

Abstract
Background
Werner syndrome (WS) results from defects in the gene encoding WRN RecQ helicase. WS fibroblasts undergo premature senescence in culture. Because cellular senescence is a tumor suppressor mechanism, we examined whether WS fibroblasts exhibited reduced tumorigenicity, in comparison to control cells, in a model of experimental conversion of normal human cells to cancer cells. The combination of oncogenic Ras (Ha-RasV12G) and SV40 large T antigen (SV40 LT) causes human cells to acquire neoplastic properties in the absence of telomerase.
Methods
Fibroblasts from WS and control donors were transduced with retroviruses encoding Ras (with GFP) and SV40 LT. Following expansion of the cell population cells were transplanted beneath the kidney capsule of RAG2-/-,γc-/- mice. Tumors were analyzed by histology and immunohistochemistry. Cells were isolated from tumors by flow cytometry and were retransplanted into secondary or tertiary hosts. In some cases cells were transduced with hTERT before retransplantation. Assays performed on isolated cells included staining for γ-H2AX nuclear foci and in situ gelatinase activity.
Results
We found that WS cells were converted to a tumorigenic state by Ras and SV40 LT, as evidenced by invasion and metastasis of cells implanted in immunodeficient mice. However, tumors could not be transplanted beyond tertiary hosts, unless transduced with hTERT, indicating that telomere-based crisis limits the continued growth of tumors. Nevertheless, individual Ras/SV40 LT-expressing cells retained invasiveness and other malignant properties even when cells reached crisis in tumors in vivo. High levels of gelatinase activity were found by an in situ assay in Ras/SV40 LT-expressing cells undergoing crisis.
Conclusions
We conclude that, despite evidence of accelerated senescence in WS cells, there is no evidence that the absence of active WRN acts as a barrier to neoplastic transformation. Moreover we find that tumorigenic human cells retain malignant properties even as they approach and reach crisis.
Background
Werner syndrome (WS) is a rare disease with segmental progeroid features [1, 2]. Apart from characteristics of accelerated aging, WS patients have a predisposition to osteosarcoma and soft tissue sarcomas, and also to other cancers, including melanoma, myeloid leukemia, myelodisplastic syndrome, thyroid carcinoma, and meningioma [3, 4]. However, cancers that are usually encountered in the later part of the life span, i.e. cancers of the lung, colon and prostate, are rare in WS [3, 4].
The gene responsible for Werner syndrome, WRN, is a RecQ helicase [5 6]. Patients with defects in any of the genes of the RecQ helicase family are cancer prone [7]. On the other hand, it is well established that fibroblasts from WS patients are susceptible to premature senescence. Although telomere erosion rates do not differ from those of control cells [8] WS cells cease replicating in culture after fewer divisions and with longer telomeres than control cells [9, 10]. This may result from replicative stress and damage to telomere DNA in WRN-deficient cells [11, 12]. WS cells also show a pronounced senescence response when infected with a retrovirus expressing as activated oncogene, Myc [13, 14]. Because WS cells exhibit premature senescence, and because senescence is an anti-cancer mechanism [15], it might be expected that WS cells would be resistant to neoplastic transformation.
Although prior experiments have shown that suppression of the pRb and p53 DNA damage checkpoint pathways by viral oncoproteins enables bypass of replicative senescence in WS cells [16 - 18], to our knowledge there have not been any experimental investigations of the neoplastic conversion of WS cells or the properties of cancer cells from WS patients. In previous studies we showed that normal human fibroblasts require only the combination of oncogenic Ras and SV40 large T antigen (SV40 LT) to be converted to fully tumorigenic cells, as evidenced by their ability to form invasive and metastatic tumors in the subrenal capsule assay in immunodeficient mice [19, 20]. Telomerase activity was not required. Here we investigated whether the increased susceptibility of WS cells to premature senescence creates a block to neoplastic conversion by the combination of Ras and SV40 LT. There are two ways in which premature senescence could potentially impact the conversion of WS cells to cancer; first, infection with Ras/SV40 LT retroviruses could result in a senescence response that prevents tumor formation; second, a greater tendency for damage at telomeres in WS cells could limit the growth or malignant properties of telomerase-negative tumors growing in an immunodeficient mouse.
Materials and Methods
Growth of human fibroblasts and retroviral transduction
Primary human fibroblasts were obtained as follows: HCA2 (MJ-90) and HCA3 (BJ) were derived in the laboratory of O. Pereira-Smith, UTHSCSA; WS fibroblasts AG00780, AG04110, AG05229 and AG06300 were obtained from Coriell Institute, Camden, NJ; control fibroblasts CRL-2707, CRL-2708 and CRL-2714 were obtained from the American Type Culture Collection, Manassas, VA. The retroviruses used are of the LX type [21] and have been described previously [22]. They were constructed from pLEGFP-N1 (BD Biosciences Clontech, Palo Alto, CA) by replacing the neo gene. In the Ras-encoding retroviral plasmid neo has been replaced with Ha-RasG12V cDNA. In the SV40 LT-encoding plasmid neo has been replaced by an intronless SV40 LT cDNA and the CMV-EGFP cassette has been removed. Retroviral plasmid pBabe-puro-hTERT was obtained from J. Campisi.
The Phoenix cell line (amphotropic) was used for production of retroviral particles [23]. Phoenix cells were transfected with retroviral plasmids by the calcium phosphate method. After 48 hours, the supernatant medium was passed through a 0.45 μm filter and added to the target cells. Cells were infected with a 1:1 mixture of Ras and SV40 LT retroviruses. Infection was performed twice for 24 hours each. Because the Ras retroviral construct also encodes GFP [22], the rate of infection of the culture was monitored by fluorescence microscopy. Following infection, the cell population was expanded to a sufficient number for cell transplantation without selection. The hTERT-encoding retrovirus was used to introduce hTERT into telomerase-negative cells following isolation from tumors (see Results). In this case cells were selected with 1 μg/ml puromycin after infection.
Transplantation of cells in RAG2-/-,γc-/- mice
RAG2-/-,γc-/- mice were purchased from Taconic (Germantown, NY). Animals (both males and females) at an age greater than 6 weeks (∼25 g body weight) were used in these experiments. Procedures were approved by the institutional animal care committee and were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals. We transplanted 2 × 106 cells beneath the kidney capsule as previously described for adrenocortical cells [22, 24 - 26].
Animals were sacrificed at various times following transplantation, as described in Results. Tumors were visualized by fluorescence using a 470 nm light source (Lightools Research, Encinitas, CA) in conjunction with a low level of white light. This combination allowed the direct visualization of the fluorescent tumor cells together with the nonfluorescent organs.
Histological and immunohistochemical analysis
The fixation, paraffin embedding, and histological examination of tissue formed from transplanted cells were carried out using standard techniques. SV40 LT was detected with monoclonal antibody PAb416 (EMD Biosciences, La Jolla, CA) used at 1:50 dilution. Additionally, tissues were fixed using the AMeX method [27]. Thick (30-50 μm) sections were prepared from AMeX-fixed, paraffin-embedded tissues and were used for immunocytochemistry without antigen retrieval. An antibody against γ-H2AX was obtained from Upstate Biotechnology (Lake Placid, NY). Bound antibody was visualized with a biotinylated secondary antibody and streptavidin-fluorescein conjugate (Vector Laboratories, Burlingame, CA). Photography was performed using a Zeiss Axiovert fluorescence microscope.
In situ gelatinase activity
For in situ measurement of gelatinase activity, cells were grown on collagen-coated glass coverslips (Becton Dickinson, Franklin Lakes, NJ) and were fixed in acetone. The fixed cells were incubated in 0.1 M HCl for 20 seconds and then transferred to PBS. This brief acid treatment destroyed GFP fluorescence but did not interfere with in situ gelatinase assays. For gelatinase assays we used DQ (dye-quenched) gelatin (Invitrogen/Molecular Probes, Carlsbad, CA). This gelatin preparation is highly conjugated with fluorescein and fluorescence is quenched until the gelatin is cleaved, releasing fluorescent peptides. We added 100 μg/ml DQ gelatin in PBS to the fixed cells. The coverslip was placed faced down on a glass slide and incubated for 2 hours at room temperature. Following the incubation the cells were photographed using a Zeiss Axiovert fluorescence microscope.
Results
Formation of invasive cancer by WS cells expressing RasG12V and SV40 large T antigen
In previous experiments we showed that normal human neonatal or fetal fibroblasts expressing RasG12V and SV40 LT acquire the ability to grow as invasive cancers in the kidney of immunodeficient mice. Neither telomerase activity nor SV40 small t antigen were required [19]. Here, we first determined whether WS cells could similarly be converted to cancer cells by expression of Ras/SV40 LT. We chose four WS fibroblast cell lines, AG00780, AG04110, AG05229, and AG06300, that could be obtained at low population doubling levels (∼7-35). Each of these lines of WS cells were co-infected with two retroviruses, once encoding Ras/GFP and the other encoding SV40 LT. Following sufficient expansion of the cell populations after infection, cells were transplanted in the subrenal capsule space of RAG2-/-,γc-/- mice. In this site they produced malignant tumors that showed invasion into the kidney to varying extents (Figure 1). Ras/SV40 LT-expressing cells from each cell line were transplanted into 4-12 mice; all mice developed tumors (Figure 1).
Figure 1. Malignant properties of WS fibroblasts expressing RasG12V and SV40 LT.
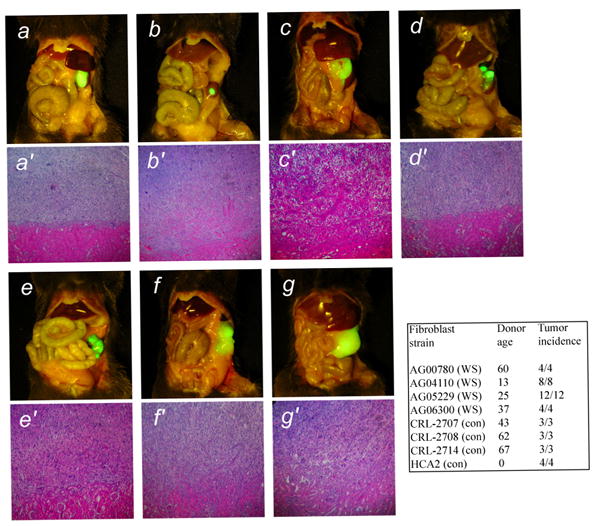
Skin fibroblasts from four WS patients and three control subjects were transduced with retroviruses encoding RasG12V and SV40 LT and and were transplanted in the subrenal capsule space of immunodeficient mice. After 40 days mice were sacrificed and the gross appearance of tumors formed were photographed in situ using fluorescence under blue light. a-d, Werner syndrome donors (AG05229, AG04110, AG00780, AG06300); e-g, control donors (CRL-2707, CRL-2708, CRL-2714). a′-g′, Histological appearance of the tumors. Hematoxylin and eosin stain. Magnification a-g, x1; a′-g′, x200. The table gives the ages of the donors and tumor incidence. HCA2 are neonatal fibroblasts used in previous studies (19).
Previously we tested this oncogene combination in neonatal and fetal human fibroblasts [19]. Neonatal or fetal fibroblasts are not available from WS patients. We therefore used fibroblasts from adult control donors of various ages as appropriate comparisons for the WS cells. These fibroblasts also produced malignant tumors when transduced with Ras and SV40 LT and then transplanted in immunodeficient mice (Figure 1). In general, tumors from WS cells and control cells produced tumors with similar biological characteristics. These characteristics are (i) extensive growth and invasion in the kidney, with eventual destruction of most of the kidney parenchyma; (ii) lung metastases in some animals (detected by fluorescence; ref. 19); and (iii) local growth in the peritoneal cavity of tumor masses that are noncontiguous with the primary tumor. Although in general the behavior of tumors from WS cells and control cells were similar, we noted that local peritoneal growth of tumor masses was more frequent in tumors formed from WS cells.
Growth of tumors derived from both WS and control cells is limited by lack of telomerase
Because the oncogene combination used here did not include telomerase (hTERT), we anticipated that tumors would not grow indefinitely in host animals [19]. In order to test this we conducted serial transplantation experiments in immunodeficient mice. Four representative series are shown in Figure 2. In order to perform serial transplantation, cells were isolated from primary tumors and GFP+ cells were selected by flow sorting; this process was used to separate the tumor cell population away from various mouse cell types. These cells were then retransplanted in secondary host animals. In many cases no secondary tumors were formed; when secondary tumors did form, they were usually smaller than the parent primary tumor (Figure 2). Cells were isolated from secondary tumors by flow sorting and were transplanted into tertiary host animals. In most cases no tumors formed in tertiary hosts; occasionally small tumors were found. In the very few cases in which tertiary tumors were large enough to permit isolation of tumor cells, no further tumors could be formed from them by retransplantation. In summary, in no serial transplantation experiment were Ras/SV40 LT-expressing cells able to be passaged beyond the tertiary host animal. Overall, the behavior of tumors from WS and control cells in serial transplantation experiments were similar.
Figure 2. Loss of growth in serially transplanted tumors derived from WS and control fibroblasts.
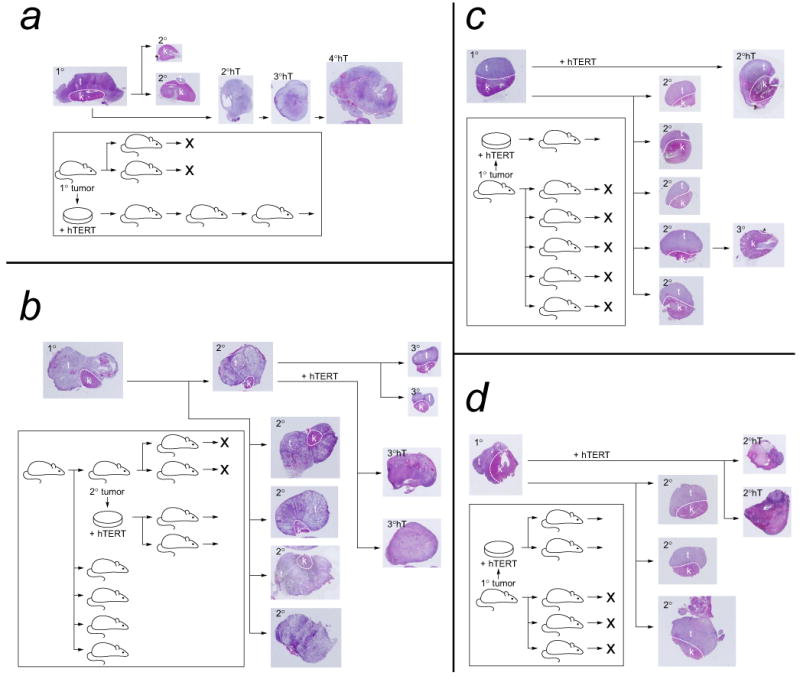
Ras/SV40 LT-transformed fibroblasts were transplanted in immunodeficient mice and allowed to grow for 40 days. Following sacrifice of the mice, the histology of the tumors was examined. Cells were also isolated from tumors and purified by flow cytometry, and then used in serial transplantation experiments. The serial transplantation histories of four reprentative tumors are shown in this figure. A hematoxylin/eosin-stained section of each tumor is shown. a and b show histories of tumors formed from control fibroblasts; c and d show histories of tumors formed from WS fibroblasts. In each case cells from a primary tumor were retransplanted into secondary host animals. Cells from secondary tumors were retransplanted into tertiary host animals. “X” indicates a failure of transplanted cells to form a detectable tumor. In some cases cells from primary or secondary tumors were infected with a retrovirus encoding hTERT, and were then retransplanted; tumors formed are indicated by 2°hT, etc., and were also serially transplanted. The dashed white line on tumor sections indicates the boundary between the kidney and the tumor tissue. In those cases where there is no dashed line the entire kidney was replaced by tumor. All tumor sections are shown at the same magnification, x1.
In order to demonstrate that the limitation on serial growth was the result of lack of telomerase, cells isolated from primary or secondary tumors were transduced with a retrovirus encoding hTERT, selected with puromycin, and then retransplanted. In all cases large tumors formed. Cells were isolated from these tumors and were retransplanted. In all cases large tumors formed on retransplantation, and in all cases these tumors could subsequently be serially retransplanted without any signs of reduction in growth. Instead, tumors formed at later in vivo passages usually grew more rapidly than those at earlier passages. We therefore conclude that the inability of tumors to be passaged indefinitely is caused by lack of telomerase and that introduction of hTERT enables cells to be passaged indefinitely.
Many cells in tumors formed from Ras/SV40 LT-expressing WS and control cells are in crisis
On the basis of the ability of Ras/SV40 LT to convert both WS and control fibroblasts to cells that produced malignant tumors in immunodeficient mice, we conclude that the absence of WRN does not provide a primary barrier to neoplastic transformation by this combination of oncogenes. We next determined whether cells in tumors formed from WS fibroblasts had evidence of more severe or altered crisis, or more evidence of DNA damage, than in tumors formed from control fibroblasts.
In the absence of introduced hTERT, cells in tumors derived from Ras/SV40 LT-expressing cells are not capable of indefinite cell division and eventually enter crisis [19]. We investigated whether cells in tumors derived from WS fibroblasts differed in this respect from those in tumors derived from control cells. In both primary and secondary WS cell tumors we noted the occurrence of cells with grossly abnormal morphology. These cells have characteristics of crisis, i.e. cell enlargement accompanied by the appearance of numerous abnormal mitotic chromosomes; abnormal cell division as evidenced by an abnormal spindle number; nuclear abnormalities such as a single enlarged nucleus (often with an abnormal shape) or multiple smaller nuclei [19, 22, 28] (Figure 3).
Figure 3. Crisis in tumors as evidenced by nuclear abnormalities.
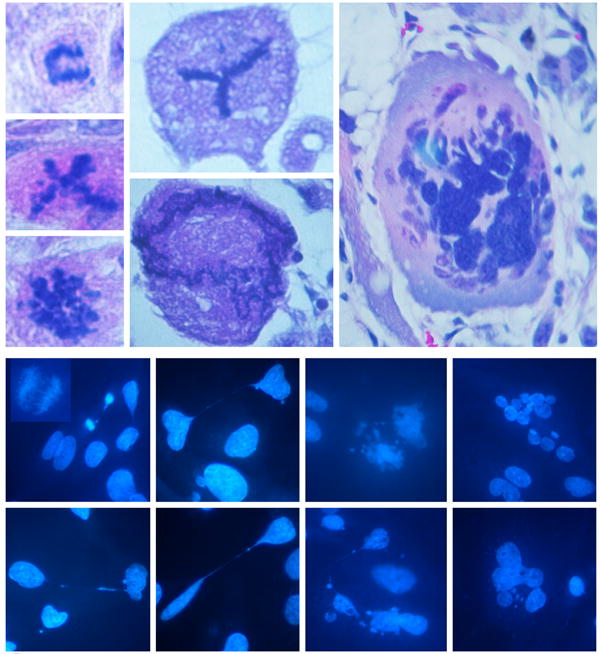
The top panel shows examples of abnormal cells in tumors derived from Ras/SV40 LT-expressing WS fibroblasts (hematoxylin- and eosin-stained sections). The top left example shows an anaphase bridge and other examples show cells with apparently abnormal numbers of spindles or other abnormal changes. The lower panel shows cells isolated from tumors and placed in culture (DAPI stain). The insert in the top left image shows an example of an anaphase bridge. Other cells show chromatin strings between separated nuclei, nuclear fragmentation, abnormal nuclear morphology or multiple nuclei. Magnifications: top panel, x500 and x1500; lower panel x500.
Cells were isolated from tumors by flow sorting and were placed back in culture for further studies. DAPI staining showed many cells with morphological evidence of crisis: abnormal nuclear morphology or nuclear number, and by the formation of chromatin strings (Figure 3). In cultures of cells isolated from primary tumors and replated in culture, ∼20 to ∼35% of the cells exhibited morphological evidence of crisis. The frequency of occurrence was similar tumors derived from both control and WS fibroblasts. The fraction increased in tumors derived from secondary and tertiary tumors.
Morphological evidence of crisis was accompanied by the occurrence of γ-H2AX-staining foci indicative of DNA double strand breaks. In sections of tumors derived from Ras/SV40 LT-expressing WS and control fibroblasts cells with morphological evidence of crisis, such as giant nuclei, also had large numbers of double strand breaks as evidenced by nuclear staining with an anti-γ-H2AX antibody (Figure 4). In cells isolated from tumors, cells with morphological evidence of crisis also showed high levels of anti-γ-H2AX staining (Figure 5). Interestingly, γ-H2AX foci were found within the chromatin strings that often formed between crisis cells attempting to divide (Figure 5). Given the fact that morphological signs of crisis are rather varied, there were no obvious differences between cells in tumors from WS and control fibroblasts.
Figure 4. γ-H2AX in tumor cells in situ.
a, b, c, Sections of tumors from Ras/SV40 LT-expressing WS fibroblasts were stained with an antibody against γ-H2AX. a′, b′, c′, DAPI stain. Magnification x500.
Figure 5. γ-H2AX in isolated tumor cells in culture.
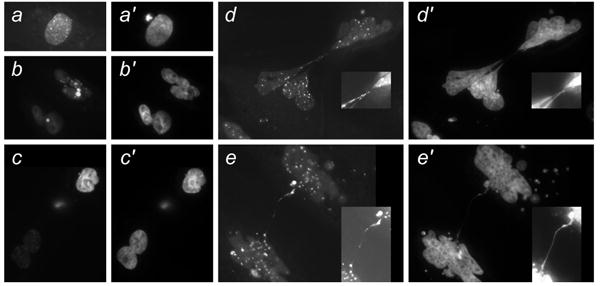
Cells were isolated from tumors from Ras/SV40 LT-expressing WS fibroblasts. a-e, stained with γ-H2AX antibody. a′-e′, DAPI stain. Insets show chromatin strings photographed with a higher exposure time.
Maintenance of invasiveness and high gelatinase activity in cells in crisis
Prior studies of the time-course of growth of Ras/SV40 LT-expressing cells in the subrenal capsule have shown that tumors begin by expanding under the capsule. The characteristic that defines the tumor as malignant is its invasion into the kidney. In the first three weeks of growth, when the tumor is still relatively small, there is a clear, relatively straight, border between the tumor and the kidney parenchyma, indicating that invasion into the kidney is not yet evident [19, 28]. At about three weeks invasion into the kidney begins. Invasion then proceeds to the extent that the entire kidney is destroyed. Very small tumors formed in secondary or tertiary hosts showed no invasion (Figure 6), but almost all primary tumors and most secondary and tertiary tumors were invasive to some extent (Figure 6). There was always a decline in invasiveness as tumors were passaged in vivo (e.g. Figure 6 b,c,d). The decline in invasiveness was less marked in tumors from WS cells than in tumors from control cells.
Figure 6. Invasive characteristics of tumors formed from Ras/SV40 LT-expressing fibroblasts.
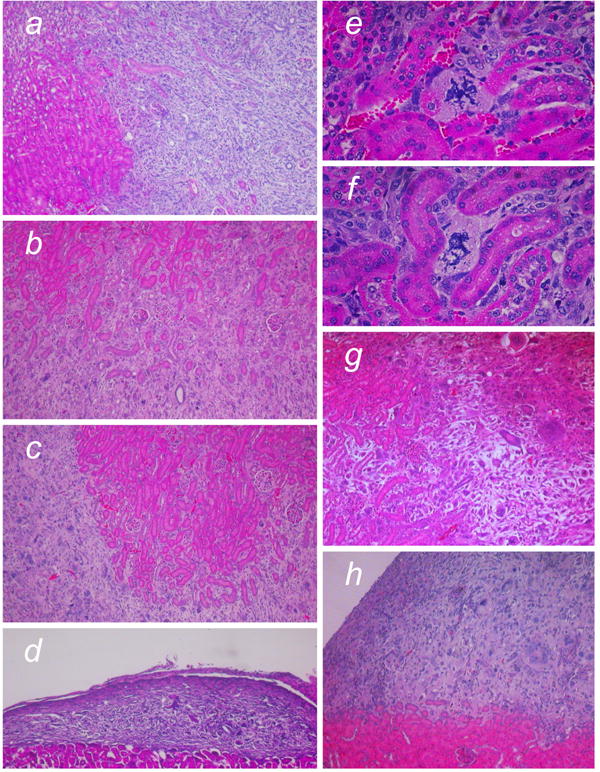
Representative examples are shown to compare invasiveness of cells from WS and control donors. Hematoxylin and eosin stain. a, Primary tumor from LT/Ras-expressing WS fibroblasts. b,c Two secondary tumors derived by re-transplantation of cells from the tumor shown in a. d, Tertiary tumor derived by re-transplantation of the cells isolated from the tumor shown in b. e,f, Cells in crisis adjacent to kidney tubules (primary tumor from Ras/SV40 LT-expressing WS fibroblasts). g Primary tumor derived from LT/Ras-expressing control fibroblasts. h Secondary tumor derived by re-transplantation of cells isolated from the tumor shown in g. Magnification, a-d x50; e-f x200; g-h x50.
On an individual cell basis, invasiveness is observed as the penetration of tumor cells between the kidney tubules. As tumors formed from WS and control cells invaded into the kidney, we noted that crisis cells could clearly be found deep within the kidney parenchyma, for example adjacent to tubules (Figure 6). In order to ensure that such cells were indeed derived from the transplanted cell population, we stained sections with anti-SV40 LT or anti-GFP (Figure 7). As evidenced by antibody staining, cells with clear morphological evidence of crisis and expressing SV40 LT or GFP were found deep in the kidney parenchyma. When we similarly examined tumors derived from Ras/SV40 LT-expressing cells that had subsequently been transduced with hTERT, we observed far fewer cells in crisis and instead cells had a uniform appearance (Figure 7h).
Figure 7. Tumor cells in crisis deep in the kidney parenchyma.
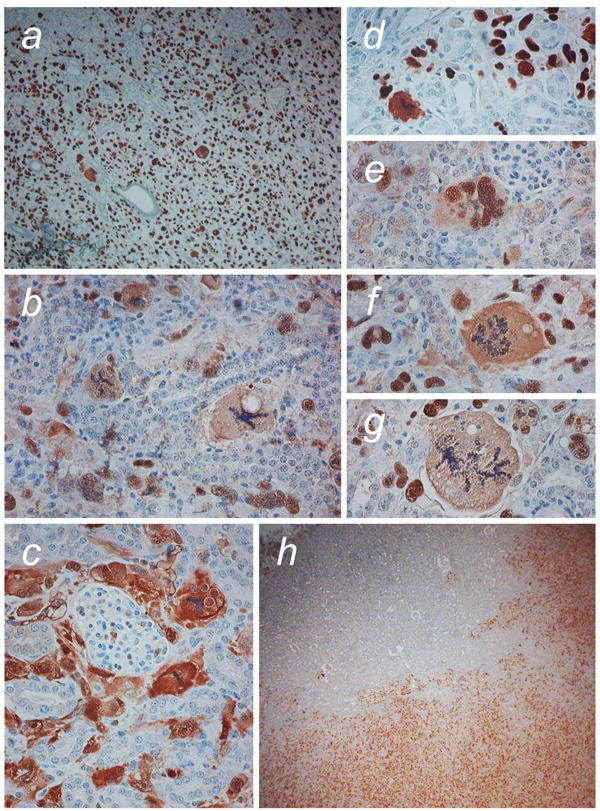
Sections of tumors derived from Ras/SV40 LT-expressing fibroblasts were stained with anti-SV40 LT to enable identification of tumor cells within the kidney. a SV40 LT staining shows scattered crisis cells throughout the tumor. b, Crisis cells observed within the kidney adjacent to tubules and glomeruli. c, GFP staining demonstrates tumor cells surrounding a glomerulus. d-g, Examples of cells with abnormal nuclear morphology within the kidney adjacent to kidney tubules and glomeruli. h SV40 LT-staining in hTERT-transduced tumor cells retransplanted into immunodeficient mouse kidney. Magnification, a x50; b-g x200; h x25.
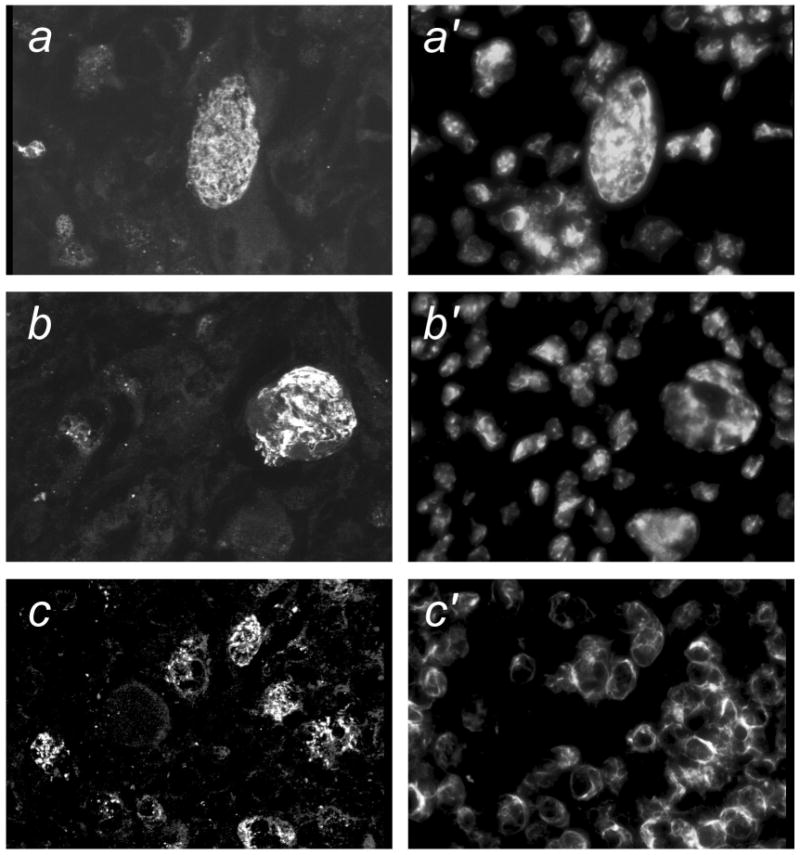
The fact that individual cells in crisis were apparently able to retain the ability to invade into the kidney indicated that they must retain biochemical properties, such as the secretion of enzymes that can degrade the extracellular matrix, that are required for invasion. Secretion of gelatinases, several forms of matrix metalloproteinases (e.g. MMP-2 and MMP-9), is a characteristic of invasive tumor cells [29]. When cells were isolated by flow sorting from tumors and placed in culture, they showed strong in situ gelatinase activity using a fluorescent gelatin substrate (Figure 8). Cells that with nuclear morphology indicative of crisis (multinucleation, blebbing and chromatin strings) showed strong activity. Control cells (not Ras/SV40 LT-expressing) had much weaker activity. The intention of this assay was to show that individual cells in crisis retained gelatinase activity rather than to provide an exact quantitation of gelatinase activity. However, we noted that cells from WS tumors appeared to have higher activity that cells from control tumors. Additionally, real-time PCR showed that MMP-9 mRNA was expressed at ∼5-fold to ∼13-fold higher in cells derived from primary tumors formed from Ras/SV40 LT-expressing WS fibroblasts than in cells derived from control tumors.
Figure 8. In situ gelatinase activity in fibroblasts before and after transduction with Ras/SV40 LT and in cells isolated from tumors.
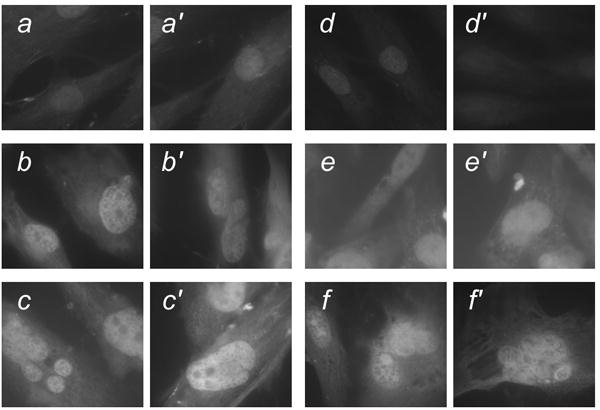
Cells were incubated with fluorescein-conjugated gelatin (DQ-gelatin) as described in Materials and Methods. All photographs were made with the same exposure time. a,a′, Nontransduced WS fibroblasts; b,b′, WS fibroblasts following transduction with Ras/SV40 LT; c,c′ Cells from tumor formed from Ras/SV40 LT-expressing WS fibroblasts; d,d′, Nontransduced control fibroblasts; e,e′, Control fibroblasts following transduction with Ras/SV40 LT; f,f′, Cells from tumor formed from Ras/SV40 LT-expressing control fibroblasts.
Discussion
In these experiments we set out to test whether the absence of the WRN RecQ helicase caused human fibroblasts to be resistant to transformation to a malignant state by a combination of oncoproteins, Ras and SV40 T antigen. Because WS cells readily enter senescence, we hypothesized that this would create a barrier to neoplastic transformation [15] or that tumors formed from WS fibroblasts would be limited in their ability to grow in immunodeficient mice. The results show that this is not the case. WS cells were indeed transformed to a malignant state by Ras and SV40 LT, and there was no indication that this was less readily accomplished than in cells from control donors, or that tumors formed from WS cells were impaired in their ability to show malignant properties in vivo. In fact, there were several hints that tumors derived from WS cells were more aggressive. MMP-9 mRNA was present at higher levels in Ras/SV40 LT-expressing WS cells than in Ras/SV40 LT-expressing control cells, which correlated with a high level of in situ gelatinase activity. Local peritoneal metastases were observed more frequently in mice that received transplants of Ras/SV40 LT-expressing WS cells than in mice with transplants of Ras/SV40 LT-expressing control cells; also invasiveness in secondary transplanted tumors was more commonly observed in tumors derived from WS cells. Larger-scale experiments would be required to test whether these trends are statistically significant. However, we may draw the conclusion that the absence of WRN does not create a barrier to neoplastic conversion because of an enhanced senescence response, for example in response to oncogenic Ras [30].
We also conclude that the absence of WRN does not retard growth in vivo because of accelerated or more severe crisis. In the absence of hTERT, Ras/SV40 LT-expressing tumor cells cannot be serially transplanted more than twice before they fail to regrow. During growth in vivo, telomerase-negative Ras/SV40 LT-expressing cells undergo extreme telomere shortening and reach the state called “crisis” [19]. In this state, short dysfunctional telomeres cause end-to-end chromosome fusions [31]; in cells with disrupted checkpoints this results in (i) breakage-fusion-bridge cycles, leading to increasing aneuploidy; and (ii) mitotic catastrophe, a failure of cytokinesis, resulting in tetraploidization, multipolar cell division, and gross aberrations in chromosome number [15]. Mitotic catastrophe leads to arrest in mitosis, or alternatively to the formation of cells with multiple nuclei or a single giant nucleus. Cells with abnormal nuclei and other features of mitotic catastrophe are often observed in human cancers [15]. Here, abnormal cells with features of crisis were observed in greater numbers in tumors as they reached the point of inability to grow and their numbers were reduced in tumors formed from cells with ectopic hTERT. The decrease in crisis cells observed by introduction of hTERT confirms that the origin of mitotic catastrophe is telomere dysfunction. However, crisis was not accelerated or more severe in tumors derived from WS cells.
We observed that crisis acted as a tumor suppressor mechanism for populations of Ras/SV40 LT-expressing cells, consistent with prior studies [19, 22]. However, here we also made the novel observation that individual cells retained malignant characteristics, even as they exhibited the morphological features of severe crisis. In both early- and late-stage tumors, we observed cells with morphological features of crisis that were also clearly malignant because they were found deep within the kidney parenchyma. In order to penetrate between the tubules of the kidney, tumor cells must be able to secrete enzymes such as matrix metalloproteinases that digest extracellular matrix, and also must be capable of migration through the digested matrix [29]. Cells retained this capability despite being in crisis. Moreover, cells isolated from tumors both exhibited features of crisis, as observed by nuclear morphology and high levels of γ-H2AX-staining foci, indicative of double strand breaks [32], and had a high level of gelatinase activity as assessed by an in situ assay. Although we did not investigate this here, it is established that Ras activates pathways that result in increased MMP expression [33]. We conclude that telomere dysfunction, resulting in crisis and prevention of proper cell division, may co-exist with activation of cellular pathways that increase invasive behavior, such as MMP expression and migratory capacity. The block to further cell division acts as a tumor suppressor mechanism for the cell population, but individual cells do not lose their malignant properties.
Whereas the susceptibility of WS cells to premature senescence did not prevent the acquisition of malignant properties by Ras/SV40 LT, it is possible that senescence-associated properties of the cells may increase pro-tumorigenic properties, such as MMP secretion. These opposing actions have indicated that senescence is a “double-edged sword,” in that senescence acts as a tumor suppressor mechanism to directly prevent cancer, yet potentially enhances cancer via the action of senescent cell products [34]. Prior studies have shown that WS cells have gene expression patterns that are characteristic of control senescent cells even when they are not actually senescent [35, 36]. We obtained suggestive but not definitive evidence that, as they reach crisis, tumor cells derived from WS fibroblasts may be more malignant than those from control fibroblasts. Further studies along these lines may provide insight into the cellular mechanisms involved and their relationship to the increased incidence of some types of cancer in Werner syndrome.
Conclusions
We conclude that the absence of WRN protein activity does not prevent the acquistion of malignant properties in fibroblasts, and does not accelerate or change eventual entry into crisis. The novel finding that individual tumor cells derived from both WS and control fibroblasts retain malignant properties even in crisis indicates that crisis acts as a tumor suppressor mechanism on the population as a whole, while not impairing the ability of individual cells to exhibit malignant properties.
Acknowledgments
This work was supported by grants from the National Institute on Aging (AG12287 and AG20752) and by a Senior Scholar Award from the Ellison Medical Foundation. Support by the San Antonio Cancer Institute Cancer Center (P30 CA54174) is also acknowledged.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Authors' contributions: FY conceived the study, participated in its design, and performed most of the experiments; MC performed one of the assays presented; PJH participated in the design of the study and wrote the manuscript.
All authors have read and approved the final manuscript.
Contributor Information
Furong Yuan, Email: fyuan@uci.edu.
Meizhen Chen, Email: chenm1@uthscsa.edu.
Peter J. Hornsby, Email: hornsby@uthscsa.edu.
References
- 1.Martin GM. The Werner mutation: Does it lead to a “public” or “private” mechanism of aging? Mol Med. 1997;3:356–358. [PMC free article] [PubMed] [Google Scholar]
- 2.Ostler EL, Wallis CV, Sheerin AN, Faragher RG. A model for the phenotypic presentation of Werner's syndrome. Exp Gerontol. 2002;37:285–92. doi: 10.1016/s0531-5565(01)00194-2. [DOI] [PubMed] [Google Scholar]
- 3.Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner syndrome (adult progeria) Cancer Epidemiol Biomarkers Prev. 1996;5:239–46. [PubMed] [Google Scholar]
- 4.Ishikawa Y, Miller RW, Machinami R, Sugano H, Goto M. Atypical osteosarcomas in Werner Syndrome (adult progeria) Jpn J Cancer Res. 2000;91:1345–9. doi: 10.1111/j.1349-7006.2000.tb00924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;6:53–61. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhillon KK, Sidorova J, Saintigny Y, Poot M, Gollahon K, Rabinovitch PS, Monnat RJ., Jr Functional role of the Werner syndrome RecQ helicase in human fibroblasts. Aging Cell. 2007;6:53–61. doi: 10.1111/j.1474-9726.2006.00260.x. [DOI] [PubMed] [Google Scholar]
- 7.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–78. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 8.Baird DM, Davis T, Rowson J, Jones CJ, Kipling D. Normal telomere erosion rates at the single cell level in Werner syndrome fibroblast cells. Hum Mol Genet. 2004;13:1515–24. doi: 10.1093/hmg/ddh159. [DOI] [PubMed] [Google Scholar]
- 9.Thompson KVA, Holliday R. Genetic effects on the longevity of cultured human fibroblasts. I. Werner's syndrome. Gerontology. 1983;29:73–82. doi: 10.1159/000213096. [DOI] [PubMed] [Google Scholar]
- 10.Schulz VP, Zakian VA, Ogburn CE, Mckay J, Jarzebowicz AA, Edland SD, Martin GM. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum Genet. 1996;97:750–754. doi: 10.1007/BF02346184. [DOI] [PubMed] [Google Scholar]
- 11.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–3. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 12.Bailey SM, Murnane JP. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006;34:2408–17. doi: 10.1093/nar/gkl303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grandori C, Wu KJ, Fernandez P, Ngouenet C, Grim J, Clurman BE, Moser MJ, Oshima J, Russell DW, Swisshelm K, Frank S, Amati B, Dalla-Favera R, Monnat RJ., Jr Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003;17:1569–74. doi: 10.1101/gad.1100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grandori C, Robinson KL, Galloway DA, Swisshelm K. Functional link between Myc and the Werner gene in tumorigenesis. Cell Cycle. 2004;3:22–5. [PubMed] [Google Scholar]
- 15.Hornsby PJ. Senescence as an anticancer mechanism. J Clin Oncol. 2007;25:1852–1857. doi: 10.1200/JCO.2006.10.3101. [DOI] [PubMed] [Google Scholar]
- 16.Huschtscha LI, Thompson KV, Holliday R. The susceptibility of Werner's syndrome and other human skin fibroblasts to SV40-induced transformation and immortalization. Proc R Soc Lond B Biol Sci. 1986;229:1–12. doi: 10.1098/rspb.1986.0070. [DOI] [PubMed] [Google Scholar]
- 17.Davis T, Faragher RG, Jones CJ, Kipling D. Investigation of the signaling pathways involved in the proliferative life span barriers in werner syndrome fibroblasts. Ann NY Acad Sci. 2004;1019:274–7. doi: 10.1196/annals.1297.046. [DOI] [PubMed] [Google Scholar]
- 18.Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci USA. 2007;104:2205–10. doi: 10.1073/pnas.0609410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun B, Chen M, Hawks CL, Pereira-Smith OM, Hornsby PJ. The minimal set of genetic alterations required for conversion of primary human fibroblasts to cancer cells in the subrenal capsule assay. Neoplasia. 2005;7:585–593. doi: 10.1593/neo.05172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun B, Chen M, Hawks CL, Hornsby PJ. Immortal ALT+ human cells do not require telomerase reverse transcriptase for malignant transformation. Cancer Res. 2005;65:6512–6515. doi: 10.1158/0008-5472.CAN-05-1210. [DOI] [PubMed] [Google Scholar]
- 21.Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–2. [PMC free article] [PubMed] [Google Scholar]
- 22.Sun B, Huang Q, Liu S, Chen M, Hawks CL, Wang L, Zhang C, Hornsby PJ. Progressive loss of malignant behavior in telomerase-negative tumorigenic adrenocortical cells and restoration of tumorigenicity by human telomerase reverse transcriptase. Cancer Res. 2004;64:6144–6151. doi: 10.1158/0008-5472.CAN-04-1376. [DOI] [PubMed] [Google Scholar]
- 23.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas M, Northrup SR, Hornsby PJ. Adrenocortical tissue formed by transplantation of normal clones of bovine adrenocortical cells in scid mice replaces the essential functions of the animals' adrenal glands. Nature Med. 1997;3:978–983. doi: 10.1038/nm0997-978. [DOI] [PubMed] [Google Scholar]
- 25.Thomas M, Yang L, Hornsby PJ. Formation of functional tissue from transplanted adrenocortical cells expressing telomerase reverse transcriptase. Nature Biotechnol. 2000;18:39–42. doi: 10.1038/71894. [DOI] [PubMed] [Google Scholar]
- 26.Thomas M, Wang X, Hornsby PJ. Human adrenocortical cell xenotransplantation: Model of cotransplantation of human adrenocortical cells and 3T3 cells in scid mice to form vascularized functional tissue and prevent adrenal insufficiency. Xenotransplantation. 2002;9:58–67. doi: 10.1046/j.0908-665x.2001.00138.x. [DOI] [PubMed] [Google Scholar]
- 27.Sato Y, Mukai K, Furuya S, Kameya T, Hirohashi S. The AMeX method: a multipurpose tissue-processing and paraffin-embedding method. Extraction of protein and application to immunoblotting. Am J Pathol. 1992;140:775–779. [PMC free article] [PubMed] [Google Scholar]
- 28.Liang S, Kahlenberg M, Rousseau D, Hornsby PJ. Neoplastic conversion of human colon smooth muscle cells: No requirement for telomerase. Mol Carcinogen. 2007 doi: 10.1002/mc.20405. in press. [DOI] [PubMed] [Google Scholar]
- 29.Bjorklund M, Koivunen E. Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta. 2005;1755:37–69. doi: 10.1016/j.bbcan.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 31.Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, Haughton M, Baird DM. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 33.Schafer R, Tchernitsa OI, Zuber J, Sers C. Dissection of signal-regulated transcriptional modules by signaling pathway interference in oncogene-transformed cells. Adv Enzyme Regul. 2003;43:379–91. doi: 10.1016/s0065-2571(02)00040-7. [DOI] [PubMed] [Google Scholar]
- 34.Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Choi D, Whittier PS, Oshima J, Funk WD. Telomerase expression prevents replicative senescence but does not fully reset mRNA expression patterns in Werner syndrome cell strains. FASEB J. 2001;15:1014–20. doi: 10.1096/fj.00-0104com. [DOI] [PubMed] [Google Scholar]
- 36.Kyng KJ, May A, Stevnsner T, Becker KG, Kolvraa S, Bohr VA. Gene expression responses to DNA damage are altered in human aging and in Werner syndrome. Oncogene. 2005;24:5026–42. doi: 10.1038/sj.onc.1208692. [DOI] [PubMed] [Google Scholar]