MV, He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006;281:38276–84. doi: 10.1074/jbc.M609509200. [DOI] [PubMed] [Google Scholar]
- 36.Weidauer SE, Schmieder P, Beerbaum M, Schmitz W, Oschkinat H, Mueller TD. NMR structure of the Wnt modulator protein Sclerostin. Biochem Biophys Res Commun. 2009;380:160–5. doi: 10.1016/j.bbrc.2009.01.062. [DOI] [PubMed] [Google Scholar]
- 37.Moustafa A, Sugiyama T, Saxon LK, Zaman G, Sunters A, Armstrong VJ, Javaheri B, Lanyon LE, Price JS. The mouse fibula as a suitable bone for the study of functional adaptation to mechanical loading. Bone. 2009;44:930–5. doi: 10.1016/j.bone.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 39.Duncan RL, Turner CH. Mechanotransduction and the functional response of bone to mechanical strain. Calcif Tissue Int. 1995;57:344–58. doi: 10.1007/BF00302070. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Duncan RL, Burr DB, Turner CH. L-type calcium channels mediate mechanically induced bone formation in vivo. J Bone Miner Res. 2002;17:1795–800. doi: 10.1359/jbmr.2002.17.10.1795. [DOI] [PubMed] [Google Scholar]
- 41.Reich KM, McAllister TN, Gudi S, Frangos JA. Activation of G proteins mediates flow-induced prostaglandin E2 production in osteoblasts. Endocrinology. 1997;138:1014–8. doi: 10.1210/endo.138.3.4999. [DOI] [PubMed] [Google Scholar]
- 42.Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci U S A. 2006;103:15463–8. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leucht P, Kim JB, Currey JA, Brunski J, Helms JA. FAK-Mediated mechanotransduction in skeletal regeneration. PLoS One. 2007;2:e390. doi: 10.1371/journal.pone.0000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buckbinder L, Crawford DT, Qi H, Ke HZ, Olson LM, Long KR, Bonnette PC, Baumann AP, Hambor JE, Grasser WA, 3rd, Pan LC, Owen TA, Luzzio MJ, Hulford CA, Gebhard DF, Paralkar VM, Simmons HA, Kath JC, Roberts WG, Smock SL, Guzman-Perez A, Brown TA, Li M. Proline-rich tyrosine kinase 2 regulates osteoprogenitor cells and bone formation, and offers an anabolic treatment approach for osteoporosis. Proc Natl Acad Sci U S A. 2007;104:10619–24. doi: 10.1073/pnas.0701421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gil-Henn H, Destaing O, Sims NA, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camilli P, Baron R, Schlessinger J. Defective microtubule-dependent podosome organization in osteoclasts leads to increased bone density in Pyk2(−/−) mice. J Cell Biol. 2007;178:1053–64. doi: 10.1083/jcb.200701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 47.Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases, and ERKs. Am J Physiol Cell Physiol. 2005;289:C633–43. doi: 10.1152/ajpcell.00278.2004. [DOI] [PubMed] [Google Scholar]
- 48.Hung CT, Allen FD, Pollack SR, Brighton CT. Intracellular Ca2+ stores and extracellular Ca2+ are required in the real-time Ca2+ response of bone cells experiencing fluid flow. J Biomech. 1996;29:1411–7. doi: 10.1016/0021-9290(96)84536-2. [DOI] [PubMed] [Google Scholar]
- 49.Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL. Fluid shear-induced ATP secretion mediates prostaglandin release in MC3T3-E1 osteoblasts. J Bone Miner Res. 2005;20:41–9. doi: 10.1359/JBMR.041009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Liu D, Ke HZ, Duncan RL, Turner CH. The P2X7 nucleotide receptor mediates skeletal mechanotransduction. J Biol Chem. 2005;280:42952–9. doi: 10.1074/jbc.M506415200. [DOI] [PubMed] [Google Scholar]
- 51.Forwood MR, Kelly WL, Worth NF. Localisation of prostaglandin endoperoxide H synthase (PGHS)-1 and PGHS-2 in bone following mechanical loading in vivo. Anat Rec. 1998;252:580–6. doi: 10.1002/(SICI)1097-0185(199812)252:4<580::AID-AR8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 52.Rawlinson SC, el-Haj AJ, Minter SL, Tavares IA, Bennett A, Lanyon LE. Loading-related increases in prostaglandin production in cores of adult canine cancellous bone in vitro: a role for prostacyclin in adaptive bone remodeling? J Bone Miner Res. 1991;6:1345–51. doi: 10.1002/jbmr.5650061212. [DOI] [PubMed] [Google Scholar]
- 53.Thorsen K, Kristoffersson AO, Lerner UH, Lorentzon RP. In situ microdialysis in bone tissue. Stimulation of prostaglandin E2 release by weight-bearing mechanical loading. J Clin Invest. 1996;98:2446–9. doi: 10.1172/JCI119061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chow JW, Chambers TJ. Indomethacin has distinct early and late actions on bone formation induced by mechanical stimulation. Am J Physiol. 1994;267:E287–92. doi: 10.1152/ajpendo.1994.267.2.E287. [DOI] [PubMed] [Google Scholar]
- 55.Forwood MR. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J Bone Miner Res. 1996;11:1688–93. doi: 10.1002/jbmr.5650111112. [DOI] [PubMed] [Google Scholar]
- 56.Pead MJ, Lanyon LE. Indomethacin modulation of load-related stimulation of new bone formation in vivo. Calcif Tissue Int. 1989;45:34–40. doi: 10.1007/BF02556658. [DOI] [PubMed] [Google Scholar]
- 57.Cullen DM, Peyton AC, Akhter MP, Gong G, Johnson ML. Mechanical loading response in EP2 receptor knockout mice. J Bone Miner Res. 2000;15:S476. [Google Scholar]
- 58.Gong G, Cullen DM, Peyton AC, Akhter MP, Recker RR. EP1 antagonist inhibition of bone formation induced by mechanical loading. J Bone Miner Res. 2000;15:S477. [Google Scholar]
- 59.Zaman G, Pitsillides AA, Rawlinson SC, Suswillo RF, Mosley JR, Cheng MZ, Platts LA, Hukkanen M, Polak JM, Lanyon LE. Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res. 1999;14:1123–31. doi: 10.1359/jbmr.1999.14.7.1123. [DOI] [PubMed] [Google Scholar]
- 60.Rawlinson SC, Pitsillides AA, Lanyon LE. Involvement of different ion channels in osteoblasts’ and osteocytes’ early responses to mechanical strain. Bone. 1996;19:609–14. doi: 10.1016/s8756-3282(96)00260-8. [DOI] [PubMed] [Google Scholar]
- 61.Watanuki M, Sakai A, Sakata T, Tsurukami H, Miwa M, Uchida Y, Watanabe K, Ikeda K, Nakamura T. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J Bone Miner Res. 2002;17:1015–25. doi: 10.1359/jbmr.2002.17.6.1015. [DOI] [PubMed] [Google Scholar]
- 62.Rahnert J, Fan X, Case N, Murphy TC, Grassi F, Sen B, Rubin J. The role of nitric oxide in the mechanical repression of RANKL in bone stromal cells. Bone. 2008;43:48–54. doi: 10.1016/j.bone.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan SD, Bakker AD, Semeins CM, Kuijpers-Jagtman AM, Klein-Nulend J. Inhibition of osteocyte apoptosis by fluid flow is mediated by nitric oxide. Biochem Biophys Res Commun. 2008;369:1150–4. doi: 10.1016/j.bbrc.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 64.Tan SD, de Vries TJ, Kuijpers-Jagtman AM, Semeins CM, Everts V, Klein-Nulend J. Osteocytes subjected to fluid flow inhibit osteoclast formation and bone resorption. Bone. 2007;41:745–51. doi: 10.1016/j.bone.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 65.Hert J, Pribylova E, Liskova M. Reaction of bone to mechanical stimuli. 3 Microstructure of compact bone of rabbit tibia after intermittent loading. Acta Anat (Basel) 1972;82:218–30. [PubMed] [Google Scholar]
- 66.Rubin CT, Lanyon LE. Regulation of bone formation by applied dynamic loads. J Bone Joint Surg Am. 1984;66:397–402. [PubMed] [Google Scholar]
- 67.Turner CH, Forwood MR, Rho JY, Yoshikawa T. Mechanical loading thresholds for lamellar and woven bone formation. J Bone Miner Res. 1994;9:87–97. doi: 10.1002/jbmr.5650090113. [DOI] [PubMed] [Google Scholar]
- 68.Bouvier M, Hylander WL. Effect of bone strain on cortical bone structure in macaques (Macaca mulatta) J Morphol. 1981;167:1–12. doi: 10.1002/jmor.1051670102. [DOI] [PubMed] [Google Scholar]
- 69.Bouvier M, Hylander WL. The effect of dietary consistency on gross and histologic morphology in the craniofacial region of young rats. Am J Anat. 1984;170:117–26. doi: 10.1002/aja.1001700109. [DOI] [PubMed] [Google Scholar]
- 70.Lanyon LE, Goodship AE, Pye CJ, MacFie JH. Mechanically adaptive bone remodelling. J Biomech. 1982;15:141–54. doi: 10.1016/0021-9290(82)90246-9. [DOI] [PubMed] [Google Scholar]
- 71.Lieberman DE, Pearson OM, Polk JD, Demes B, Crompton AW. Optimization of bone growth and remodeling in response to loading in tapered mammalian limbs. J Exp Biol. 2003;206:3125–38. doi: 10.1242/jeb.00514. [DOI] [PubMed] [Google Scholar]
- 72.Mori S, Burr DB. Increased intracortical remodeling following fatigue damage. Bone. 1993;14:103–9. doi: 10.1016/8756-3282(93)90235-3. [DOI] [PubMed] [Google Scholar]
- 73.Hillam RA, Skerry TM. Inhibition of bone resorption and stimulation of formation by mechanical loading of the modeling rat ulna in vivo. J Bone Miner Res. 1995;10:683–9. doi: 10.1002/jbmr.5650100503. [DOI] [PubMed] [Google Scholar]
- 74.Bacabac RG, Smit TH, Mullender MG, Dijcks SJ, Van Loon JJ, Klein-Nulend J. Nitric oxide production by bone cells is fluid shear stress rate dependent. Biochem Biophys Res Commun. 2004;315:823–9. doi: 10.1016/j.bbrc.2004.01.138. [DOI] [PubMed] [Google Scholar]
- 75.Mullender MG, Dijcks SJ, Bacabac RG, Semeins CM, Van Loon JJ, Klein-Nulend J. Release of nitric oxide, but not prostaglandin E2, by bone cells depends on fluid flow frequency. J Orthop Res. 2006;24:1170–7. doi: 10.1002/jor.20179. [DOI] [PubMed] [Google Scholar]
- 76.Fan X, Roy E, Zhu L, Murphy TC, Ackert-Bicknell C, Hart CM, Rosen C, Nanes MS, Rubin J. Nitric oxide regulates receptor activator of nuclear factor-kappaB ligand and osteoprotegerin expression in bone marrow stromal cells. Endocrinology. 2004;145:751–9. doi: 10.1210/en.2003-0726. [DOI] [PubMed] [Google Scholar]
- 77.Turner CH, Owan I, Jacob DS, McClintock R, Peacock M. Effects of nitric oxide synthase inhibitors on bone formation in rats. Bone. 1997;21:487–90. doi: 10.1016/s8756-3282(97)00202-0. [DOI] [PubMed] [Google Scholar]
- 78.Fan X, Rahnert JA, Murphy TC, Nanes MS, Greenfield EM, Rubin J. Response to mechanical strain in an immortalized pre-osteoblast cell is dependent on ERK1/2. J Cell Physiol. 2006;207:454–60. doi: 10.1002/jcp.20581. [DOI] [PubMed] [Google Scholar]
- 79.Rubin J, Murphy TC, Fan X, Goldschmidt M, Taylor WR. Activation of extracellular signal-regulated kinase is involved in mechanical strain inhibition of RANKL expression in bone stromal cells. J Bone Miner Res. 2002;17:1452–60. doi: 10.1359/jbmr.2002.17.8.1452. [DOI] [PubMed] [Google Scholar]
- 80.Gross TS, Akeno N, Clemens TL, Komarova S, Srinivasan S, Weimer DA, Mayorov S. Selected Contribution: Osteocytes upregulate HIF-1alpha in response to acute disuse and oxygen deprivation. J Appl Physiol. 2001;90:2514–9. doi: 10.1152/jappl.2001.90.6.2514. [DOI] [PubMed] [Google Scholar]
- 81.Gross TS, King KA, Rabaia NA, Pathare P, Srinivasan S. Upregulation of osteopontin by osteocytes deprived of mechanical loading or oxygen. J Bone Miner Res. 2005;20:250–6. doi: 10.1359/JBMR.041004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Basso N, Heersche JN. Effects of hind limb unloading and reloading on nitric oxide synthase expression and apoptosis of osteocytes and chondrocytes. Bone. 2006;39:807–14. doi: 10.1016/j.bone.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 83.Verborgt O, Tatton NA, Majeska RJ, Schaffler MB. Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation? J Bone Miner Res. 2002;17:907–14. doi: 10.1359/jbmr.2002.17.5.907. [DOI] [PubMed] [Google Scholar]
- 84.Simpson AE, Stoddart MJ, Davies CM, Jahn K, Furlong PI, Gasser JA, Jones DB, Noble BS, Richards RG. TGFbeta3 and loading increases osteocyte survival in human cancellous bone cultured ex vivo. Cell Biochem Funct. 2009;27:23–9. doi: 10.1002/cbf.1529. [DOI] [PubMed] [Google Scholar]
- 85.Zaman G, Jessop HL, Muzylak M, De Souza RL, Pitsillides AA, Price JS, Lanyon LL. Osteocytes use estrogen receptor alpha to respond to strain but their ERalpha content is regulated by estrogen. J Bone Miner Res. 2006;21:1297–306. doi: 10.1359/jbmr.060504. [DOI] [PubMed] [Google Scholar]
- 86.Rubin J, Murphy T, Nanes MS, Fan X. Mechanical strain inhibits expression of osteoclast differentiation factor by murine stromal cells. Am J Physiol Cell Physiol. 2000;278:C1126–32. doi: 10.1152/ajpcell.2000.278.6.C1126. [DOI] [PubMed] [Google Scholar]
- 87.Liu J, Zhao Z, Zou L, Li J, Wang F, Li X, Zhang J, Liu Y, Chen S, Zhi M, Wang J. Pressure-Loaded MSCs During early osteodifferentiation promote osteoclastogenesis by increase of RANKL/OPG ratio. Ann Biomed Eng. 2009;37:794–802. doi: 10.1007/s10439-009-9638-9. [DOI] [PubMed] [Google Scholar]
- 88.Mehrotra M, Saegusa M, Wadhwa S, Voznesensky O, Peterson D, Pilbeam C. Fluid flow induces Rankl expression in primary murine calvarial osteoblasts. J Cell Biochem. 2006;98:1271–83. doi: 10.1002/jcb.20864. [DOI] [PubMed] [Google Scholar]
- 89.Kim CH, You L, Yellowley CE, Jacobs CR. Oscillatory fluid flow-induced shear stress decreases osteoclastogenesis through RANKL and OPG signaling. Bone. 2006;39:1043–7. doi: 10.1016/j.bone.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 90.Sanchez C, Gabay O, Salvat C, Henrotin YE, Berenbaum F. Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarthritis Cartilage. 2009;17(4):473–81. doi: 10.1016/j.joca.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 91.David V, Laroche N, Boudignon B, Lafage-Proust MH, Alexandre C, Ruegsegger P, Vico L. Noninvasive in vivo monitoring of bone architecture alterations in hindlimb-unloaded female rats using novel three-dimensional microcomputed tomography. J Bone Miner Res. 2003;18:1622–31. doi: 10.1359/jbmr.2003.18.9.1622. [DOI] [PubMed] [Google Scholar]
- 92.Tanaka S, Sakai A, Tanaka M, Otomo H, Okimoto N, Sakata T, Nakamura T. Skeletal unloading alleviates the anabolic action of intermittent PTH(1-34) in mouse tibia in association with inhibition of PTH-induced increase in c-fos mRNA in bone marrow cells. J Bone Miner Res. 2004;19:1813–20. doi: 10.1359/JBMR.040808. [DOI] [PubMed] [Google Scholar]
- 93.Martin A, de Vittoris R, David V, Moraes R, Begeot M, Lafage-Proust MH, Alexandre C, Vico L, Thomas T. Leptin modulates both resorption and formation while preventing disuse-induced bone loss in tail-suspended female rats. Endocrinology. 2005;146:3652–9. doi: 10.1210/en.2004-1509. [DOI] [PubMed] [Google Scholar]
- 94.Ichinose Y, Tanaka H, Inoue M, Mochizuki S, Tsuda E, Seino Y. Osteoclastogenesis inhibitory factor/osteoprotegerin reduced bone loss induced by mechanical unloading. Calcif Tissue Int. 2004;75:338–43. doi: 10.1007/s00223-004-0028-x. [DOI] [PubMed] [Google Scholar]
- 95.Bateman TA, Dunstan CR, Ferguson VL, Lacey DL, Ayers RA, Simske SJ. Osteoprotegerin mitigates tail suspension-induced osteopenia. Bone. 2000;26:443–9. doi: 10.1016/S8756-3282(00)00256-8. [DOI] [PubMed] [Google Scholar]
- 96.Umemura Y, Ishiko T, Yamauchi T, Kurono M, Mashiko S. Five jumps per day increase bone mass and breaking force in rats. J Bone Miner Res. 1997;12:1480–5. doi: 10.1359/jbmr.1997.12.9.1480. [DOI] [PubMed] [Google Scholar]
- 97.Turner CH. Three rules for bone adaptation to mechanical stimuli. Bone. 1998;23:399–407. doi: 10.1016/s8756-3282(98)00118-5. [DOI] [PubMed] [Google Scholar]
- 98.Forwood MR, Turner CH. The response of rat tibiae to incremental bouts of mechanical loading: a quantum concept for bone formation. Bone. 1994;15:603–9. doi: 10.1016/8756-3282(94)90307-7. [DOI] [PubMed] [Google Scholar]
- 99.Srinivasan S, Weimer DA, Agans SC, Bain SD, Gross TS. Low-magnitude mechanical loading becomes osteogenic when rest is inserted between each load cycle. J Bone Miner Res.l. 2002;17:1613–20. doi: 10.1359/jbmr.2002.17.9.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robling AG, Burr DB, Turner CH. Recovery periods restore mechanosensitivity to dynamically loaded bone. J Exp Biol. 2001;204:3389–99. doi: 10.1242/jeb.204.19.3389. [DOI] [PubMed] [Google Scholar]
- 101.Robling AG, Hinant FM, Burr DB, Turner CH. Shorter, more frequent mechanical loading sessions enhance bone mass. Med Sci Sports Exerc. 2002;34:196–202. doi: 10.1097/00005768-200202000-00003. [DOI] [PubMed] [Google Scholar]
- 102.Saxon LK, Robling AG, Alam I, Turner CH. Mechanosensitivity of the rat skeleton decreases after a long period of loading, but is improved with time off. Bone. 2005;36:454–64. doi: 10.1016/j.bone.2004.12.001. [DOI] [PubMed] [Google Scholar]