

Abstract
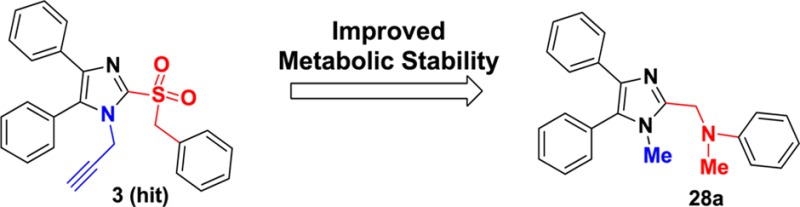
Targeting host cell factors required for virus replication provides an alternative to targeting pathogen components and represents a promising approach to develop broad-spectrum antiviral therapeutics. High-throughput screening (HTS) identified two classes of inhibitors (2 and 3) with broad-spectrum antiviral activity against ortho- and paramyxoviruses including influenza A virus (IAV), measles virus (MeV), respiratory syncytial virus (RSV), and human parainfluenza virus type 3 (HPIV3). Hit-to-lead optimization delivered inhibitor 28a, with EC50 values of 0.88 and 0.81 μM against IAV strain WSN and MeV strain Edmonston, respectively. It was also found that compound 28a delivers good stability in human liver S9 fractions with a half-life of 165 min. These data establish 28a as a promising lead for antiviral therapy through a host-directed mechanism.
Keywords: Myxovirus inhibitor, host-directed, influenza A, respiratory syncytial virus, benzimidazole, metabolic stability
The orthomyxovirus and paramyxovirus families comprise many pathogens that cause common respiratory illnesses; among them are measles virus, mumps virus, respiratory syncytial virus, human parainfluenza virus (paramyxoviruses), and influenza viruses A and B (orthomyxoviruses). Together, the acute respiratory illnesses caused by these viruses represent a major medical need.1 In particular, the likelihood of a pandemic from influenza A makes developing new and broadly active therapies a pressing global need.2 Our research group has undertaken a program of discovering and developing antivirals that target host factors implicated in virus reproduction. Although this runs counter to the prevailing virus-targeted mechanistic approach to discovering antivirals, the potential benefits of such a strategy include decreased incidence of resistance because host factors mutate less readily than viral factors and a broadened spectrum of antiviral activity because many of the same host factors are co-opted by different viruses.3,4 A potential drawback to this approach is the nontrivial effort required to define mechanism of action since we use a whole-cell phenotypic screen to identify these antivirals.5 To date, our most successful compound series has been exemplified by the broadly active benzimidazole 1 (JMN3-003), which has low nanomolar activity against a number of different viruses in cell culture.6,7 In our efforts to develop a backup to 1, we have re-examined two different hits (scaffolds 2 and 3, Figure 1) that were described in the same high-throughput screening paper in which we first disclosed benzimidazole 1.5 This report details our attempts to simultaneously improve potency against both an orthomyxovirus (influenza A) and paramyxovirus (measles) representative and to understand the metabolic profile in liver S9 fractions for 2 and 3.
Figure 1.

Structures of host-directed antivirals and their biological profiles.
To confirm the biological activity of series 2 and 3, these two hits along with a small set of analogues were synthesized and tested in reporter gene assays. In the case of IAV assays, a firefly luciferase minireplicon reporter was driven by superinfection of cells with influenza A/sw/Texas/2009 or influenza A/Aichi/1968. EC50 (50% inhibitory concentrations) were calculated by means of four-parameter nonlinear regression-fitting; values in parentheses represent 95% confidence intervals. Syntheses of 2 and closely related analogues were initiated by treatment of commercially available benzimidazolylacetonitrile 4 with sodium nitrite in acetic acid. The resulting cyano oxime 5 was combined with hydroxylamine and cyclized to afford furazan 6. The latter was further coupled with acyl chlorides 7 to deliver 8, followed by alkylation to give compound 2 and its analogues (Scheme 1). Analogues with a thiazole, pyrazole, or pyrazine instead of the furazan moiety were also synthesized (Schemes S2–5, Supporting Information).
Scheme 1.

Compound 2 and analogues were retested against both IAV and MeV. Replacement of the ethyl ester with a 4-aminophenyl moiety (2b) led to a slight loss of activity, while replacement with hydrogen caused a complete loss of potency against both viruses (2a). All series 2 analogues featuring an altered central ring system lost bioactivity (2c–g, Table 1). Active compounds 2 and 2b were subjected to further chemical and metabolic stability testing against human and hamster liver S9 fractions (T1/2 < 5 min in the presence or absence of cofactors). Although the potency of compound 2 is good, we speculate that the biological activity of this compound might arise from its metabolic or chemical instability, which ultimately led us to drop pursuit of this series in favor of alternative scaffold 3.
Table 1. Analogues of Hit Series 2.
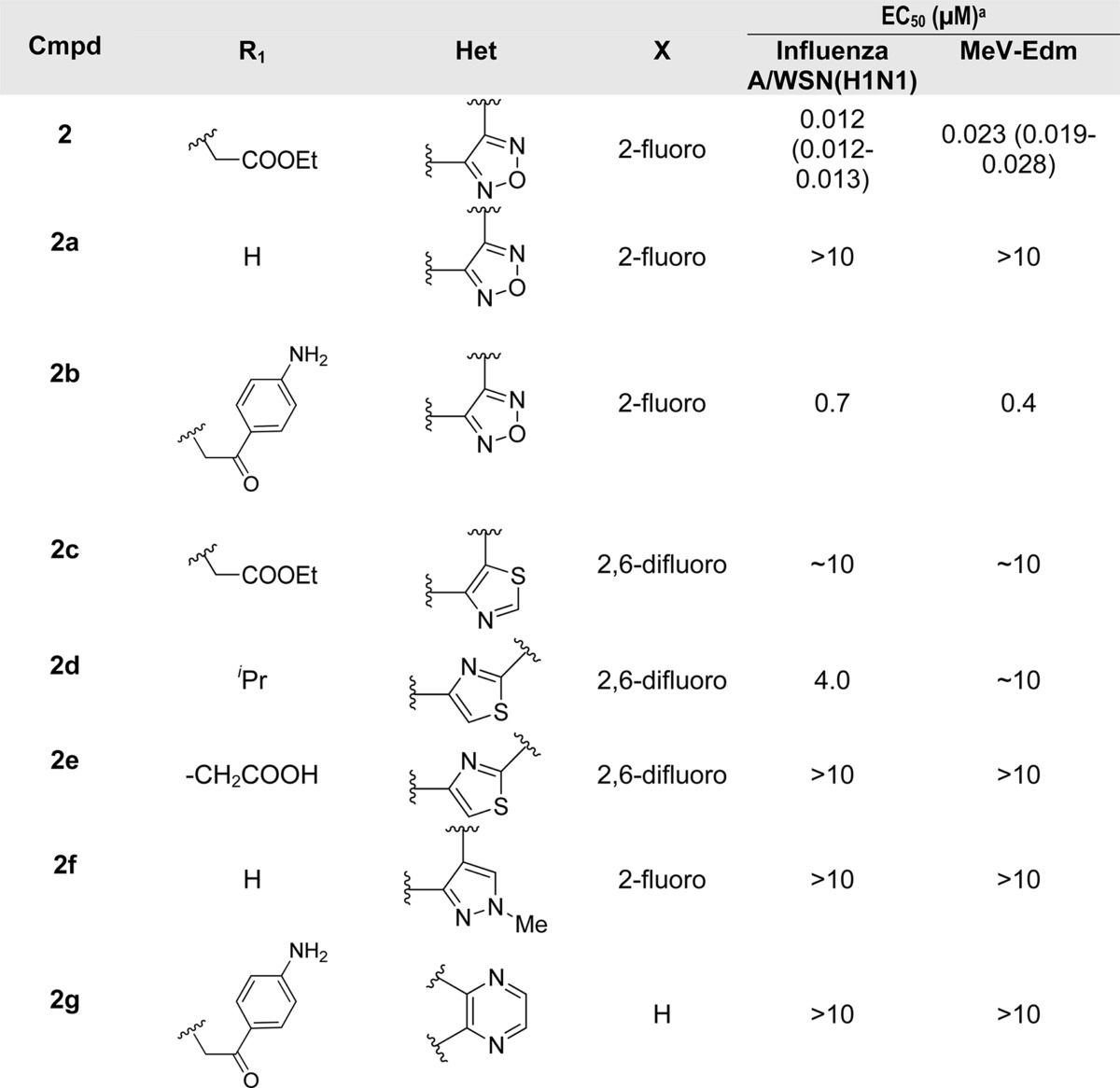
EC50 values were calculated using the variable slope (four parameters) nonlinear regression-fitting algorithm embedded in the Prism 5 software package (GraphPad Software). Values represent averages of four assessments; highest concentration assessed, 10 μM.
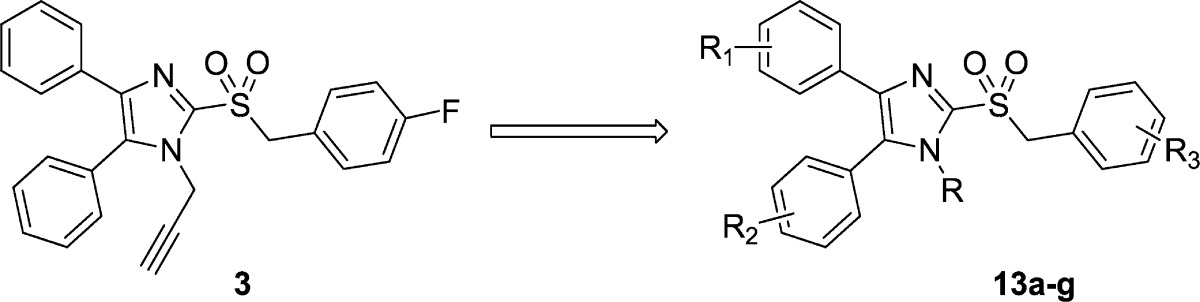
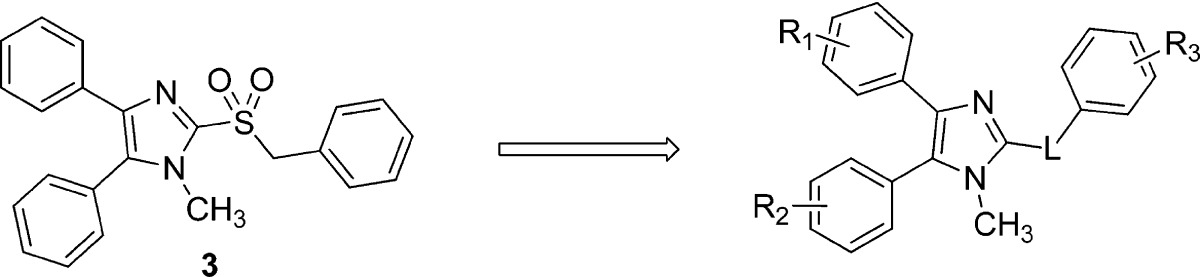
The syntheses of analogues of 3 were modified based on the procedures of Laufer et al.8 Briefly, 4,5-diphenyl-2-thioimidazole was treated with alkyl bromides to give thioethers 10. The thioether was either oxidized to the sulfone 11 first and alkylated9 or, alternatively, alkylated and then oxidized to yield 13.10 Deprotonating the benzylic α-carbon and fluorinating once or twice gave analogues 14a and 14b (Scheme 2).
Scheme 2.

To obtain differentially substituted analogues, the acetophenones 15 were brominated 16, coupled with methylamine, and acidified to give α-ammonium ketones 17.8 These analogues were cyclized using sodium thiocyanate to give the thioimidazoles 18.8,11 Alkylation delivered the thioethers 19, and bromination gave the 5-bromoimidazoles 20.8,12mCPBA oxidation provided the sulfone 21, and Suzuki coupling yielded the tetra-substituted imidazoles 22 (Scheme 3). The synthesis of 28a and analogues is shown in Scheme 4 starting with treatment of 1-methyl-4,5-diphenylimidazole 29 with n-butyllithium and DMF to afford 2-formyl imidazole 30.13 Reductive amination of 30 with different aromatic amines delivered amines 31, which underwent either a second reductive amination with formaldehyde or benzaldehyde or LiHMDS-mediated alkylation to furnish 28a–d, respectively.
Scheme 3.

Scheme 4.

In exploring the SAR around 3, we found that replacing the propargyl group with a hydrogen atom (13a) or a benzyl group (13e) led to a complete loss of potency against both IAV and MeV (13a), replacement with an ethyl (13d) or allyl group (13f) led to reduced potency, and replacement with a methyl group (13b) yielded analogues with comparable potency to unmodified 3 against influenza, particularly, indicating a preference for small alkyl substituents at the 1-position. Tris-fluoro analogue 13c was synthesized and found to have reduced potency against IAV and MeV as compared to 3. In an attempt to increase potency, solubility, and metabolic stability, two of the three phenyl rings were independently replaced with pyridine (13g and 13h), and both compounds completely lost activity.
Having demonstrated promising activity within this series, we turned our attention to the metabolic stability of the compounds. We assayed a subset of the compounds for metabolic stability in human and hamster liver S9 fractions, the latter species chosen for its likely use in a potential efficacy study. We selected a panel of compounds for this study to probe likely metabolic transformations: nucleophilic addition into the heterocyclic sulfone and subsequent elimination of the sulfinate, oxidation of the aromatic rings, oxidation of the N-alkyl group, or oxidation of the methylene group α to the sulfonyl. While rapidly metabolized in pooled hamster liver S9 fractions (T1/2 < 2.5 min), compounds 3, 13a, and 13b showed a slightly prolonged half-life in the pooled human liver S9 fractions with half-lives of 19, 14, and 2–12 min, respectively (Table 2). Surprisingly, the tris-fluorophenyl analogue 13c showed rapid breakdown in human and hamster liver S9 fractions with both half-lives less than 2.5 min (Table 2). Common to these four analogues is a sulfonylmethylene moiety, which links the left-most imidazole and right-most aryl rings of the molecule, presenting a potential metabolic instability. We observed no metabolism in the absence of cofactors, implying that the compounds were chemically stable and that elimination of the sulfone as a sulfinate, were it to occur, must be cofactor-mediated. Toward understanding the role of the methylene carbon in the metabolism, we introduced one or two fluorines into the molecule at the methylene carbon. Although incorporation of one fluorine at that position (14a) conferred no additional S9 stability, introduction of a second fluorine (15) resulted in good stability in pooled hamster S9 fractions (T1/2 = 54 min) and excellent stability in the human system (T1/2 > 90 min). These results suggest that the sulfonylmethylene represents a substantial metabolic liability for this compound class (Table 2).
Table 2. Antiviral EC50 Values for Selected Sulfone Analogues.
| EC50 (μM)a |
S9 stability T1/2 (min) |
||||
|---|---|---|---|---|---|
| compd | R, R1, R2, R3 | Influenza | MeV | human | hamster |
| 3 | 0.5 | 0.74 | 19 | <2.5 | |
| 13a | R = H, R1 = R2 = R3 = H | >10 | >10 | 14 | <2.5 |
| 13b | R = Me, R1 = R2 = R3 = H | 0.3 | 4.0 | 2–12b | <2.5b |
| 13c | R = Me, R1 = R2 = R3 = p-F | >10 | >10 | <2.5b | <2.5b |
| 13d | R = Et, R1 = R2 = R3 = H | 2.0 | 1.0 | NDc | NDc |
| 13e | R = Bn, R1 = R2 = R3 = H | 10 | >10 | NDc | NDc |
| 13f | R = allyl, R1 = R2 = H, R3 = p-F | 4.2 | 4.6 | NDc | NDc |
| 13g | R = Me, R1 = R2 = H, R3 = 4-py | >10 | >10 | NDc | NDc |
| 13h | R = Me, R1 = R2 = H, R3 = 3-py | >10 | 9 | NDc | NDc |
| 22a | R = Me, R1 = R3 = p-F, R2 = 4-py | >10 | >10 | NDc | NDc |
| 22b | R = Me, R1 = R3 = p-F, R2 = 3-py | >10 | >10 | NDc | NDc |
EC50 were calculated using the variable slope (four parameters) nonlinear regression-fitting algorithm embedded in the Prism 5 software package (GraphPad Software). Values represent averages of four assessments; highest concentration assessed, 10 μM.
Values represent the range of two experiments carried out on different dates. Where only one value is given, the half-lives were calculated to be the same.
Not determined (ND) when EC50 > 10 μM.
We attempted to determine the identity of the metabolite(s) using LC–MS/MS. After incubating 13b (100 μM) with pooled human liver S9 fractions for 20 min, we observed a peak (91% of total area) in the LC–MS that corresponded to a mass with m/z of 576 (+187 Da) and 531 (+142 Da; data not shown). Enhanced product ion (EPI) of the metabolite with m/z 576 produced a fragment with m/z 298, which is consistent with the mass of the 4,5-diphenylimidazole sulfone structure. Taken together, these data are consistent with our hypothesis of an unstable species, perhaps a reactive metabolite that arises from hydrolysis of an α-hydroxysulfone intermediate or a product of secondary metabolism.
Unfortunately, having repaired the metabolic liability in 13b, we found that fluorinating the metabolically labile methylene resulted in complete loss of antiviral activity (14b and 15, Table 3). Understanding this key weakness within the molecule, we replaced the sulfonylmethylene moiety altogether with a variety of two-atom linkers. Replacing the sulfone with a thioether moiety led to analogue 12, which exhibited not only reduced antiviral activity against IAV, but also shortened compound half-life in both hamster and human liver S9 fractions (12, Table 3), a curious finding when one considers the difference in electron density at the α-carbons of 13a and 12. Further modification by replacing the sulfone with a carbonyl group led to analogue 25, which showed much improved stability in human liver S9 fractions (human S9 T1/2 > 90 min) but diminished potency (Table 3). Replacing the sulfonylmethylene with a sulfonamide 23 or a 1,2-disubstituted ethanol 26 caused not only complete loss of activity but also failed to confer metabolic stability. Compounds 24a and 24b, in which an amide linker replaces the sulfonylmethylene, were about 100-fold less potent than compound 3. A similar result was found with replacement by an ethane linker (27, Table 3). Strikingly, we found analogue 28a, with an aminomethyl linker, to return good potency with EC50 values of 0.88 μM against IAV-WSN and 0.81 μM against MeV-Edm. Compound 28a was also found to be metabolically stable in pooled human liver S9 fractions with a half-life of 165 min. While the potency and metabolic stability of 28a are very promising, the solubility of 28a is quite poor (<15 μg/mL at pH = 7.4). We attempted to address this issue by either generating salts of 22 or incorporating hydrophilic groups in the right-hand portion of the molecule. Compound 28b with a hydroxyl group, 28c with a carboxyl group, and 28d with pyridine instead of a phenyl ring were synthesized and subjected to solubility testing based on nephelometry. Analogue 28c showed superb solubility of greater than 300 μg/mL at pH = 7.4 (Table 3).
Table 3. Analogues of 3 Containing Different Linker Moieties.

Lacks 1-methyl group on imidazole ring.
EC50 were calculated using the variable slope (four parameters) nonlinear regression-fitting algorithm embedded in the Prism 5 software package (GraphPad Software). Values represent averages of four assessments; highest concentration assessed,10 μM.
Not determined when EC50 >10 μM.
In conclusion, we have described two new series of analogues that may have broad-spectrum antimyxovirus activity, albeit with higher EC50 values than with our previously reported scaffold 1.6,7,14 While the first of these series, based on furazan 2, delivered potent compounds, this series was chemically and metabolically unstable. The second series, based on imidazole 3, yielded bioactive compounds that were compromised, however, by metabolic liabilities likely arising from the benzylic methylene group α to the sulfone. Replacing the sulfonylmethylene with a methyleneamine moiety leads to a bioactive (EC50 value for 28a approximately 0.8–0.9 μM against different myxovirus family members) and metabolically stable analogue (human S9 T1/2 = 165 min). Once solubility is further improved, the series would appear suitable for in vivo efficacy testing studies.
Supporting Information Available
Experimental details for the synthesis and characterization of 2, 2a-h, and 3–32. Experimental details for biology assays. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported, in part, by Public Health Service Grants AI071002 and AI057157 (to R.K.P.) from the NIH/NIAID.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- World Health Organization. World Health Statistics, 2013.
- Why should influenza be a public health priority? Vaccine 2012, 30, 7418–7420. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Watanabe S.; Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 2010, 7, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R.; Stertz S.; Zhou Y.; Inoue A.; Hoffmann H. H.; Bhattacharyya S.; Alamares J. G.; Tscherne D. M.; Ortigoza M. B.; Liang Y. H.; Gao Q. S.; Andrews S. E.; Bandyopadhyay S.; De Jesus P.; Tu B. P.; Pache L.; Shih C.; Orth A.; Bonamy G.; Miraglia L.; Ideker T.; Garcia-Sastre A.; Young J. A. T.; Palese P.; Shaw M. L.; Chanda S. K. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. J.; Chawla D.; Paal T.; Ndungu M.; Du Y. H.; Kurtkaya S.; Sun A. M.; Snyder J. P.; Plemper R. K. High-throughput screening-based identification of paramyxovirus inhibitors. J. Biomol. Screening 2008, 13, 591–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T. W.; Sana K.; Yan D.; Thepchatri P.; Ndungu J. M.; Saindane M. T.; Lockwood M. A.; Natchus M. G.; Liotta D. C.; Plemper R. K.; Snyder J. P.; Sun A. M. Asymmetric synthesis of host-directed inhibitors of myxoviruses. Beilstein J. Org. Chem. 2013, 9, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A. M.; Ndungu J. M.; Krumm S. A.; Yoon J. J.; Thepchatri P.; Natchus M.; Plemper R. K.; Snyder J. P. Host-directed inhibitors of myxoviruses: Synthesis and in vitro biochemical evaluation. ACS Med. Chem. Lett. 2011, 2, 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer S. A.; Hauser D. R. J.; Liedtke A. J. Regiospecific and highly flexible synthesis of 1,4,5-trisubstituted 2-sulfanylimidazoles from structurally diverse ethanone precursors. Synth.-Stuttgart 2008, 253–266. [Google Scholar]
- Guravaiah N.; Rao V. R. Stereoselective synthesis of substituted 2-(Z-styrylsulfonyl)-imidazoles and benzothiazole. Synth. Commun. 2010, 40, 808–813. [Google Scholar]
- Sugimoto H.; Nakamura S.; Watanabe Y.; Toru T. Enantioselective hydrogen atom transfer to alpha-sulfonyl radicals controlled by selective coordination of a chiral Lewis acid to an enantiotopic sulfonyl oxygen. Tetrahedron: Asymmetry 2003, 14, 3043–3055. [Google Scholar]
- Dodson R. M.; Ross F. The preparation of 2-alkylthioimidazoles. J. Am. Chem. Soc. 1950, 72, 1478–1480. [Google Scholar]
- Rao K. V. P.; Sundaramurthy V. Regioselective electrophilic substitutions of 4h-imidazo[2,1-C][1,4]benzoxazine and 4h-imidazo[2,1-C][1,4]benzthiazine. J. Org. Chem. 1992, 57, 2737–2739. [Google Scholar]
- Zhou Y. R.; Gong Y. F. Asymmetric copper(II)-catalysed nitroaldol (Henry) reactions utilizing a chiral C-1-symmetric dinitrogen ligand. Eur. J. Org. Chem. 2011, 6092–6099. [DOI] [PubMed] [Google Scholar]
- Krumm S. A.; Ndungu J. M.; Yoon J. J.; Dochow M.; Sun A. M.; Natchus M.; Snyder J. P.; Plemper R. K. Potent host-directed small-molecule inhibitors of myxovirus RNA-dependent RNA-polymerases. PLoS One 2011, 6, e20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.