

Abstract
Highly parallel experimental biology is offering opportunities to not just accomplish work more easily, but to explore for underlying governing principles. Recent analysis of the large-scale organization of gene expression has revealed its complex and dynamic nature. However, the underlying dynamics that generate complex gene expression and cellular organization are not yet understood. To comprehensively and quantitatively elucidate these underlying gene expression dynamics, we have analyzed genome-wide gene expression in many experimental conditions in Escherichia coli, Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster, Mus musculus, and Homo sapiens. Here we demonstrate that the gene expression dynamics follows the same and surprisingly simple principle from E. coli to human, where gene expression changes are proportional to their expression levels, and show that this “proportional” dynamics or “rich-travel-more” mechanism can regenerate the observed complex and dynamic organization of the transcriptome. These findings provide a universal principle in the regulation of gene expression, show how complex and dynamic organization can emerge from simple underlying dynamics, and demonstrate the flexibility of transcription across a wide range of expression levels.
Keywords: systems biology, DNA microarray, gene expression dynamics, transcription, transcriptional organization
The past decade has seen a growing number of model organisms with available draft or complete genome sequences including Escherichia coli, Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster, Mus musculus, and Homo sapiens. The availability of these resources and the development of tools such as DNA arrays that allow for high-throughput experimental biology have been driving a paradigm shift in the life sciences, from the molecular “one-gene-at-a-time” level to a network or systems level (1-5).
Genome-wide experimental biology approaches offer the opportunity to work more efficiently (6), and also to explore for universal governing principles underlying the physiology. Several recent microarray studies have investigated the large-scale organization of gene expression, revealing complex networks that capitulate physiological processes such as the cell cycle, responses to environmental change, circadian rhythms, and developmental and tissue-specific gene regulation (6-10). However, universal features of gene expression dynamics that produce such complex and dynamic cellular organizations have largely gone unexplored. To comprehensively and quantitatively elucidate these underlying gene expression dynamics, we analyzed genome-wide gene expression data collected from experimental studies in E. coli, S. cerevisiae, A. thaliana, D. melanogaster, M. musculus, and H. sapiens samples by using high-density oligonucleotide arrays. Here we show that the gene expression dynamics follows the same and surprisingly simple principle from E. coli to H. sapiens, where gene expression changes are proportional to their initial expression levels. Furthermore, we show that this “proportional dynamics” or “rich-travel-more” mechanism can regenerate the observed complex and dynamic organization of the transcriptome.
Materials and Methods
E. coli. Cells of E. coli strain YMC21 lacking glutamine synthetase gene was collected under normal or symbiotic conditions as described (11). Total RNA was isolated by using MasterPure RNA purification kit (Epicentre Technologies, Madison, WI).
S. cerevisiae. Cells of S. cerevisiae strain FY1679 were grown in yeast extract/peptone/dextrose medium with aeration at 20 ± 0.5°C to maintain an optical density of 0.5 at 600 nm (OD600) by using a continuous culturing system. The culture was adapted to 12-h light/12-h dark cycles (LD) for 2 days before samples were obtained under LD conditions every 4 h starting at Zeitgeber time (ZT) 0 over 2 days. Total RNA was isolated from 10-ml cultures by using the hot phenol method.
A. thaliana. Wild-type Arabidopsis seeds of the Col-1 ecotype were sown on Murashige and Skoog (MS) agar plates containing 3% sucrose. Seeds were stratified at 4°C for 2 days and then placed in growth chambers held at 22°C. Plants were grown in LD cycles for 7 days and then released into constant light (60 μEinsteins·m-2·s-1). Starting at subjective dawn on day 9 [circadian time (CT) 0], plants were harvested every 4 h over 2 days. Total RNA was prepared from the staged tissue samples by using the Qiagen RNeasy Plant Mini kit (Valencia, CA).
D. melanogaster. white1118 flies were reared to LD and collected under LD or constant darkness conditions every 4 h starting at ZT1 or CT1 over 2 days. Total RNA was prepared from 100 heads of 1-week-old adult males and females by using the Fast RNA kit (BIO 101, Carlsbad, CA).
M. musculus Time Course Sampling. BALB/c mice (male), purchased 5 weeks postpartum, were adapted to LD cycles for 2 weeks before samples were obtained under LD or constant darkness conditions every 4 h starting at ZT0 or CT0 over 2 days. Slices (0.5 mm thick) of mouse brain were generated by using Mouse Brain Matrix (Neuroscience) under LD or constant darkness condition, and then the suprachiasmatic nuclei (SCNs) were punched out bilaterally from the frozen slices under a stereomicroscope with a microdissecting needle (gauge, 0.5 mm). Livers were dissected and frozen in liquid nitrogen. Total RNA was prepared from 50 pooled pairs of SCNs and four pooled livers at each time point by using by TRIzol reagent (GIBCO/BRL).
M. musculus and H. sapiens Tissue Sampling. Forty-seven human tissue samples and cell lines were obtained from commercial sources and previously published research collaborations, and 45 mouse tissue samples were derived from dissections. Detailed sample descriptions can be obtained on the web site (http://expression.gnf.org) (6).
Microarray Experiments. We examined genome-wide RNA expression of E. coli, S. cerevisiae, A. thaliana, D. melanogaster, M. musculus, and H. sapiens by using Affymetrix high-density oligonucleotide probe arrays (GeneChip) representing ≈4,200 E. coli, ≈6,400 S. cerevisiae, ≈8,200 A. thaliana, ≈13,500 D. melanogaster, ≈10,000 M. musculus, and ≈12,000 H. sapiens genes, respectively (7, 8). We extracted total RNA from the organisms, prepared biotinylated cRNA, hybridized samples to GeneChip arrays, and obtained hybridization signals as described (6, 8-10).
cDNA synthesis and cRNA labeling reactions were performed as described in the Affymetrix technical manual (Affymetrix, Santa Clara, CA). We used Affymetrix high-density oligonucleotide arrays: E. coli Genome Array for E. coli, Yeast Genome S98 Array for S. cerevisiae, Arabidopsis Genome Array for A. thaliana, Drosophila Genome Array for D. melanogaster, Murine Genome Array U74A for M. musculus (Murine Genome Array U74A), and Human Genome Array U95A for H. sapiens. To facilitate comparisons between samples, the obtained data were scaled globally such that the average of signal intensity for probe sets of each GeneChip array is 1/30. We defined this scaled signal intensity values as an “expression level” of a gene and used it in all subsequent analyses.
ANOVA. Statistical analysis (one-way ANOVA, P < 0.01) was performed on gene expression data above from E. coli to human. Expected false positives derived from the multiple testing are ≈1% of the tested genes.
Calculation of the Absolute Expression Change. We calculated the absolute expression change 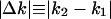 for individual genes from an expression level (k1) to another expression level (k2), along two different conditions of E. coli, along the different ZT and/or CT in S. cerevisiae, A. thaliana, heads of D. melanogaster, liver and SCN of M. musculus, or along 45 M. musculus tissues and 47 H. sapiens tissues. We calculated the absolute expression change in all distinct pairs of different conditions.
for individual genes from an expression level (k1) to another expression level (k2), along two different conditions of E. coli, along the different ZT and/or CT in S. cerevisiae, A. thaliana, heads of D. melanogaster, liver and SCN of M. musculus, or along 45 M. musculus tissues and 47 H. sapiens tissues. We calculated the absolute expression change in all distinct pairs of different conditions.
Results and Discussion
To investigate systems-level features of RNA expression, we analyzed the distribution of gene expression levels from our data sets obtained from E. coli to H. sapiens. Fig. 1 shows that the distribution of gene expression levels exhibits a power-law distribution in which the probability that a gene has amount of expression, k, decays as a power law, P(k) ∝ k-r. Furthermore, this genome-wide transcriptional organization is conserved from E. coli to H. sapiens with the exponent r of the distribution of gene expression levels close to 2 (1.69-2.09). This power-law distribution is not artifactual because the hybridization signal intensity of each probe on the arrays is linear along the cRNA concentration (Fig. 5, which is published as supporting information on the PNAS web site) and as an analysis of serial analysis of gene expression (SAGE) data (12-15) revealed a similar power-law distribution and exponent from S. cerevisiae to H. sapiens (Fig. 6, which is published as supporting information on the PNAS web site). These results are also consistent with a previous report on power-law distribution of gene expression levels (16, 17). Collectively, these results indicate that network regulation of genome-wide transcriptional organization is conserved between prokaryotes and eukaryotes.
Fig. 1.

Evolutional conservation of transcriptional organization. The distributions of gene expression levels in E. coli (A), S. cerevisiae (B), A. thaliana (C), D. melanogaster (D), M. musculus (E), and H. sapiens (F) exhibit a power-law distribution in which the probability that a gene has an expression level k, decays as a power law, P(k) ∝ k-r. A straight line in each panel represents the estimated power-law distribution. The estimated value of exponent r is indicated in the lower left corner of each panel.
To confirm these results and examine whether this quantitative property of transcription extends to temporal and spatial gene regulation, we analyzed RNA expression from diverse datasets at different ZT and/or CT or tissues in S. cerevisiae, A. thaliana, D. melanogaster, M. musculus, and H. sapiens. Fig. 7A, which is published as supporting information on the PNAS web site, shows that the distribution of gene expression levels exhibits a power-law distribution P(k) ∝ k-r with an exponent r close to 2 (Fig. 7 C-J). Importantly, statistical analysis (ANOVA) revealed that 3% of genes in S. cerevisiae, 16% of genes in A. thaliana, 17% of genes in heads of D. melanogaster, and 8% of genes in liver and 8% of genes in SCN of M. musculus are dynamically expressed along different ZT and/or CT (P < 0.01). Mammalian tissue specific gene expression displays an even more dynamic pattern of expression; statistical analysis (ANOVA) revealed that 95% and 88% of genes are differentially expressed in 47 H. sapiens and 45 M. musculus tissues, respectively (P < 0.01). Therefore, we analyzed the RNA expression in 47 tissues of H. sapiens and 45 tissues of M. musculus. Despite differential expression of the vast majority of genes in these data sets, Fig. 8A, which is published as supporting information on the PNAS web site, shows that the distribution of gene expression levels exhibits a power-law distribution P(k) ∝ k-r with an exponent r close to 2 (see also Fig. 8 B and C). Therefore, both temporal and spatial gene expression regulation follows a power law distribution despite the differential dynamic expression of a substantial fraction of genes.
To investigate the type of dynamics that generates this evolutionary and spatio-temporally conserved power-law transcriptional organization, we analyzed the systems-level features in expression changes of individual genes. We calculated the transition probability T(k2, k1) of expression change by counting the incidence of expression changes for individual genes from an expression level (k1) to another expression level (k2), along two different conditions of E. coli (Fig. 2A), or for variouis conditions in S. cerevisiae (Fig. 2B), A. thaliana (Fig. 2C), heads of D. melanogaster (Fig. 2D), liver and SCN of M. musculus (Fig. 2 E and F), or along 45 M. musculus tissues (Fig. 2G) and 47 H. sapiens tissues (Fig. 2H). This analysis revealed that the transition probabilities are not random, but rather are highly dependent on the before-transition expression level (k1). For example, transition from the higher expression levels spread more widely, whereas transition from the lower expression levels is confined to the lower expression levels (Fig. 2 A-H). To exclude the possibility that the narrower expression changes from lower expression levels are caused by limit of detection for genes of low intensity or “dead probe sets,” we applied the same analyses to probe sets that pass a statistical criteria (ANOVA, P < 0.01), and we obtained the similar results (data not shown).
Fig. 2.

Characteristics in gene expression dynamics. (A-H) Transition probability T(k2, k1), where a gene with a certain expression level k1 changes its expression level to k2, calculated from expression data in E. coli (A), S. cerevisiae (B), A. thaliana (C), heads of D. melanogaster (D), liver and suprachiasmatic nucleus of M. musculus (E and F), 45 tissues of M. musculus (G), and 47 tissues of H. sapiens (H). Colors from red to yellow to green represent transition probability of descending values. Gray indicates the value of zero (the lack of the transition data). (I-P) Proportionality in gene expression dynamics. The absolute expression level change (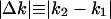 ) is plotted along the before-transition expression level k1 in E. coli (I), S. cerevisiae (J), A. thaliana (K), heads of D. melanogaster (L), liver and suprachiasmatic nucleus of M. musculus (M and N), 45 tissues of M. musculus (O), and 47 tissues of H. sapiens (P) expression data. The estimated values for exponent s from a log-log plot of absolute expression change against the before-transition expression level (i.e.,
) is plotted along the before-transition expression level k1 in E. coli (I), S. cerevisiae (J), A. thaliana (K), heads of D. melanogaster (L), liver and suprachiasmatic nucleus of M. musculus (M and N), 45 tissues of M. musculus (O), and 47 tissues of H. sapiens (P) expression data. The estimated values for exponent s from a log-log plot of absolute expression change against the before-transition expression level (i.e., 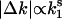 ) are indicated in the lower right corner of each panel.
) are indicated in the lower right corner of each panel.
To extract the quantitative characteristics of these gene expression dynamics, we analyzed relationships between the before-transition expression level (k1) and the absolute expression change 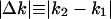 . Fig. 2 I-P shows that the absolute expression change is proportional to the before-transition expression level from E. coli to H. sapiens. Stated differently, although the absolute change from higher expression levels is larger, it is proportional to the initial before transition value. To confirm this proportionality, we measured an exponent s from log-log plot of absolute expression change (
. Fig. 2 I-P shows that the absolute expression change is proportional to the before-transition expression level from E. coli to H. sapiens. Stated differently, although the absolute change from higher expression levels is larger, it is proportional to the initial before transition value. To confirm this proportionality, we measured an exponent s from log-log plot of absolute expression change (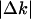 ) against the before-transition expression level k1 (i.e.,
) against the before-transition expression level k1 (i.e., 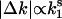 ) and found it to be close to 1 (Fig. 2 I-P). We also note that the absolute expression change becomes asymptotically a small but nonzero value with lower expression levels (Fig. 9, which is published as supporting information on the PNAS web site). These results suggest that the proportional gene expression dynamics underlie genetic networks from E. coli to H. sapiens.
) and found it to be close to 1 (Fig. 2 I-P). We also note that the absolute expression change becomes asymptotically a small but nonzero value with lower expression levels (Fig. 9, which is published as supporting information on the PNAS web site). These results suggest that the proportional gene expression dynamics underlie genetic networks from E. coli to H. sapiens.
We then tested whether this proportional gene expression dynamics can generate the observed power-law transcriptional organization. We calculated the stationary distribution of gene expression levels by using transition probability matrices of E. coli to H. sapiens (Fig. 2 A-H) from arbitrary initial distributions of gene expression levels. We found that the simulated distribution of gene expression levels exhibits the power-law distribution P(k) ∝ k-r, regardless of the initial distribution, with an exponent r close to 2 (Fig. 3). These results suggest that gene expression dynamics are conserved from E. coli to H. sapiens and that this universal gene expression dynamic can regenerate the observed power-law organization of gene expression levels.
Fig. 3.

Proportional gene expression dynamics can regenerate the observed transcriptional organization. Shown are the stationary distributions of gene expression levels calculated by using transition probability matrices in Fig. 2 A-H from arbitrary initial distribution of gene expression levels. The stationary distributions of E. coli (A), S. cerevisiae (B), A. thaliana (C), heads of D. melanogaster (D), liver and suprachiasmatic nucleus of M. musculus (E and F), 45 tissues of M. musculus (G), and 47 tissues of H. sapiens (H) exhibit a power-law decay in which the probability that a gene has an expression level k, is P(k) ∝ k-r. The straight line in each panel represents the estimated power-law distribution. The estimated value of exponent r is indicated in the lower left corner of each panel.
To explore the relationship between proportional gene expression dynamics and power-law transcriptional organization, we modeled these gene expression dynamics. We hypothesized proportionality in gene expression dynamics; where the standard deviation of gene expression change, 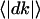 , increases in proportional to the before-transition gene expression level k. We next hypothesized that the average of gene expression changes
, increases in proportional to the before-transition gene expression level k. We next hypothesized that the average of gene expression changes 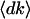 is zero, indicating that the expression level of each gene may increase or decrease and it has no systematic tendency (see Supporting Text, which is published as supporting information on the PNAS web site). From these assumptions, we can derive the stationary distribution of gene expression levels and find that it exhibits the power-law distribution with an exponent -2, i.e., P(k) ∝ k-2 (see Supporting Text and Fig. 10, which is published as supporting information on the PNAS web site, for details).
is zero, indicating that the expression level of each gene may increase or decrease and it has no systematic tendency (see Supporting Text, which is published as supporting information on the PNAS web site). From these assumptions, we can derive the stationary distribution of gene expression levels and find that it exhibits the power-law distribution with an exponent -2, i.e., P(k) ∝ k-2 (see Supporting Text and Fig. 10, which is published as supporting information on the PNAS web site, for details).
To validate this model, we implemented the model with parameters estimated from S. cerevisiae expression analysis (a = 0.25, b = 0.0006, s = 1 for 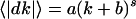 , see Supporting Text). Fig. 4A shows the transition probability matrix representing the model gene expression dynamics (see Supporting Text and Fig. 11, which is published as supporting information on the PNAS web site). The modeled transition probability matrix highly resembles the transition probability matrix calculated from S. cerevisiae expression data (Figs. 4A and 2B). Moreover, we demonstrated that the stationary distribution generated by the model transition probability matrix from any initial distributions exhibits the power-law distribution P(k) ∝ k-r with an exponent r close to 2 (Figs. 4B and 3B). For comparison, we also implemented the model with other parameter values such as s = 0.5 or s = 2 and found much less agreement with observed distributions, indicating the importance of proportional dynamics in generating observed power-law distributions (Fig. 12, which is published as supporting information on the PNAS web site). Based on these results, we concluded that gene expression dynamics from E. coli to H. sapiens are governed by the same proportional dynamics, where the expression level of each gene is dynamically changed in proportion to its initial expression level, and that this proportionality in gene expression dynamics has a critical role in generating the power-law organization of gene expression levels.
, see Supporting Text). Fig. 4A shows the transition probability matrix representing the model gene expression dynamics (see Supporting Text and Fig. 11, which is published as supporting information on the PNAS web site). The modeled transition probability matrix highly resembles the transition probability matrix calculated from S. cerevisiae expression data (Figs. 4A and 2B). Moreover, we demonstrated that the stationary distribution generated by the model transition probability matrix from any initial distributions exhibits the power-law distribution P(k) ∝ k-r with an exponent r close to 2 (Figs. 4B and 3B). For comparison, we also implemented the model with other parameter values such as s = 0.5 or s = 2 and found much less agreement with observed distributions, indicating the importance of proportional dynamics in generating observed power-law distributions (Fig. 12, which is published as supporting information on the PNAS web site). Based on these results, we concluded that gene expression dynamics from E. coli to H. sapiens are governed by the same proportional dynamics, where the expression level of each gene is dynamically changed in proportion to its initial expression level, and that this proportionality in gene expression dynamics has a critical role in generating the power-law organization of gene expression levels.
Fig. 4.

Theoretical model of proportional gene expression dynamics. (A) Transition probability matrix representing the theoretical model of proportional gene expression dynamics. The model transition probability T(k2, k1) represents the probability of expression change from a certain expression level k1 to other expression level k2 during unit time interval. Colors from red to yellow to green represent transition probabilities of descending values. (B) The stationary distribution of gene expression calculated by using the modeled transition probability matrix from arbitrary initial distribution of gene expression levels. The stationary distributions of model proportional exhibit a power-law distribution in which the probability that a gene has an expression level k, decays as a power law. The straight line represents the estimated power-law distribution P(k) ∝ k-2.
Collectively, these results indicate that gene expression dynamics follows the same and surprisingly simple principles from bacteria to humans in which gene expression changes are proportional to their initial expression levels. Importantly, this proportionality assures the same level of flexibility of expression for highly expressed genes as well as for lower expressed genes. This flexibility is vital for transcriptional regulation because both highly expressed genes such as structural genes as well as more rare mRNAs such as G protein-coupled receptors have to change their expression levels to adapt an environmental or developmental change.
Power-law distributions arise in the expression levels with proportional dynamics where highly expressed genes change their expression levels more dynamically than less expressed genes. This “rich-travel-more” mechanism generates power-law distributions in the fixed networks with constant number of components and resembles the “rich-get-richer” mechanism (e.g., preferential attachment) that can generate the power-law distributions in growing networks such as the World Wide Web, in which highly connected nodes have a higher chance than less connected nodes to linking to new nodes. Interestingly, we have analyzed the evolution of metabolic networks with similar number of constituents, and found that the proportional dynamics also underlies the evolution of metabolic networks with a fixed or similar number of components (H.R.U., J.B.H., and M.I., unpublished data).
Recently, several studies have investigated the genome-wide network structure of the metabolome (18-20), transcriptome (21-24), and proteome (25), and have revealed the universality of the scale-free, modular, and hierarchical topology of biological networks (26-30). Moreover, several recent microarray studies (4, 5) have identified the specific dynamics of specialized systems including the cell cycle, environmental responses, circadian rhythms, development, and tissue-specific gene regulation. This present study illustrates generic gene expression dynamics underlying regulation of transcription, complementing previous work on generic genome-wide network structure or on specific dynamics of specialized systems. A next challenge is, thus, the understanding of the systems-level features in the network structure that generates this proportional dynamic, as well as the investigation of the generality of proportional dynamics in other physiological processes, such as the regulation of metabolic processes. The development of predictive models that accurately represent biological processes in conjunction with highly parallel experimental data should accelerate the understanding of additional underlying principles governing the complex and dynamic systems of life.
Supplementary Material
Acknowledgments
We thank Hideo Iwasaki and Takao Kondo for a continuous culturing system and Sumio Sugano and Yutaka Suzuki for stimulating discussion. We also thank Koichi Shiozuka, Stancey L. Harmer, Akira Matsumoto, Teiichi Tanimura, Miho Kawamura, Wenbin Chen, Hisanori Wakamatsu, Akihito Adachi, Yasufumi Shigeyoshi, Andrew I. Su, Michael P. Cooke, Keith A. Ching, Yaron Hakak, John R. Walker, Tim Wiltshire, and Anthony P. Orth for yeast, Arabidopsis, fly, mouse, and human genome-scale expression measurement. We thank Tomoko Kojima, Toshie Katakura, and Hiromi Urata for technical assistance. This study was performed as a part of a research and development project of the Industrial Science and Technology Program supported by New Energy and Industrial Technology Development Organization (NEDO). This work was also supported in part by Grants-in-Aid 11CE2006, 15207020, 12002003, and 15013235 and The 21st Century Center of Excellence Program of the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and by a grant from Takeda Science Foundation.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ZT, zeitgeber time; CT, circadian time; LD, 12-h light/12-h dark; SCN, suprachiasmatic nucleus.
References
- 1.Hartwell, L. H., Hopfield, J. J., Leibler, S. & Murray, A. W. (1999) Nature 402, C47-C52. [DOI] [PubMed] [Google Scholar]
- 2.Kitano, H. (2002) Science 295, 1662-1664. [DOI] [PubMed] [Google Scholar]
- 3.Oltvai, Z. N. & Barabasi, A. L. (2002) Science 298, 763-764. [DOI] [PubMed] [Google Scholar]
- 4.Brown, P. O. & Botstein, D. (1999) Nat. Genet. 21, 33-37. [DOI] [PubMed] [Google Scholar]
- 5.Lipshutz, R. J., Fodor, S. P., Gingeras, T. R. & Lockhart, D. J. (1999) Nat. Genet. 21, 20-24. [DOI] [PubMed] [Google Scholar]
- 6.Su, A. I., Cooke, M. P., Ching, K. A., Hakak, Y., Walker, J. R., Wiltshire, T., Orth, A. P., Vega, R. G., Sapinoso, L. M., Moqrich, A., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 4465-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lockhart, D. J., Dong, H., Byrne, M. C., Follettie, M. T., Gallo, M. V., Chee, M. S., Mittmann, M., Wang, C., Kobayashi, M., Horton, H. & Brown, E. L. (1996) Nat. Biotechnol. 14, 1675-1680. [DOI] [PubMed] [Google Scholar]
- 8.Harmer, S. L., Hogenesch, J. B., Straume, M., Chang, H. S., Han, B., Zhu, T., Wang, X., Kreps, J. A. & Kay, S. A. (2000) Science 290, 2110-2113. [DOI] [PubMed] [Google Scholar]
- 9.Ueda, H. R., Matsumoto, A., Kawamura, M., Iino, M., Tanimura, T. & Hashimoto, S. (2002) J. Biol. Chem. 277, 14048-14052. [DOI] [PubMed] [Google Scholar]
- 10.Ueda, H. R., Chen, W., Adachi, A., Wakamatsu, H., Hayashi, S., Takasugi, T., Nagano, M., Nakahama, K., Suzuki, Y., Sugano, S., et al. (2002) Nature 418, 534-539. [DOI] [PubMed] [Google Scholar]
- 11.Todoriki, M., Oki, S., Matsuyama, S. I., Ko-Mitamura, E. P., Urabe, I. & Yomo, T. (2002) Biosystems 65, 105-112. [DOI] [PubMed] [Google Scholar]
- 12.Velculescu, V. E., Zhang, L., Zhou, W., Vogelstein, J., Basrai, M. A., Bassett, D. E., Jr., Hieter, P., Vogelstein, B. & Kinzler, K. W. (1997) Cell 88, 243-251. [DOI] [PubMed] [Google Scholar]
- 13.Lash, A. E., Tolstoshev, C. M., Wagner, L., Schuler, G. D., Strausberg, R. L., Riggins, G. J. & Altschul, S. F. (2000) Genome Res. 10, 1051-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hishiki, T., Kawamoto, S., Morishita, S. & Okubo, K. (2000) Nucleic Acids Res. 28, 136-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones, S. J., Riddle, D. L., Pouzyrev, A. T., Velculescu, V. E., Hillier, L., Eddy, S. R., Stricklin, S. L., Baillie, D. L., Waterston, R. & Marra, M. A. (2001) Genome Res. 11, 1346-1352. [DOI] [PubMed] [Google Scholar]
- 16.Kuznetsov, V. A., Knott, G. D. & Bonner, R. F. (2002) Genetics 161, 1321-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoyle, D. C., Rattray, M., Jupp, R. & Brass, A. (2002) Bioinformatics 18, 576-584. [DOI] [PubMed] [Google Scholar]
- 18.Karp, P. D., Krummenacker, M., Paley, S. & Wagg, J. (1999) Trends Biotechnol. 17, 275-281. [DOI] [PubMed] [Google Scholar]
- 19.Kanehisa, M., Goto, S., Kawashima, S. & Nakaya, A. (2002) Nucleic Acids Res. 30, 42-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overbeek, R., Larsen, N., Pusch, G. D., D'Souza, M., Selkov, E., Jr., Kyrpides, N., Fonstein, M., Maltsev, N. & Selkov, E. (2000) Nucleic Acids Res. 28, 123-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wingender, E., Chen, X., Fricke, E., Geffers, R., Hehl, R., Liebich, I., Krull, M., Matys, V., Michael, H., Ohnhauser, R., et al. (2001) Nucleic Acids Res. 29, 281-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costanzo, M. C., Crawford, M. E., Hirschman, J. E., Kranz, J. E., Olsen, P., Robertson, L. S., Skrzypek, M. S., Braun, B. R., Hopkins, K. L., Kondu, P., et al. (2001) Nucleic Acids Res. 29, 75-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., Hannett, N. M., Harbison, C. T., Thompson, C. M., Simon, I., et al. (2002) Science 298, 799-804. [DOI] [PubMed] [Google Scholar]
- 24.Guelzim, N., Bottani, S., Bourgine, P. & Kepes, F. (2002) Nat. Genet. 31, 60-63. [DOI] [PubMed] [Google Scholar]
- 25.von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S. G., Fields, S. & Bork, P. (2002) Nature 417, 399-403. [DOI] [PubMed] [Google Scholar]
- 26.Jeong, H., Tombor, B., Albert, R., Oltvai, Z. N. & Barabasi, A. L. (2000) Nature 407, 651-654. [DOI] [PubMed] [Google Scholar]
- 27.Wagner, A. & Fell, D. A. (2001) Proc. R. Soc. London B 268, 1803-1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong, H., Mason, S. P., Barabasi, A. L. & Oltvai, Z. N. (2001) Nature 411, 41-42. [DOI] [PubMed] [Google Scholar]
- 29.Maslov, S. & Sneppen, K. (2002) Science 296, 910-913. [DOI] [PubMed] [Google Scholar]
- 30.Ravasz, E., Somera, A. L., Mongru, D. A., Oltvai, Z. N. & Barabasi, A. L. (2002) Science 297, 1551-1555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.