

1. Introduction
Asymmetric transformationsi and transition metal-catalyzed cross-coupling reactionsii are critical components of modern organic synthesis. The impact of these reactions on synthetic chemistry and related fields can be measured in many ways. For example, in 2001 the Nobel Prize in chemistry was awarded to Dr. William Knowles, Professor Ryoji Noyori, and Professor K. Barry Sharpless for their contributions to catalytic asymmetric synthesis. Then, in 2010, Professors Richard Heck, Ei-ichi Negishi, and Akira Suzuki were awarded the Nobel Prize for pioneering the development of catalytic cross-coupling reactions. The intersection of these two areas of research, the development of cross-coupling reactions that construct stereogenic centers though incorporation and control of secondary sp3 hybridized fragments, has emerged as an important frontier in reaction design.iii,iv The first stereochemical investigations of such cross-coupling reactions were primarily performed as part of stoichiometric mechanistic studies. The field has since evolved to encompass target-oriented endeavors and enable precise formation and manipulations of stereogenic centers. We now have a growing pool of cross-coupling reactions from which to construct tertiary stereogenic centers with excellent stereoselectivity.
Stereochemical control in alkyl cross-coupling reactions can be accomplished in several ways. In a substrate-controlled stereoselective reaction, also referred to as a stereospecific reaction, stereochemical information is transferred from one of the starting materials to the product. For example, during a stereospecific cross-coupling reaction an enantioenriched electrophile will provide enantioenriched product in the presence of an achiral catalyst. Alternatively, an enantioenriched alkyl metal reagent can also be used to achieve a stereospecific cross-coupling. These two scenarios are illustrated in Scheme 1a. The success of the reaction can be determined by measuring enantiospecificity (es), which is calculated by dividing the enantiomeric excess (ee) of the product by the ee of the starting material.v In contrast, stereoselectivity can be controlled by chiral ligands on the metal in a catalyst-controlled stereoselective reaction. These reactions occur through a stereoconvergent process, utilizing racemic electrophiles or transmetalating agents (Scheme 1b).
Scheme 1.

General types of asymmetric cross-coupling reactions.
This review will focus on the cross-coupling reactions of secondary alkyl reagents that afford enantioenriched tertiary carbon centers. The main sections will cover the two approaches outlined above; first, stereospecific reactions will be discussed, then stereoselective reactions that are controlled by chiral catalysts will be reviewed. Asymmetric substitution reactions of cuprates with alkyl halides and tosylates will also be presented to provide relevant context for subsequent development of catalytic methods. While there are many excellent diastereoselective cross-coupling reactions, we will focus on enantioselective variants that cannot be influenced by thermodynamic preference for formation of one diastereomer. Allylic displacement reactions,vi asymmetric Heck reactionsvii and cross-coupling reactions that result in products with axial chiralityviii will not be discussed in this review. Additionally, desymmetrization reactions of achiral compounds in cross-coupling reactions to construct tertiary and quaternary stereocenters are outside the scope of this review.ix We refer the reader to other recent reviews of asymmetric cross-coupling reactionsx and, for general discussions of cross-coupling reactions with alkyl partners, we refer the reader to recent reviews on cross-coupling reactions with alkyl halidesiii and alkyl metal complexes.iv
2. Stereospecific Reactions
Stereospecific reactions of enantioenriched electrophiles use the inherent stereochemical information in the substrate to dictate the configuration of the product. This method can take advantage of the many excellent asymmetric additions, oxidations, reductions, or resolutions to construct chiral starting materials with C–X bonds. Asymmetric formation of tertiary carbon centers is arguably more challenging, particularly when the substituents about the chiral center are very similar. One advantage of stereospecific reactions is that a chiral catalyst is not required to differentiate between very similar substituents about the chiral center. This section will cover reactions of enantioenriched electrophiles and nucleophiles.
2.1 Enantioenriched Alkyl Electrophiles
While historically and pedagogically important, the classical SN2 reaction has limited synthetic utility, especially with carbon nucleophiles, due to competing racemization and elimination pathways. The reactions discussed in this section provide alternative solutions to this problem. Metal-mediated reactions have emerged to facilitate the formation of C–C bonds under mild reaction conditions. Despite the challenges of alkyl–alkyl cross-coupling reactions, such as sluggish oxidative addition and facile β-hydride elimination, significant progress has been made in this area. In general, examination of copper-, zinc-, palladium-, and nickel-catalyzed reactions has enabled oxidative addition to otherwise unreactive substrates. Additionally, bulky ligands on the metal can be used to disfavor interactions with β-hydrogens that result in elimination. Analogous to SN2 reactivity, these reactions all maintain stereochemical information during the coupling process and result in inversion of configuration. Iron-based catalysts are also known to facilitate robust coupling reactions with alkyl partners, however these catalysts favor stereorandom processes and thus have not yet been shown to be viable catalysts for stereospecific reactions.xi
2.1.1. Organocuprates
A synthetically and historically important stereospecific transformation that predated modern cross-coupling reactions is the displacement of halides and pseudohalides by organocuprates. While primarily featured in addition reactions to π-systems, the role of cuprates in substitution reactions of enantioenriched alkyl halides is also well-known. Cuprates are uniquely successful where other organometallic nucleophiles such as organomagnesium and organolithium reagents fail. In Whitesides’ seminal publication, the viability of organocuprates to give coupled products in a stereospecific manner was demonstrated.xii Enantioenriched (R)-bromobutane 1, synthesized from the requisite secondary alcohol, was subjected to standard reaction conditions with Ph2CuLi to produce (+)-2 in 67–68% ee (eq 1). Comparison to known optical rotations indicated 84–89% enantiospecificity with inversion of configuration.
 |
(1) |
 |
(2) |
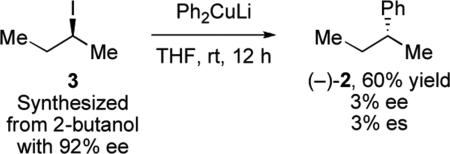
This demonstration of stereochemical fidelity spurred many other investigations of reactions of organocuprates. It has since been shown, however, that the outcome is highly substrate-dependent. For instance, reactions of enantioenriched alkyl iodides result in racemic products.xiii Equation 2 illustrates the analogous reaction with enantioenriched iodobutane 3 to produce 2-phenylbutane with only 3% enantiospecificity. Furthermore, the yield of the reaction can be variable with certain cuprates, including methylcuprate, which typically provides low yields. Lipshutz and co-workers reported systematic studies that provide a general rationale to explain these contradictory outcomes. One possibility was that the method of cuprate preparation could have a strong influence on the reaction. Organocuprates are classified as “lower order” or “higher order” depending on the number of ligands they possess. While they often have very different reactivity, in the case of reactions with alkyl halides, the degree of enantiospecificity was not affected by the cuprate employed in the reaction (Table 1). For example, both higher and lower order organocuprates gave low es with an alkyl iodide (entries 1 and 2) and high es with an alkyl bromide (entries 3 and 4). More highly mixed organocuprates Et(Me)Cu(CN)Li2 and Et(Me)CuLi were necessary for the reactions with alkyl bromides to diminish competing reduction side pathways. The degree of enantiospecificity, however, generally correlated with the identity of the halide, irrespective of the cuprate employed.
Table 1.
Reactions of 2-octyl iodides and bromides with higher and lower-order cuprates.

| ||||||
|---|---|---|---|---|---|---|
| Entry | X | Alkyl Cuprate | Conditions | Yield (%) | ee (%) | es (%) |
| 1 | I | Et2Cu(CN)Li2 | THF, –78 °C, 2 h | 92 | 1 | 1 |
| 2 | I | Et2CuLi | THF, –50 °C, 3 h | 82 | 0 | 0 |
| 3 | Br | Et(Me)Cu(CN)Li2 | THF, 0 °C, 3 h | 72 | >97 | >97 |
| 4 | Br | Et(Me)CuLi | THF, 0 °C, 10 h | 59 | >97 | >97 |
In the early 1970s Johnson and co-workers reported the first mechanistic studies pertaining to alkyl tosylate displacement. Analogous studies with (+)-2-butyl tosylate were consistent with >99% inversion of configuration, however the yield could not be improved above 45%.xiv To address this limitation, a directing group strategy was employed by Hanessian and co-workers. They demonstrated that stereospecific reactions of acyclic alkyl tosylates with lithium dialkyl cuprates proceed in good yields when the strategic placement of a thioether at a distance of two methylene units from the leaving group is employed.xv Specifically, thioethers were found to increase reactivity and suppress elimination and reduction byproducts, as seen with the formation of 4 in 90% yield, compared to 6 in 40% yield. Furthermore, the preference for sulfur over oxygen is apparent in the enhanced reactivity of (methylthio)methyl ether 5 as compared to methoxymethyl ether 7.
Gmeiner and co-workers reported a recent example of stereospecific alkyl tosylate displacement in target-oriented synthesis.xvi All stereoisomers of the dopamine receptor ligand 10 were prepared; therefore, the flexibility of the route with respect to stereochemical control was of utmost importance. An example featuring one stereochemical permutation is shown in Scheme 3. Beginning with readily accessible tosyl pyrrolidine 8, lithium dimethyl cuprate was employed to introduce the methyl group in 70% yield and >99% es. The ester remained intact with no epimerization of the C-2 stereocenter. This compound was carried forward to prepare dopamine receptor ligand 10.
Scheme 3.

Synthesis of dopamine receptor ligand via stereospecific cuprate displacement.
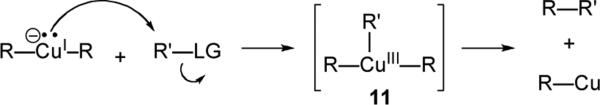
A general mechanism for cuprate displacement is shown in Scheme 4. The reaction is thought to begin with oxidative addition to the substrate by the copper center, followed by reductive elimination of the unstable Cu(III) species 11 to afford the new C–C bond. For a more rigorous discussion of the mechanism of the reactions of nucleophilic organocopper complexes, we refer the reader to a recent review by Nakamura.xvii It is noteworthy that this redox mechanism is one possible reason why cuprates are uniquely suited for displacement reactions at saturated centers as compared to organomagnesium or organolithium reagents.
Scheme 4.
Proposed mechanism for cuprate substitution.
2.1.2. Transition Metal-Catalyzed Reactions
2.1.2.1. Cu-catalyzed
Recent advances in cuprate chemistry have included catalytic variants. Ready and co-workers reported the stereospecific coupling of α-chloro ketones with organozinc reagents in the presence of a copper catalyst and stoichiometric magnesium salts.xviii While most of the reported substrates are racemic, the stereochemical course of the reaction was investigated by coupling enantioenriched chloro ketone 12 with isopropylzinc chloride (eq 3). The substitution proceeds in 77% yield to provide 13 with clean inversion of configuration.
 |
(3) |
Liu and co-workers have recently disclosed the copper-catalyzed cross-coupling of primary and secondary Grignard reagents with alkyl tosylates.xix This reaction is facilitated by a three-component catalyst system consisting of CuI, N,N,N’,N’-tetramethylethylenediamine (TMEDA), and LiOMe. Enantioenriched secondary tosylates were found to couple with cyclohexyl and n-hexyl Grignard reagents with excellent transfer of chirality (Scheme 5).
Scheme 5.

Cu(I)-catalyzed stereospecific cross-coupling of alkyl Grignard reagents with secondary tosylates.
2.1.2.2. Zn-catalyzed
In 2008, Briet and co-workers demonstrated that α-hydroxy ester triflates can undergo stereospecific sp3–sp3 cross-coupling reactions with Grignard reagents in the presence of catalytic zinc chloride.xx In this reaction copper salts are less effective catalysts, producing side reactions such as homocoupling or reduction of the triflate. This reaction proceeds well with a variety of α-hydroxy ester triflates and Grignard reagents (Scheme 6). Steric encumbrance is well-tolerated as the coupling proceeds in good yields with isopropylmagnesium chloride and a γ-substituted electrophile to produce 18. Alkenes, esters, and ethers are all well tolerated in the reaction (19, 20 and 21). Generation of a highly nucleophilic zincate complex and Lewis acid activation of the triflate leaving group by magnesium salts are proposed to facilitate this cross-coupling.
Scheme 6.

Stereospecific displacement of α-hydroxy ester triflates with Grignard reagents.
Briet and co-workers adapted this method for the interative synthesis of polyketide-derived (oligo)deoxypropionates.xxi The first cross-coupling reaction with tert-butyl ester triflate 23, prepared from commercially available enantiopure lactic acid, affords the enantioenriched intermediate ester 24. Reduction, conversion to the chloride, and exposure to Mg yields Grignard reagent 25 for the next coupling reaction. Proper choice of triflate enantiomer allows for synthesis of all possible diastereomers (28–31) in excellent diastereoselectivity and enantioselectivity without the use of chiral ligands or auxiliaries.
2.1.2.3. Pd-catalyzed
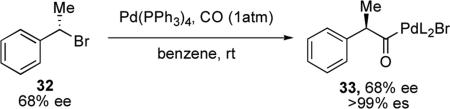
Palladium-catalyzed cross-coupling reactions have become indispensable methods in organic synthesis, particularly in reactions of aryl and vinyl halides. The stereochemical outcome of palladium-catalyzed cross-coupling reactions with sp3 electrophiles was first reported in the 1970s by the Stille lab.xxii Enantioenriched (S)-phenethyl bromide 32, synthesized from the requisite (R)-phenethyl alcohol, was treated with Pd(PPh3)4 under an atmosphere of CO. The resulting palladium complex was indirectly determined to have approximately 68% ee (eq 4). The reaction proceeds with overall inversion of configuration, confirmed by comparison to known optical rotations after derivatization of 33.
 |
(4) |
In 1995, Legros and co-workers reported the palladium-catalyzed stereospecific substitution of benzylic carbonates with malonate nucleophiles.xxiii Optimal yields were achieved using 0.5 mol % Pd(dba)2 and achiral bidentate ligand bis(diphenylphosphino)ethane (dppe), to furnish substitution product 35 in 80% yield and 97% es (Scheme 8). The reaction is proposed to proceed through a double-inversion process in analogy to Tsuji–Trost allylic substitution reactions; first displacement of the leaving group by Pd(0), then nucleophilic attack of subsequent Pd(II) intermediate by sodium malonate. Curiously, when the catalyst loading was increased to 5 mol %, the stereospecificity decreased significantly to 55% es. Racemization is proposed to occur through attack of an exogenous Pd(0) complex on the π-benzyl Pd(II) intermediate 36. This mechanism is consistent with the observation that the rate of racemization is dependent on the catalyst concentration. Indeed, in further studies, researchers found that a chiral ligand controlled stereoselective reaction could also be achieved through increasing the catalyst loading to 2 mol % and employing chiral (R,R)-Me-DuPhos as the ligand.xxiv Under these new conditions, racemic mixtures of naphthylmethanol derivative 38 could be cross-coupled with sodium malonate in up to 64% ee (Scheme 8).
Scheme 8.

Stereospecific and stereoconvergent reactions of benzylic carbonates and sodium malonate.
In 2009, Carretero reported the palladium-catalyzed coupling of aryl Grignard reagents with secondary alkyl halides.xxv The stereochemical course of the reaction was investigated with (S)-phenethyl bromide 32. Exposure of 32 to p-methoxymagnesium bromide in the presence of Pd(CH3CN)2Cl2 and the achiral ligand Xantphos affords the corresponding cross-coupled adduct 39 in 97% yield and >98% es (eq 5).
 |
(5) |
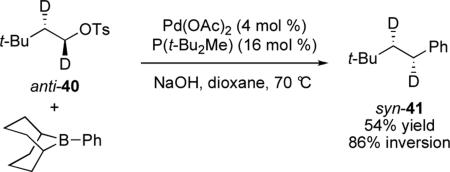
Three examples of stereospecific Suzuki-Miyaura reactions with alkyl electrophiles have been reported. First, Fu and co-workers carried out mechanistic experiments of alkyl–alkyl cross-couplings that indicated that the palladium-catalyzed cross-coupling reaction of stereochemically defined alkyl electrophiles proceeds principally with inversion.xxvi Using a strategy pioneered by Whitesides,xxvii Fu and co-workers used the anti-diasteromer of deuterium-labeled primary alkyl tosylate to elucidate the stereochemical outcome of the reaction. As shown in equation 6, alkyl tosylate 40 was subjected to reaction conditions with phenyl-9-borabicyclo[3.3.1]nonane (phenyl-9-BBN) in the presence of catalytic Pd(OAc)2 to afford 41 in 54% yield and 86% inversion at the carbon bearing the leaving group, corresponding to a syn:anti ratio of 6:1. Since the major product obtained is of the syn configuration, this experiment is consistent with predominate inversion of configuration during the reaction.
 |
(6) |
Asensio and co-workers reported the first stereospecific Suzuki-Miyaura cross-coupling reaction of secondary alkyl electrophiles.xxviii They utilized bromosulfoxides as substrates, which were synthesized by bromination of enantioenriched ethyl-p-tolyl sulfoxide. During optimization of the reaction conditions, t-amyl alcohol was identified as the optimal solvent because it efficiently suppressed competing β-H elimination. Bulky t-amyl alcohol is proposed to fill vacant sites on the palladium complex, thus disfavoring agostic interactions between the palladium and the neighboring C–H bond. The reaction proceeds with inversion of configuration at the stereogenic carbon, confirmed by X-ray analysis of 43 (Scheme 9a). In general, the reaction tolerates a variety of aryl boronic acids, however strong electron-withdrawing groups slow the reaction considerably. Interestingly, no reaction occurs with the anti diastereomer of 44 (Scheme 9b), postulated to be the result of unfavorable steric interactions that prohibit oxidative addition.
Scheme 9.

Suzuki-Miyaura cross-coupling reactions of bromosulfoxides and phenylboronic acid.
In 2010, Falck and He reported the stereospecific Suzuki-Miyaura cross-coupling of alkyl α-cyanohydrin triflates.xxix The electron-withdrawing cyano group helps facilitate an oxidative addition of the alkyl triflate with palladium catalysts. Palladium sources with bulky, electron-rich phosphine ligands, such as P(t-Bu)3, were found to be the best catalysts. The sterically-demanding ligands are proposed to facilitate reductive elimination over β-H elimination. A range of vinyl and aryl boronic acids, including thiophene- and ether-containing substrates, were shown to undergo cross-coupling in 21-94% yield. To illustrate the stereospecifity of the reaction three optically active triflates were subjected to standard reactions conditions to afford 48, 49 and 50 with good translation of stereochemical information (Scheme 10).
Scheme 10.

Stereospecific cross-coupling reactions of α-cyanohydrin triflates with aryl boronic acids.
2.1.2.4. Ni-catalyzed
Nickel catalysts have been key players in the development of catalytic cross-coupling reactions.xxx Early studies with nickel complexes set the stage for many modern cross-coupling reactions, however, their use was rapidly eclipsed by more reliable palladium complexes. Nickel catalysts present several attractive features when compared to palladium complexes, including lower cost, higher activity toward oxidative addition of difficult substrates, and lower tendency toward β-hydride elimination. However, unlike palladium catalysts, nickel catalysts are prone to production of alkyl radicals, which complicates analysis of reaction mechanisms and can lead to racemization of the stereogenic center of the electrophilic carbon.xxxi This proclivity toward radical reactions has enabled the development of stereoconvergent reactions (vide infra).xxxii For stereospecific reactions, however, this type of reactivity is undesirable as stereochemical information can be lost with the formation of an alkyl radical intermediate.
Encouragingly, reactions with allylic alcohols have been shown to be stereospecific in the presence of nickel catalysts,xxxiii indicating that the activation of a C–O bond, rather than a C–X bond, may effectively disfavor the one-electron pathways that lead to racemization. Based on these findings, our laboratory has developed nickel-catalyzed stereospecific cross-coupling reactions of alkyl ethers. We have reported that alkyl ethers can be effectively cross-coupled with methylmagnesium iodide in a stereospecific processxxxiv to construct a new tertiary stereogenic center with high stereochemical fidelity (Scheme 11). Catalysts prepared in situ from Ni(cod)2 and achiral ligands DPEphos, Xantphos or racemic BINAP were found to be optimal for this transformation. Reactions are highly stereospecific, typically proceeding with >95% es. Under our optimized conditions, the reaction tolerates benzofuran- and benzothiophene-containing substrates. Additionally, the reaction is chemoselective for benzylic ethers: aryl ethers do not undergo cross-coupling under the reaction conditions.
Scheme 11.

Stereospecific Ni-catalyzed cross-coupling reactions of alkyl ethers with MeMgI.
The successful transfer of stereochemical information from a secondary alcohol to a tertiary carbon was applied to the synthesis of enantioenriched diarylethanes, a class of compounds with diverse biological activity.xxxv Asymmetric arylation of requisite aldehyde 55xxxvi and subsequent alkylation of the alcohol furnished desired heterocyclic substrate 56 in 90% ee (Scheme 12). The cross-coupling reaction was found to proceed in good yield and excellent stereospecificity to give 57, which was further elaborated over 3 steps to produce 58, an H1 antihistamine shown to have anti-insomnia properties.xxxvii An enantioenriched anticancer agent was also synthesized using our cross-coupling method.xxxviii
Scheme 12.
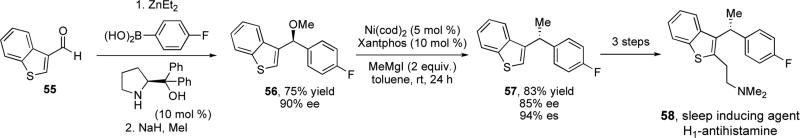
Enantioselective synthesis of sleep inducing agent 58 via stereospecific cross-coupling reaction.
During our studies we found that these reactions were limited to substrates with extended aromatic systems due to their ability to stabilize the putative π-benzylnickel intermediate. To address this limitation, a traceless directing group was used to promote cross-coupling reactions with previously poorly performing reagents.39 The directing group was designed to activate the benzylic C–O bond for oxidative addition through chelation of magnesium salts, thus accelerating the reaction. We have applied this strategy to reactions with non-extended aromatic systems and aryl Grignard reagents to access diarylalkanes (e.g., 59 and 60) and triarylmethanes (e.g., 61). Both of these substructures have demonstrated utility in medicinal chemistry40,41 yet are not easily prepared using existing methods. The optimal phosphine ligand for coupling of benzhydryl ethers with methyl Grignard reagent is DPEphos; aryl Grignard reagents perform best with the diphenylphosphinoalkane class of ligands (e.g., DPPO or DPPH). Representative examples of both reactions are shown in Scheme 13.
Scheme 13.

Directing group assisted stereospecific cross-coupling reactions of benzylic ethers with Grignard reagents.
2.2. Enantioenriched Alkyl Nucleophiles
Stereospecific reactions with configurationally stable, α-chiral alkylmetals have been an exciting area of research for many years. Development of such reactions has been demanding because secondary alkylmetal reagents present an interesting combination of challenges to the synthetic chemist. First, these reagents are traditionally not straightforward to prepare in an enantioenriched form and subsequent purification is often difficult, although the number of reported preparations is continually growing. Second, the configurational stability of the alkylmetal reagent varies depending on the identity of the metal (vide infra). Third, alkylmetal reagents can be plagued by decomposition through proto-demetalation and β-hydride elimination during the cross-coupling reaction, which can complicate cross-coupling reactions of elaborated alkylmetal reagents. Lastly, for alkylmetal reagents with known toxicity such as alkyltin reagents, a balance of toxicity with ease of synthesis and handling of the reagent must be achieved, particularly when multiple equivalents of the alkylmetal reagent are necessary. Advances with a wide variety of organometallic reagents over the last few decades, however, have indicated that these problems are surmountable and that reactions with α-chiral alkylmetals can offer unique access to chiral materials for complex synthesis.
One of the most important factors to consider in reactions of alkylmetal reagents is their configurational stability under reaction conditions. With the exception of organozincs, which display unusually high configurational stability, a general trend can be applied across transmetalating agents that the rate of racemization decreases as the covalent character of the C–M bond increases (Scheme 14).42 For example, secondary organomagnesium reagents racemize at –10 °C with a half life of about 5 h.43 By comparison, enantioenriched secondary alkylboron reagents are reported to be configurationally stable to reaction temperatures above 70 °C for over 16 h.44 The rate of racemization must be taken into consideration when synthesizing the reagent as well as when identifying conditions for cross-coupling reactions.
Scheme 14.

Configurational stability of alkylmetal reagents.
Among the reported cross-coupling reactions of secondary alkylmetal reagents the stereochemical outcome is not homogeneous; reactions have been reported with retention or inversion of configuration depending on several factors. While this variability complicates prediction of stereochemical outcome, on the other hand, it allows for the possibility of development of methods to access either enantiomer of product from a single enantiomer of organometallic reagent, a feature that has only recently been realized for stereospecific cross-coupling reactions of alkyl electrophiles.45 Alkylboron reagents in particular have garnered much interest recently. In addition to being relatively non-toxic and functional group tolerant, they are configurationally stable and can be prepared by a number of asymmetric hydroboration reactions of alkenes.46 The following section is ordered approximately by increasing electronegativity of the metal which thereby increases covalent character of the carbon–metal bond (Mg<Zn<Cu<Si<Sn<B). Synthesis and reactions of chiral alkyllithium reagents will not be covered here explicitly, but we direct the reader to recent reviews on this topic.47
2.2.1. Organomagnesium Reagents
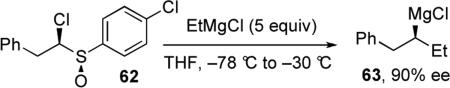
The study of enantioenriched stereogenic Grignard reagents in cross-coupling reactions is challenging because most routes to access these reagents involve radical pathways that result in racemic products. Additionally, Grignard reagents racemize quickly above –10 °C. Interestingly, Hoffmann and co-workers accomplished the first synthesis of enantioenriched Grignard reagents where the sole chiral center is at the carbon bearing the magnesium.43 The reagent was accessed through sulfoxide/magnesium exchange followed by a carbenoid homologation reaction beginning from α-haloalkyl sulfoxide 62 (eq 7). Reagent 63 was obtained in 90% ee and found to be configurationally stable when stored at –78 °C.
 |
(7) |
Exposure of the Grignard reaction mixture to cross-coupling reaction conditions at low temperature with vinyl bromide results in the corresponding cross-coupling adducts in varying optical purity, depending on the catalyst.48 Cross-coupling under palladium and nickel catalysis affords 64 with excellent stereochemical fidelity, while cross-coupling reactions mediated by iron and cobalt complexes exhibit significant erosion in enantiospecificity (Table 2). The loss of stereochemical information in entries 3 and 4 is attributed to single electron transfer processes that occur during the transmetalation step.
Table 2.
Reactions of enantioenriched Grignard reagents with vinyl bromide.

| |||
|---|---|---|---|
| Entry | Catalyst | Yield (%) | es (%) |
| 1 | NiCl2dppf | 60 | 98 |
| 2 | PdCl2dppf | 58 | 98 |
| 3 | Fe(acac)3 | 35 | 59 |
| 4 | Co(acac)2 | 30 | 61 |
2.2.2. Organozinc Reagents
Enantioenriched organozinc reagents have also been generated in situ and found to be viable reagents for the synthesis of chiral materials. While studying the α-arylation of N-Boc pyrrolidines, researchers at Merck utilized a sparteine-mediated asymmetric deprotonation49 to afford the corresponding enantioenriched organolithium intermediate. In contrast to the work of Dieter and co-workers (Section 2.2.3), they found that transmetalation to copper gave low enantiospecificity. They discovered, however that transmetalation to zinc resulted in configurationally stable, enantioenriched alkylzinc reagent 66 capable of undergoing Negishi cross-coupling reactions.50 Notably, this zinc reagent could be coupled at temperatures up to 60 °C without loss of stereochemical integrity. The reaction proceeds smoothly with less than 5 mol % Pd(OAc)2 and air-stable t-Bu3P–HBF4. Results obtained when varying the equivalents of ZnCl2 indicate that the reaction is compatible with all types of Negishi reagents (RZnX, R2Zn, and R3ZnLi), although 0.6 equivalents of ZnCl2 result in optimal yields. The scope was shown to be quite broad with respect to the electrophile, with coupling demonstrated for aryl bromides containing carbonyl, pyridyl, and unprotected indole functionalities (Scheme 15).
Scheme 15.

Cross-coupling reaction of in situ generated alkylzinc reagents.
The same group has applied the Negishi cross-coupling of 66 to the kilogram scale synthesis of 73, a glucokinase activator.51 The zinc reagent was generated in situ and then exposed to aryl bromide 71 under standard reaction conditions. Intermediate 72 was attained in good yield with 92% ee and carried through subsequent manipulations to produce 1.4 kg of bioactive benzimidizole 73.
An analogous reaction was reported by Gawley and Beng for the arylation and vinylation of piperidines.52 A catalytic dynamic resolution53 of N-Boc-2-lithiopiperidine was used to generate the necessary enantioenriched organolithium intermediate, requiring only 5 mol % of chiral ligand 75. Transmetalation to zinc at –78 °C, followed by palladium-catalyzed cross-coupling at ambient temperature furnished the desired arylated products in 60–75% yield with 70–94% ee. The reaction was found to be sufficiently general to tolerate o-substitution, anilines, and ketones. Heteroaryl bromides also gave desired products with heating to 60 °C, although in slightly diminished yields (Scheme 17). Notably, Knochel and co-workers have recently reported the diastereoselective arylation of substituted piperidines, an extension of the excellent cross-coupling methods developed by their laboratory for reactions with diastereomeric alkylzinc reagents.54
Scheme 17.

Synthesis of α-arylated N-Boc piperidines via stereospecific Pd-catalyzed cross-coupling reactions.
2.2.3. Organocopper Reagents
Organocopper reagents bearing a stereogenic center bound to copper have been successfully synthesized in situ by transmetalation from other alkylmetals. Dieter and co-workers disclosed the first stereospecific substitution reactions of enantioenriched organocuprates (Scheme 18).55 They demonstrated that the previously mentioned (–)-sparteine-mediated asymmetric deprotonation49 can be used to form α-chiral organocuprates upon treatment of the enantioenriched N-Boc-2-lithiopyrrolidine with CuCN•2LiCl. The resulting organocopper reagent was found to be configurationally stable below –50 °C; warming the cuprate reagent above –50 °C for 15 minutes affords only racemic products. Examination of substitution reactions with various electrophiles revealed strong dependencies on solvent and substrate. For instance, the reaction of cuprate 80 with vinyl iodide 81 in THF results in racemic products, while the same reaction in mixed THF/Et2O affords the desired product in 80–86% ee. These results are consistent with the configurational stability of the cuprate displaying a solvent dependence. Substitution reactions with vinyl halides, tosylates, and nonafluorobutanesulfonates (nonaflates) were realized in good yields and with similar levels of enantioselectivity. Propargylic halides could also be coupled to give allenyl-substituted pyrrolidines such as 85, although the enantiospecificity is significantly diminished for these substrates. Derivatization of the product from reaction with electrophile 81 and comparison to a known proline derivative is consistent with retention of configuration during cross-coupling. Dieter and co-workers have applied this methodology to the synthesis of various alkaloids,56 including the indolizidine alkaloid elaeokanine A.57
Scheme 18.

Alkyl cuprates generated via asymmetric deprotonation and reactions with representative electrophiles.
2.2.4. Organosilicon Reagents
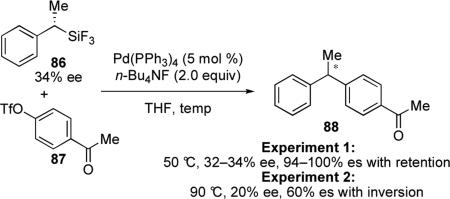
In 1990, Hiyama reported the interesting stereochemical outcome of the reaction of (S)-1-phenyl-1-(trifluorosilyl)ethane (86) with an aryl triflate.58 At temperatures below 75 °C in THF, the reaction proceeds with retention at the stereogenic center; however, upon heating, the stereochemical outcome of the reaction changes to give the product that results from inversion of configuration (eq 8). A nearly linear relationship was found between ee and reaction temperature. At moderate temperatures, solvent can also affect the stereochemical outcome of this reaction with THF/DMSO or THF/DMF yielding retention of configuration, while the addition of HMPA in THF results in overall inversion.
 |
(8) |
The stereochemical outcome is thought to be the result of changes in the transition state that allow for different geometries during the transmetalation step (Scheme 19). Retention of configuration (via complex 89) can originate from a cyclic four-centered transition state, SE2 (cyclic), where the fluoride coordinates to both the silyl group and the organopalladium species. At higher temperatures or in more polar solvents, disruption of the Si–F bond results in an open transition state, SE2 (open), by which backside attack of the palladium gives inversion of configuration, leading to complex 90. This is an interesting example where simple changes in temperature and solvent control the configuration of the resulting cross-coupling adduct. This phenomenon has not yet been documented in other enantioenriched organometallic reagents.
Scheme 19.

Transition state geometries that would result in either retention or inversion of configuration during transmetalation of chiral alkyl silanes.
2.2.5. Organotin Reagents
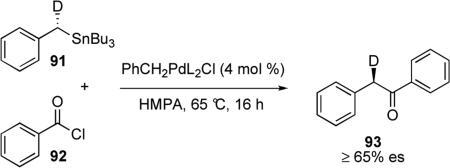
In the early 1980s Stille and Labadie reported the palladium-catalyzed cross-coupling reactions of acid chlorides with enantioenriched organostannanes.59 The reaction of (S)-(α-deuteriobenzyl)tributyltin 91 with benzoyl chloride furnished the corresponding phenyl ketone 93 (eq 9). Subsequent derivatization and comparison of optical rotation to literature values indicated that 93 had R configuration and thus the cross-coupling must have proceeded with inversion of configuration. Since their lab had previously demonstrated that reductive elimination occurs with retention,60 it was proposed that the transmetalation step in the catalytic cycle occurs with inversion.
 |
(9) |
In 1990, Liebeskind and co-workers reported that the addition of copper (I) salts to palladium-catalyzed Stille reactions greatly enhances reactivity.61 Utilizing this strategy, Falck and co-workers showed that enantioenriched secondary α-alkoxyalkylstannanes could be cross-coupled with various electrophiles using Pd/Cu co-catalysts to give enantioenriched α-hetero ketones. In their initial report, they investigated the stereochemical course of this reaction using enantioenriched stannane 94.62 Surprisingly, in contrast to the previous report by Stille, retention of configuration was observed for this reaction (eq 10). It was noted that participation of the neighboring Lewis basic functional group could be responsible for this difference.
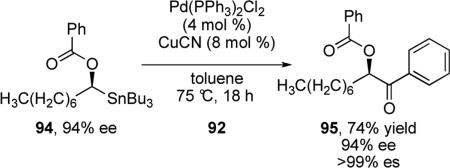 |
(10) |
Since Falck's seminal report, cross-coupling reactions of a variety of α-alkoxylalkylstannanes have also been found to proceed with retention of configuration at the stereogenic center. The scope of these reactions has been expanded to replace acyl chlorides with allylic and propargylic halides,63 and recently, thioesters (Scheme 20a,b).64 Pyrrolidinylthionocarbamoyl (PTC)-protected α-alkoxyalkylstannanes (e.g. 96 and 99) were found to exhibit increased reactivity towards cross-coupling with a broader range of electrophiles, postulated to be the result of special stabilization of the putative Cu intermediate via coordination shown in complex 98. Reaction conditions for cross-coupling with aryl iodides have been reported using catalytic palladium and simple α-(acetyloxy)stannane 102 (Scheme 20c).65 Stoichiometric copper(I) thiophene-2-carboxylate (CuTC) also effectively promotes cross-coupling of a wide range of vinyl and aryl iodides with (PTC)-protected α-alkoxyalkylstannanes, although under these reaction conditions, additional Newman-Kwart O-to-S rearrangements were found to accompany the desired cross-coupling reaction.66
Scheme 20.

Reactions of α-alkoxylalkylstannanes catalyzed by copper and palladium.
Diastereoselective cross-couplings have been reported for stannyl pyranosides67 and epoxides.68 In 2008, Hoppe reported the Cu(I)-catalyzed cross-coupling of α-stannylated benzyl carbamates (e.g. 104) with acid chlorides (eq 11) and allyl bromides.69
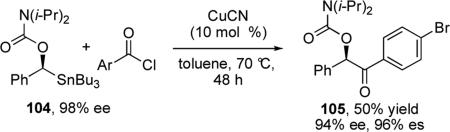 |
(11) |
The utility of stereospecific Stille cross-coupling to assemble hetereoatom-containing stereogenic centers has been showcased in the synthesis of biologically relevant molecules (Scheme 21). For example, this strategy provided a key step in the synthesis of the anticancer agent (+)-goniofufurone (106).70 Stereospecific Stille cross-coupling with enantioenriched α-alkoxylalkylstannane 107 in the presence of palladium and copper co-catalysts was used to set a key stereogenic center in the target structure. Endothelium-derived vasodilator 11,12,15-THETA (108) was also synthesized via cross-coupling of an enantioenriched alkylstannane.71
Scheme 21.

Application of stereospecific Stille cross-coupling for the synthesis of (+)-goniofufurone and 11,12,15-THETA.
Kells and Chong reported that α-(sulfonamido)benzylstannanes react with benzoyl chloride under very similar conditions to those reported by Falck and co-workers, with the exception that electron-rich tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP) was found to be superior to Ph3P for this transformation (eq 12).72 Interestingly, the reaction proceeds with selective inversion at the stereogenic center, consistent with Stille's initial report, but in contrast to previous reports by Falck and Liebeskind with α-alkoxystannanes.
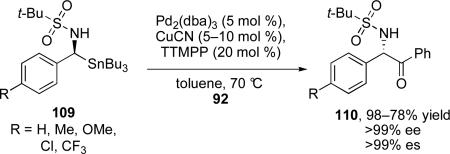 |
(12) |
An overarching mechanistic rational for the observed stereochemical outcome of cross-coupling of tin reagents has not yet been disclosed. However, the data in Scheme 20 and equations 9–12 are consistent with a correlation between the Lewis basicity of the neighboring functional group and the stereochemical outcome: strong donors result in retention of configuration at the stereogenic center and weak donors provide inversion. Interestingly, this trend appears to be the opposite of that observed with alkylboron reagents (vide infra); continued mechanistic investigation could clarify the differences between these reagents.
2.2.6. Organoboron Reagents
The stereochemical outcome of cross-coupling reactions of α-chiral alkylboron reagents was first elucidated using deuterium-labeled reagents. The deuterium label was employed to overcome the difficulty of coupling secondary alkylboron regents, which was not well-established at the time. Incorporation of deuterium at the α and β position of primary alkylboranes allows for determination of configuration by analysis of 1H NMR coupling constants, while introducing little thermodynamic preference for formation of one diastereomer over the other.xxvii The Soderquist and Woerpel groups independently showed that alkylboron reagents undergo palladium-catalyzed cross-coupling reactions with aryl and vinyl halides with retention of configuration (Scheme 22a,b).73 In a recent report by our laboratory we have demonstrated that the same holds true for nickel-catalyzed cross-coupling reactions with alkyl halides (Scheme 22c).74
Scheme 22.

Reactions of deuterium labeled primary alkylboranes.
In the last five years cross-coupling reactions of enantioenriched secondary alkylboron reagents have emerged. These reactions possess several attractive features. For example, enantioenriched boron reagents may be prepared directly from styrenes by catalytic asymmetric hydroboration.46 Alkylboron reagents are not prone to racemization, even at elevated temperatures that may be required for certain cross-coupling reactions. Furthermore, in comparison to alkylstannanes, the byproducts are relatively non-toxic. Therefore, their participation in stereospecific cross-coupling reactions would be a powerful strategy for asymmetric synthesis of C–C bonds. However, achieving this goal requires suppression of β-hydride elimination, which is especially important in these reactions due to isomerization of secondary organoboron intermediates through reinsertion into the resultant alkene. Early work towards this end involved the diastereoselective cross-coupling reactions of cyclopropylboranes.75 These substrates do not undergo β-hydride elimination. Cyclopentylborane was also successfully applied to cross-coupling reactions shortly thereafter.76 Cyclopentyl reagents avoid rearrangement to primary linear reagents, another common side reaction of secondary alkylboranes.
In 2009, Crudden and co-workers reported the first successful example of acyclic secondary enantioenriched alkylboron reagents undergoing stereospecific Suzuki-Miyaura cross-coupling reactions.44 Rhodium-catalyzed hydroboration was used to generate the requisite alkylboronate esters in high optical purity from styrene.77 These reagents were found to be configurationally stable, transferring the chiral alkyl component with excellent stereospecificity even at elevated temperatures and with minor decomposition to elimination or rearranged products. Silver oxide was found to be the optimal base and critical in promoting reactions of secondary alkylboranes. This improvement is postulated to be due to its ability to promote sluggish transmetalation. Other typical bases such as K3PO4 and Cs2CO3 failed to produce any cross-coupled product. A variety of aryl iodides were subjected to reaction conditions (Scheme 23). The reactions proceeded with retention of configuration with moderate to good enantiospecificity.78 Interestingly, primary alkylboranes were unreactive under the optimized reaction conditions.
Scheme 23.

Stereospecific cross-coupling of enantioenriched alkylboronate esters.
Similar to reactions with organostannanes and organosilicon reagents, the stereochemical course of cross-coupling reactions of alkylboron reagents can be influenced by neighboring Lewis basic functional groups. In 2010, the first stereospecific Suzuki-Miyaura coupling with inversion of configuration was reported by Ohmura and Suginome.79 The cross-coupling of enantioenriched α-(acylamino)benzylboronic esters was found to proceed with high enantiospecificity when a sterically demanding pivolyl group was employed. The reaction proceeds smoothly at 80 °C in toluene with a variety of aryl bromides and aryl chlorides to furnish diarylmethanamines with high enantiopurity (Scheme 24).
Scheme 24.

Reactions of aryl bromides and benzylic boronate esters with overall inversion of configuration.
Participation of the neighboring carbonyl group is proposed to be critical in achieving inversion of configuration during the transmetalation event. Coordination of the pendant carbonyl to the empty p-orbital of the boron blocks approach of the palladium catalyst and results in an open SE2 transition state (Figure 1). This mechanism is in close analogy to the mechanism proposed for transmetalation of organosilanes in polar solvents (c.f Scheme 19).
Figure 1.

Proposed transition state that results in inversion during the reaction of α-aminoboronate esters.
In further studies, Suginome has demonstrated that the stereochemical outcome can be controlled with Lewis acid additives.80 A variety of Lewis acids were evaluated and it was found that PhOH was the best acid for achieving high levels of inversion and Zr(Oi-Pr)4•i-PrOH was optimal for providing the arylated product with retention of configuration (Scheme 25). The acid additive is believed to change the intramolecular coordination mode at boron. Activation of the pinacol ester by the Brønsted acid, phenol, removes electron density from boron, strengthening coordination to the neighboring carbonyl (130) and favoring inversion. Alternatively, coordination of the Lewis acid, Zr(Oi-Pr)4, to the carbonyl disrupts intramolecular coordination of the carbonyl to boron (131). This favors a traditional four-centered transition state for transmetalation that results in retention at the stereogenic center. These reaction conditions allow for access to either enantiomer of product from a single enantiomer of alkylboron reagent.
Scheme 25.

Role of Lewis acid coordination in cross-coupling adduct geometry.
Molander and co-workers have recently shown that non-benzylic secondary alkyltrifluoroborate salts can be effectively coupled with aryl chlorides and bromides (eq 13).81 The optimal reaction conditions were found to be a combination of Pd(OAc)2 with Xphos in a mixed solvent system of cyclopentyl methyl ether (CPME) and water. This reaction proceeds with inversion at the stereogenic center. The authors invoke coordination of the carbonyl to the empty p-orbital on boron, similar to the Suginome model shown in Figure 1. Molander has also suggested that restricted rotation of the organopalladium intermediate as a result of this coordination enhances desired reactivity by disfavoring β–H elimination. Interestingly, only β-amides such as 132 were found to be effective; ketones and esters do not produce cross-coupled products under similar conditions.
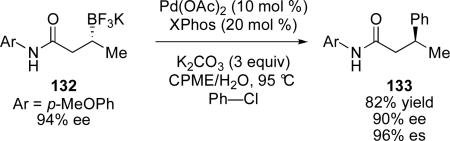 |
(13) |
A unique approach to stereospecific Suzuki-Miyaura cross-coupling has been recently reported by Hall and co-workers in which 1,1-diboron compounds bearing two different boronyl units undergo chemoselective cross-coupling reactions.82 The geminal diboron reagent 134 was prepared in high optical purity from a copper-catalyzed conjugate addition of bis(pinacolato)diboron to the requisite α,β-unsaturated ester. Chiral ligand Walphos-1 was found to give the highest level of enantioselectivity for this transformation. Attempts toward direct cross-coupling of this boronic ester were unsuccessful; however, subsequent conversion to the trifluoroborate salt resulted in a more active coupling reagent. The reaction proceeds exclusively with the trifluoroborate salt in the presence of the 1,8-diaminonaphthalenyl boronyl moiety. A variety of aryl bromides were found to be effective electrophilic coupling partners for this reaction to furnish the desired product in up to 99% ee and 92% yield (Scheme 26). Vinyl bromides also undergo cross-coupling, albeit in lower yields. Comparison to known optical rotations indicates that the reaction proceeds with overall inversion at the stereogenic center, consistent with previous reports by Molander and Suginome (vide supra). In addition to providing the stereogenic center and chemoselective basis for the coupling reaction, the second boron is proposed to promote transmetalation during the catalytic cycle by stabilizing the Pd(II) intermediate.
Scheme 26.

Chemoselective reactions of 1,1-diboron compounds with aryl and vinyl bromides.
To establish the reactivity of the cross-coupling adducts, studies were undertaken to cross-couple the second boronate. Conversion to the trifluoroborate salt and alteration of the methyl ester to an amide was necessary to allow smooth conversion to the final diarylethane 140. The second cross-coupling also proceeds with inversion.
3. Catalyst-controlled Stereoselective Reactions
In a catalyst-controlled stereoselective cross-coupling process chiral ligands on the metal control the stereochemical outcome of the reaction. One advantage of this method is that only substoichiometric amounts of chiral compounds are necessary. Beginning with racemic substrates, the success of the reaction is measured by the ee of the product obtained. Stereoconvergent reactions are the most common method. These reactions convert both stereoisomers of racemic starting material into a single stereoisomer of product. Stereoconvergent reactions encompass dynamic kinetic resolutions83 or reactions that proceed though a common achiral intermediate (Scheme 28a). Dynamic kinetic resolutions can occur if the rate of isomerization between the enantiomers of starting material is faster than their respective rates of reaction with the chiral catalyst. Catalyst preference for reaction with one enantiomer over the other yields enantioenriched products. Enantioselective cross-coupling reactions that are consistent with a traditional kinetic resolution are less common (Scheme 28b).
Scheme 28.

Catalyst-controlled stereoselective reactions. ML* = metal complex with chiral ligand.
3.1. Racemic Alkyl Electrophiles
Since 2004, Fu and co-workers have pioneered the field of stereoconvergent cross-couplings of secondary alkyl electrophiles. The cross-coupling reactions proceed using one of three general nitrogen ligand types (Figure 2). The chelating ability of these ligands is presumed to help occupy vacant sites on the alkylnickel intermediate, effectively suppressing β-H elimination. Other types of ligands such as phosphines and N-heterocyclic carbenes fail to produce the desired reactivity. Notably, most couplings require 10% or less catalyst and proceed at room temperature with excellent enantioselectivities.
Figure 2.

Typical ligand types for stereoselective cross-coupling reactions.
Reactions of a wide array of substrates have been developed; general examples are summarized in Tables 3 and 4. Table 3 catalogs reactions of α-halo carbonyl compounds. A range of transmetalating agents have been disclosed including aryl, vinyl, and primary alkyl reagents. Table 4 features electrophiles without special activation through a neighboring carbonyl group. Notably, each alkyl halide contains a functional group that serves as a directing group that allows the catalyst to discriminate between the faces of the putative alkyl radical and form a single diastereomer of the intermediate organonickel complex (vide infra). The variety of directing groups that can be used to induce enantioselectivity has grown rapidly to encompass an impressive array of functional groups, increasing potential for broad applications in synthesis. These reactions also encompass several sources of aryl, vinyl, and primary alkyl transmetalating agents as well as the first example of a secondary alkyl reagent, cyclopropyl-9-BBN (Table 4, entry 6), with a secondary electrophile in a stereoconvergent cross-coupling reaction.
Table 3.
Stereoselective cross-coupling of α-halo carbonyl compounds.
| Entry | Electrophilea | Nucleophile | Ligand typeb | Conditions | ee Range (%) | Yield Range (%) | Reference |
|---|---|---|---|---|---|---|---|
| 1 |

|
Ar–ZnI | A | 5% NiCl2•glyme glyme/THF, –30 °C | 72–96 | 71–93 | lxxxiv |
| 2 |
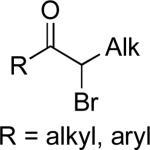
|
Ar–MgX X = Br, Cl |
B | 7% NiCl2•glyme DME, –40 or –60 °C | 72–95 | 70–91 | lxxxv |
| 3 |
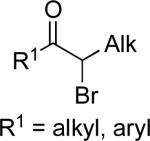
|
|
B | 3% NiCl2•glyme DME/THF, 10 °C | 80–98 | 74–95 | lxxxvi |
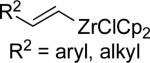
| 4 |
|
°1 Alk –ZnBr | A | 10% NiCl2•glyme DMI/THF, 0 °C | 77–96 | 51–90 | lxxxvii |
| 5 |
|
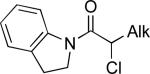
Ar–(9-BBN) | C | 8% NiBr2•glyme, 1.3 equiv KOt-Bu, 1.5 equiv i-BuOH | 84–94 | 70–88 | lxxxviii |
| 6 |

|
R–Si(OMe)3 R = aryl, vinyl |
C | 10% NiCl2•glyme, 2.0 equiv TBAT, dioxane, rt | 75–99 | 64–84 | lxxxix |
Alk = alkyl.
See Figure 2.
Table 4.
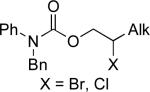
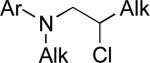
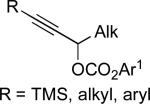
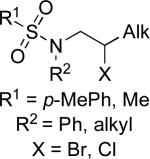
Stereoselective cross-coupling reactions of non-activated electrophiles.
| Entry | Electrophilea | Nucleophile | Ligand typeb | Conditions | ee Range (%) | Yield Range (%) | Reference |
|---|---|---|---|---|---|---|---|
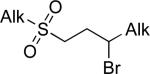
| 1 |
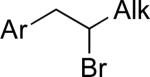
|
°1 Alk –(9-BBN) | C | 10% Ni(cod)2 KOt-Bu, i-BuOH, i-Pr2O, rt or 5 °C | 40–94 | 64–86 | xc |
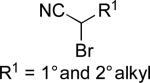
| 2 |
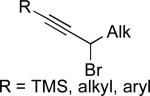
|
Ar–ZnEt | A | 3% NiCl2•glyme, glyme, –20 °C | 84–94 | 39–92 | xci |
| 3 |
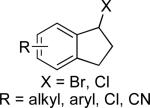
|
°1 Alk –ZnBr | A | 10% NiBr2•glyme, DMA, 0 °C | 91–99 | 39–89 | xcii |
| 4 |
|
°1 Alk –(9-BBN) | C | 10% NiBr2•diglyme, KOt-Bu, hexanol, i-Pr2O, rt | 90–98 | 54–82 | xciii |
| 5 |
|
°1 Alk –(9-BBN) | C | 10% NiBr2•diglyme, KOt-Bu, hexanol, i-Pr2O, rt | 71–96 | 52–86 | xciv |
| 6 |

|
R–(9-BBN) R = 1°alkyl, cyclopropyl |
C | 10% NiBr2•diglyme, KOt-Bu, hexanol, Et2O/Hexane (1:1), rt | 69–91 | 51–83 | xcv |
| 7 |
|
Ar–ZnI | A | 10% NiCl2(PCy3)2 DME/THF (1:1) 10 °C | 84–93 | 57–94 | xcvi |
| 8 |

|
°1 Alk–(9-BBN) | C | 10% NiBr2•diglyme, KOt-Bu, hexanol, i-Pr2O, rt | 80–91 | 56–83 | xcvii |
| 9 |
|
R3–(9-BBN) R3 = °1 alkyl, Ph |
C | 10% NiBr2•diglyme, KOt-Bu, hexanol, i-Pr2O, rt | 72–95 | 54–88 | xcvii |
| 10 |
|
°1 Alk–(9-BBN) | C | 10% NiBr2•diglyme, KOt-Bu, hexanol, i-Pr2O, rt | 87–90 | 79–84 | xcvii |
| 11 |
|
(R2)2Zn R2 = aryl, vinyl |
B | 10% NiBr2•diglyme 20% TMEDA THF, –78 °C | 76–94 | 59–99 | xcviii |
Alk = alkyl.
See Figure 2.
Work is ongoing to elucidate the mechanism of this transformation. The reaction is proposed to proceed through a two-step oxidative addition process as shown in the general catalytic cycle in Scheme 29. The first oxidative addition step generates an achiral radical intermediate from the racemic alkyl halide. Selective formation of one diastereomer of the alkylnickel complex sets the configuration at the new stereogenic center (oxidative addition step 2). Retention of configuration during reductive elimination affords the enantioenriched C–C bond formation. Radical intermediates in nickel-catalyzed cross-coupling reactions are precedented by mechanistic studies performed by Stille,xxxi Kochi,xcix and Vicic.c
Scheme 29.

General catalytic cycle for Ni-catalyzed stereoselective cross-coupling reactions of secondary alkyl halides.
A few important reactions have been utilized to probe this mechanism. For instance, when subjecting either enantiomer of electrophile 141 to standard reaction conditions, no loss of enantiopurity in the recovered starting material is observed (Scheme 30). This result indicates that the interconversion of starting material enantiomers is not facile and thus rules out a dynamic kinetic resolution-type mechanism. Through kinetic experiments it has been determined that the reaction is first order with respect to the nucleophile and catalyst, and zero order with respect to the electrophile, consistent with transmetalation being the rate determining step.ci
Scheme 30.

Recovery of enantiopure substrates after partial conversion.
Fu and co-workers have demonstrated that oxygen, carbon, or nitrogen functionalities can be used to direct the facial selectivity on the substrate. In the reaction shown in Scheme 31a, the aryl ring effectively directs the catalyst to differentiate between benzyl and homobenzyl moieties on the electrophile to afford the product in 90% ee. Omitting or moving the directing group results in significantly diminished enantioselectivity (Scheme 31b). Remarkably, in some recent examples, the directing group can be up to four carbons away from the reaction center and chiral recognition can still occur, for example in reactions with δ-halocarbonyl compounds (Scheme 31c).
Scheme 31.

Select reactions of alkyl halides.
Caeiro, Pérez Sestelo, and Sarandeses have reported an additional example of an enantioselective nickel-catalyzed cross-coupling. Using a ligand of type A (Figure 2), they have reported the cross-coupling reaction of trialkynylindium reagents with secondary racemic benzyl bromides.cii This reaction proceeds in good yields and enantioselectivites for benzyl bromide and 1-bromoindane with a range of sp-hybridized indium reagents, affording cross-coupling products in 35–70% yield and 77–87% ee (eq 14).
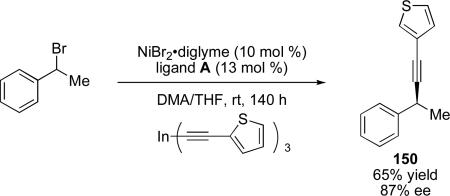 |
(14) |
3.2 Racemic Alkyl Nucleophiles
In contrast to stereospecific cross-coupling with Grignard reagents as reported by Hoffman (Table 2), secondary alkyl Grignard reagents can also be coupled in a catalyst-controlled stereoconvergent manner. At reaction temperatures above –10 °C, Grignard reagents racemize, opening the possibility for a dynamic kinetic resolution. A challenging but interesting goal has been the identification of a chiral catalyst that can select for coupling of one enantiomer of the Grignard reagent.
The first examples of catalyst-controlled enantioselective cross-coupling reactions of secondary alkyl Grignard reagents were reported in the early 1970s. Independently, the groups of Consiglio and Botteghiciii and Kumadaciv reported nickel-catalyzed cross-coupling of secondary Grignard reagents with vinyl and aryl halides that employed (–)-diop as a ligand (eq 15 and 16). Although the stereoselectivity for these reactions was low (up to 17% ee), these results provided important proof of concept that resolution of the Grignard can be achieved.
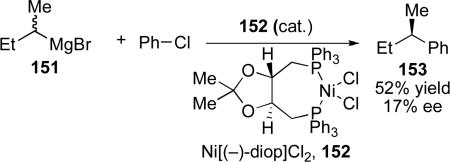 |
(15) |
 |
(16) |
The basis of stereoselectivity in these reactions is hypothesized to be the relatively fast racemization of the Grignard reagent, seen in Figure 3 with arbitrary rate krac. If the racemization occurs faster than cross-coupling, i.e. krac > kR and kS, and the chiral catalyst can effectively select one enantiomer of the organomagnesium reagent, i.e. kR ≠ kS, then high conversion to the corresponding enantioenriched product can be attained.
Figure 3.
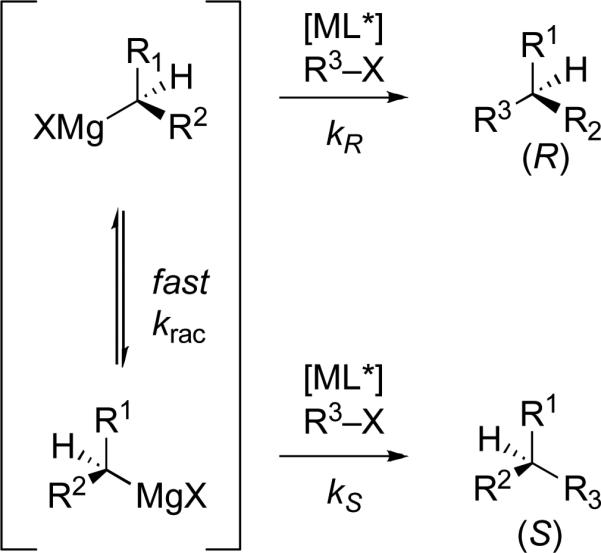
Relative rates of isomerization versus cross-coupling to yield enantioenriched products.
Since these early reports, several chiral ligands have been applied to this transformation with varying success. A representative summary of effective catalysts is shown in Scheme 32. Vinyl bromide and (E)-bromostyrenecv are the most well-studied electrophiles. Structural alterations to the ligand have provided insight into the geometric requirements of the enantioselective transformation. Hayashi and co-workers noted that the removal of the carbon-centered stereogenic center α to the amine in ligand 156, to produce analog 157, did not impede the effectiveness of the ligand in directing the reaction.cvi This implies that the planar chirality of these ligands is the most important feature for enantiodiscrimiation. Removal of the amino group results in significantly diminished stereoselectivity. Alkyl amino phosphine ligands 159 and 160 have also been shown to serve as competent ligands for this transformation yielding enantioselectivities up to 85%.cvii,cviii Additionally, Kellogg and co-workers demonstrated that alkyl sulfide amino ligands (e.g. 161) are effective in nickel-catalyzed systems.cix As evidenced by the narrow substrate scope, this area of investigation remains a challenge.
Scheme 32.

Catalysts for the stereoselective reaction of phenethyl Grignard with vinyl bromide.
4. Conclusions
The asymmetric cross-coupling of configurationally stable, enantioenriched electrophiles or nucleophiles has presented many novel synthetic advantages for the construction of complex molecules. These reactions have been applied to the synthesis of a wide variety of biologically relevant molecules. Additionally, catalytic enantioselective reactions of racemic substrates have expanded the boundaries of known asymmetric induction beyond our usual toolbox.
This growing field will continue to stimulate mechanistic and synthetic experiments for organic synthesis. In the coming years, applications of known cross-coupling strategies and evaluation of reaction conditions are necessary to expand the substrate scope broadly. Additionally, further mechanistic probes are needed to elucidate the elemental steps of cross-coupling reactions with alkyl partners, as in many cases it is apparent that these are quite different from the far more familiar steps in aryl and vinyl cross-couplings. Ultimately, the utilization and careful manipulation of alkyl electrophilic coupling partners and alkyl organometallic reagents will continue to expand the limits of efficiency and specificity in modern organic synthesis.
Scheme 2.

Stereospecific reactions of alkyl tosylates with methyl cuprate.
Scheme 7.

Iterative synthesis of (oligo)deoxypropionates.
Scheme 16.

Application of the stereospecific Negishi cross-coupling reaction to synthesize bioactive compound 73.
Scheme 27.

Transformation of chemoselective cross-coupling product to give diarymethane 140.
Acknowledgments
This work was supported by NIH NIGMS (R01GM100212).
Biography
 Elizabeth R. Jarvo was born in Halifax, NS (Canada) in 1975. She obtained her B.Sc. Honours) from Acadia University working in the laboratory of Michael A. Kerr and was a summer NSERC student at Concordia University with Youla Tsantrizos. She carried out her PhD studies under the direction of Scott J. Miller at Boston College, developing new peptide-based catalysts for kinetic resolution of secondary and tertiary alcohols. In 2002 she began postdoctoral studies with Eric N. Jacobsen at Harvard University, and developed enantioselective quinone Diels-Alder reactions. In 2005 she began her independent career at the University of the California, Irvine, where her research program focuses on the development of new catalytic reactions including nickel-catalyzed stereospecific cross-coupling reactions.
Elizabeth R. Jarvo was born in Halifax, NS (Canada) in 1975. She obtained her B.Sc. Honours) from Acadia University working in the laboratory of Michael A. Kerr and was a summer NSERC student at Concordia University with Youla Tsantrizos. She carried out her PhD studies under the direction of Scott J. Miller at Boston College, developing new peptide-based catalysts for kinetic resolution of secondary and tertiary alcohols. In 2002 she began postdoctoral studies with Eric N. Jacobsen at Harvard University, and developed enantioselective quinone Diels-Alder reactions. In 2005 she began her independent career at the University of the California, Irvine, where her research program focuses on the development of new catalytic reactions including nickel-catalyzed stereospecific cross-coupling reactions.
 Elizabeth Swift was born in San Diego, CA. She obtained her undergraduate degree from UC Santa Barbara in 2008. While there she worked in the lab of Professor Bruce Lipshutz studying heterogeneous catalysis. She is now a fifth year graduate student in the lab of Professor Elizabeth Jarvo. Her doctoral research is focused on developing stereospecific cross-coupling reactions of alkyl electrophiles.
Elizabeth Swift was born in San Diego, CA. She obtained her undergraduate degree from UC Santa Barbara in 2008. While there she worked in the lab of Professor Bruce Lipshutz studying heterogeneous catalysis. She is now a fifth year graduate student in the lab of Professor Elizabeth Jarvo. Her doctoral research is focused on developing stereospecific cross-coupling reactions of alkyl electrophiles.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- i.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. Springer; Berlin: 1999. pp. 1–3. [Google Scholar]
- ii.de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd Ed. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- iii.For alkyl halides see: Rudolph A, Lautens M. Angew. Chem., Int. Ed. 2009;48:2656–2670. doi: 10.1002/anie.200803611.Frisch AC, Beller M. Angew. Chem., Int. Ed. 2005;44:674–688. doi: 10.1002/anie.200461432.
- iv.For alkyl metals see: Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p.
- v.a Denmark SE, Vogler T. Chem.—Eur. J. 2009;15:11737–11745. doi: 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]; b Denmark SE, Burk MT, Hoover AJ. J. Am. Chem. Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]
- vi.a Trost BM, Van Vranken DL. Chem. Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; B Trost BM, Crawley ML. Chem. Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- vii.a Mc Cartney D, Guiry PJ. Chem. Soc. Rev. 2011;40:5122–5150. doi: 10.1039/c1cs15101k. [DOI] [PubMed] [Google Scholar]; b Dounay AB, Overman LE. Chem. Rev. 2003;103:2945–2964. doi: 10.1021/cr020039h. [DOI] [PubMed] [Google Scholar]
- viii.Bringmann G, Price Mortimer AJ, Keller PA, Gresser MJ, Garner J, Breuning M. Angew. Chem., Int. Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- ix.For Suzuki reactions see: Willis MC, Powell LHW, Claverie CK, Watson SJ. Angew. Chem., Int. Ed. 2004;43:1249–1251. doi: 10.1002/anie.200352648.Cho SY, Shibasaki M. Tetrahedron: Asymmetry. 1998;9:3751–3754. For C–H activation see: Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273. and references therein. For cross-coupling of anhydrides see: Johnson JB, Rovis T. Acc. Chem. Res. 2008;41:327–338. doi: 10.1021/ar700176t. and references therein.
- x.For additional asymmetric cross-coupling reviews see: Taylor BLH, Jarvo ER. Synlett. 2011;19:2761–2765.Glorius F. Angew. Chem., Int. Ed. 2008;47:8347–8349. doi: 10.1002/anie.200803509. c See also section 8 within reference iv.
- xi.a Dongol KG, Koh H, Sau M, Chai CLL. Adv. Synth. Catal. 2007;349:1015–1018. [Google Scholar]; b Martin R, Fürstner A. Angew. Chem., Int. Ed. 2004;43:3955–3957. doi: 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]
- xii.Whitesides GM, Fischer WF, Jr., San Filippo J, Jr., Bashe BW, House HO. J. Am. Chem. Soc. 1969;91:4871–4882. See also Corey EJ, Posner GH. J. Am. Chem. Soc. 1967;89:3911–3912.
- xiii.a Lipshutz BH, Wilhelm RS. J. Am. Chem. Soc. 1982;104:4696–4698. [Google Scholar]; b Lipshutz BH, Wilhelm RS, Kozlowski JA. Tetrahedron. 1984;24:5005–5038. [Google Scholar]; c Lipshutz BH, Sengupta S. Org. React. 1992;41:135–631. [Google Scholar]
- xiv.Johnson CR, Dutra GA. J. Am. Chem. Soc. 1973;95:7783–7788. [Google Scholar]
- xv.Hanessian S, Thavonekham B, DeHoff B. J. Org. Chem. 1989;54:5831–5833. [Google Scholar]
- xvi.Heindl C, Hubner H, Gmeiner P. Tetrahedron: Asymmetry. 2003;14:3153–3172. [Google Scholar]
- xvii.Yoshikai N, Nakamura E. Chem. Rev. 2012;112:2339–2372. doi: 10.1021/cr200241f. and references therein.
- xviii.Malosh CF, Ready JM. J. Am. Chem. Soc. 2004;126:10240–10241. doi: 10.1021/ja0467768. [DOI] [PubMed] [Google Scholar]
- xix.Yang C-T, Zhang Z-Q, Liang J, Liu J-H, Lu X-Y, Chen H-H, Liu L. J. Am. Chem. Soc. 2012;134:11124–11127. doi: 10.1021/ja304848n. [DOI] [PubMed] [Google Scholar]
- xx.Studte C, Breit B. Angew. Chem., Int. Ed. 2008;47:5451–5455. doi: 10.1002/anie.200800733. [DOI] [PubMed] [Google Scholar]
- xxi.Brand GJ, Studte C, Breit B. Org. Lett. 2009;11:4668–4670. doi: 10.1021/ol901944b. [DOI] [PubMed] [Google Scholar]
- xxii.Lau KSY, Fries RW, Stille JK. J. Am. Chem. Soc. 1974;96:4983–4986. [Google Scholar]
- xxiii.a Legros J-Y, Toffano M, Fiaud J-C. Tetrahedron. 1995;51:3235–3246. [Google Scholar]; b Legros J-Y, Boutros A, Fiaud J-C, Toffano M. J. Mol. Catal. A: Chem. 2003;196:21–25. [Google Scholar]
- xxiv.Assie M, Meddour A, Fiaud J-C, Legros J-Y. Tetrahedron: Asymmetry. 2010;21:1701–1708. [Google Scholar]
- xxv.Lopez-Perez A, Adrio J, Carretero JC. Org. Lett. 2009;11:5514–5517. doi: 10.1021/ol902335c. [DOI] [PubMed] [Google Scholar]
- xxvi.Netherton MR, Fu GC. Angew. Chem., Int. Ed. 2002;41:3910–3912. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- xxvii.Bock PL, Boschetto DM, Rasmussen JR, Demers JP, Whitesides GM. J. Am. Chem. Soc. 1974;96:2814–2825. [Google Scholar]
- xxviii.Rodriquez N, de Arellano CR, Asensio G, Medio-Simon M. Chem.—Eur. J. 2007;13:4223–4229. doi: 10.1002/chem.200601488. [DOI] [PubMed] [Google Scholar]
- xxix.He A, Falck JR. J. Am. Chem. Soc. 2010;132:2524–2525. doi: 10.1021/ja910582n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxx.Takahashi T, Kanno K. Nickel-catalyzed Cross-coupling Reactions. In: Tamaru Y, editor. Modern Organonickel Chemistry. Wiley-VCH; Weinheim: 2005. p. 47. [Google Scholar]
- xxxi.Stille JK, Cowell AB. J. Organomet. Chem. 1977;124:253–261. [Google Scholar]
- xxxii.See Section 3.1 for examples.
- xxxiii.a Consiglio G, Morandini F, Piccolo O. J. Am. Chem. Soc. 1981;103:1846–1847. [Google Scholar]; b Kobayashi Y, Ikeda E. J. Chem. Soc., Chem. Commun. 1994:1789–1790. [Google Scholar]; c Didiuk MT, Morken JP, Hoveyda AH. Tetrahedron. 1998;54:1117–1130. [Google Scholar]
- xxxiv.Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J. Am Chem. Soc. 2011;133:389–391. doi: 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]
- xxxv.a Huang Z, Ducharme Y, MacDonald D, Robichaud A. Curr. Opin. Chem. Biol. 2001;5:432–438. doi: 10.1016/s1367-5931(00)00224-6. [DOI] [PubMed] [Google Scholar]; b Cheltsov AV, Aoyagi M, Aleshin A, Yu EC-W, Gilliland T, Zhai D, Bobkov AA, Reed JC, Liddington RC, Abagyan R. J. Med. Chem. 2010;53:3899–3906. doi: 10.1021/jm901446n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxvi.Bolm C, Rudolph J. J. Am. Chem. Soc. 2002;124:14850–14851. doi: 10.1021/ja028518l. [DOI] [PubMed] [Google Scholar]; b Braga AL, Paixão MW, Westermann B, Schneider PH, Wessjohann LA. J. Org. Chem. 2008;73:2879–2882. doi: 10.1021/jo702413n. [DOI] [PubMed] [Google Scholar]
- xxxvii.a Moree WJ, et al. J. Med. Chem. 2009;52:5307–5310. doi: 10.1021/jm900933k. [DOI] [PubMed] [Google Scholar]; b Beaton G, Moree WJ, Jovic F, Coon T, Yu J. 2006 Jan 19; U.S. Patent 11,156,252.
- xxxviii.Alami M, Messaoudi S, Hamze A, Provot O, Brion J-D, Liu J-M, Bignon J, Bakala J. 2009 Dec 10; Patent WO/2009/147217 A1.
- 39.a Greene MA, Yonova IM, Williams FJ, Jarvo ER. Org. Lett. 2012;14:4293–4296. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]; b Taylor BLH, Harris MR, Jarvo ER. Angew. Chem., Int. Ed. 2012;51:7790–7793. doi: 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]
- 40.For examples of biologically relevant diarylalkanes see: Nuclear receptor ligand: Kainuma M, Kasuga J.-i., Hosoda S, Wakabayashi K.-i., Tanatani A, Nagasawa K, Miyachi H, Makishima M, Hashimoto Y. Bioorg. Med. Chem. Lett. 2006;16:3213–3218. doi: 10.1016/j.bmcl.2006.03.075. Anti-lung cancer agent: Alami M, Messaoudi S, Hamze A, Provot O, Brion J-D, Liu J-M, Bignon J, Bakala J. 2009 Dec 10; Patent WO/2009/147217 A1. c Anti-viral agent: See ref. xxxvb; Anti-prostate cancer agent: Hu QZ, Yin LN, Jagusch C, Hille UE, Hartmann RW. J. Med. Chem. 2010;53:5049–5053. doi: 10.1021/jm100400a. Anti-tubulin polymerization agent: Messaoudi S, Hamze A, Provot O, Tréguier B, De Losada JR, Bignon J, Liu J-M, Wdzieczak-Bakala J, Thoret S, Dubois J, Brion J-D, Alami M. Chem. Med. Chem. 2011;6:488–497. doi: 10.1002/cmdc.201000456. Anticancer agent: Pathak TP, Gligorich KM, Welm BE, Sigman MS. J. Am. Chem. Soc. 2010;132:7870–7871. doi: 10.1021/ja103472a.
- 41.For examples of biologically relevant triarylmethanes see: Antitubercular agent: Parai MK, Panda G, Chaturvedi V, Manju YK, Sinha S. Bioorg. Med. Chem. Lett. 2008;18:289–292. doi: 10.1016/j.bmcl.2007.10.083.Ellsworth BA, Ewing WR, Jurica E. 2011 Apr 7; U.S. Patent Application 2011/0082165A1. Anti-breast cancer agent: Shagufta, Srivastava AK, Sharma R, Mishra R, Balapure AK, Murthy PSR, Panda G. Bioorg. Med. Chem. 2006;14:1497–1505. doi: 10.1016/j.bmc.2005.10.002.
- 42.Dieter RK, Oba G, Chandupatla KR, Topping CM, Lu K, Watson RT. J. Org. Chem. 2004;69:3076–3086. doi: 10.1021/jo035845i. [DOI] [PubMed] [Google Scholar]
- 43.Hoffmann RW, Hölzer B, Knopff O, Harms K. Angew. Chem., Int. Ed. 2000;39:3072–3074. doi: 10.1002/1521-3773(20000901)39:17<3072::aid-anie3072>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 44.Imao D, Glasspoole BW, Laberge VS, Crudden CM. J. Am. Chem. Soc. 2009;131:5024–5025. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 45.Harris MR, Hanna LE, Greene MA, Moore CE, Jarvo ER. J. Am. Chem. Soc. 2013;135:3303–3306. doi: 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.a Hayashi T, Matsumoto Y, Ito Y. J. Am. Chem. Soc. 1989;111:3426–3428. [Google Scholar]; b Hayashi T, Matsumoto Y, Ito Y. Tetrahedron: Asymmetry. 1991;2:601–612. [Google Scholar]; c Crudden CM, Hleba YB, Chen AC. J. Am. Chem. Soc. 2004;126:9200–9201. doi: 10.1021/ja049761i. [DOI] [PubMed] [Google Scholar]; d Chea H, Sim H-S, Yun J. Adv. Synth. Catal. 2009;351:855–858. [Google Scholar]
- 47.a Basu A, Thayumanavan S. Angew. Chem., Int. Ed. 2002;41:716–738. doi: 10.1002/1521-3773(20020301)41:5<716::aid-anie716>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; b Gawley RE. Top. Stereochem. 2010;26:93–133. doi: 10.1002/9783906390628.ch3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hölzer B, Hoffmann RW. Chem. Commun. 2003:732–733. doi: 10.1039/b300033h. [DOI] [PubMed] [Google Scholar]
- 49.Beak P, Kerrick ST, Wu S, Chu J. J. Am. Chem. Soc. 1994;116:3231–3239. For a general review of asymmetric deprotonation with alkyllithium-(–)-sparteine see: Hoppe D, Christoph G. In: The Chemistry of Organolithium Compounds. Part 2. Rappoport Z, Marek I, editors. Wiley; Chichester: 2004. pp. 1055–1164.
- 50.Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C.-y. J. Am. Chem. Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- 51.Klapars A, Campos KR, Waldman JH, Zewge D, Dormer PG, Chen C.-y. J. Org. Chem. 2008;73:4986–4993. doi: 10.1021/jo8006804. [DOI] [PubMed] [Google Scholar]
- 52.Beng TK, Gawley RE. Org. Lett. 2011;13:394–397. doi: 10.1021/ol102682r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.For more information on catalytic dynamic resolution see: Beng TK, Gawley RE. J. Am. Chem. Soc. 2010;132:12216–12217. doi: 10.1021/ja105772z. and references therein.
- 54.a Seel S, Thaler T, Takatsu K, Zhang C, Zipse H, Straub BF, Mayer P, Knochel P. J. Am. Chem. Soc. 2011;133:4774–4777. doi: 10.1021/ja201008e. [DOI] [PubMed] [Google Scholar]; b Boudier A, Darcel C, Flachsmann F, Micouin L, Oestreich M, Knochel P. Chem.—Eur. J. 2000;6:2748–2761. doi: 10.1002/1521-3765(20000804)6:15<2748::aid-chem2748>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 55.Dieter RK, Topping CM, Chandupatla KR, Lau K. J. Am. Chem. Soc. 2001;123:5132–5133. doi: 10.1021/ja0156587. b See also ref. 42.
- 56.Dieter RK, Chen N, Watson RT. Tetrahedron. 2005;61:3221–3230. [Google Scholar]
- 57.Dieter RK, Chen N. J. Org. Chem. 2006;71:5674–5678. doi: 10.1021/jo060717q. [DOI] [PubMed] [Google Scholar]
- 58.Hatanaka Y, Hiyama T. J. Am. Chem. Soc. 1990;112:7793–7794. See also: Hiyama T. J. Organomet. Chem. 2002;653:58–61.
- 59.LaBadie JW, Stille JK. J. Am. Chem. Soc. 1983;105:669–670. [Google Scholar]
- 60.Milstein D, Stille JK. J. Am. Chem. Soc. 1979;101:4981–4991. [Google Scholar]
- 61.a Liebeskind LS, Fengl RW. J. Org. Chem. 1990;55:5359–5364. [Google Scholar]; b Farina V, Kapadia S, Krishnan B, Wang C, Liebeskind LS. J. Org. Chem. 1994;59:5905–5911. [Google Scholar]
- 62.Ye J, Bhatt RK, Falck JR. J. Am. Chem. Soc. 1994;116:1–5. [Google Scholar]
- 63.Falck JR, Bhatt RK, Ye J. J. Am. Chem. Soc. 1995;117:5973–5982. [Google Scholar]
- 64.Li H, He A, Falck JR, Liebeskind LS. Org. Lett. 2011;13:3682–3685. doi: 10.1021/ol201330j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goli M, He A, Falck JR. Org. Lett. 2011;13:344–346. doi: 10.1021/ol102863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Falck JR, Patel PK, Bandyopadhyay A. J. Am. Chem. Soc. 2007;129:790–793. doi: 10.1021/ja064948q. [DOI] [PMC free article] [PubMed] [Google Scholar]; J. Am. Chem. Soc. 2008;130:2372. Correction. [Google Scholar]
- 67.Belosludtsev YY, Bhatt RK, Falck JR. Tetrahedron Lett. 1995;36:5881–5882. [Google Scholar]
- 68.Falck JR, Bhatt RK, Reddy KM, Ye J. Synlett. 1997:481–482. [Google Scholar]
- 69.Lange H, Fröhlich R, Hoppe D. Tetrahedron. 2008;64:9123–9135. [Google Scholar]
- 70.Ye J, Bhatt RK, Falck JR. Tetrahedron Lett. 1993;34:8007–8010. [Google Scholar]
- 71.Falck JR, Barma DK, Mohapatra S, Bandyopadhyay A, Reddy KM, Qi J, Campbell WB. Bioorg. Med. Chem. Lett. 2004;14:4987–4990. doi: 10.1016/j.bmcl.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 72.Kells KW, Chong JM. J. Am. Chem. Soc. 2004;126:15666–15667. doi: 10.1021/ja044354s. [DOI] [PubMed] [Google Scholar]
- 73.a Ridgway BH, Woerpel KA. J. Org Chem. 1998;63:458–460. doi: 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]; b Matos K, Soderquist JA. J. Org Chem. 1998;63:461–470. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]
- 74.Taylor BLH, Jarvo ER. J. Org. Chem. 2011;76:7573–7576. doi: 10.1021/jo201263r. [DOI] [PubMed] [Google Scholar]
- 75.a Doucet H. Eur. J. Org. Chem. 2008;201:2013–2030. [Google Scholar]; b Rubina M, Rubin M, Gevorgyan V. J. Am. Chem. Soc. 2003;125:7198–7199. doi: 10.1021/ja034210y. [DOI] [PubMed] [Google Scholar]
- 76.a Littke AF, Dai CY, Fu GC. J. Am. Chem. Soc. 2000;122:4020–4028. [Google Scholar]; b Dreher SD, Dormer PG, Sandrock DL, Molander GA. J. Am. Chem. Soc. 2008;130:9257–9259. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; c van den Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Visser M, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2008;49:4122–4124. [Google Scholar]
- 77.Chen AC, Ren L, Crudden CM. J. Org. Chem. 1999;64:9704–9710. [Google Scholar]
- 78.Reported values for retention of er were converted to es values for purposes of consistency throughout the data presented.
- 79.Ohmura T, Awano T, Suginome M. J. Am. Chem. Soc. 2010;132:13191–13193. doi: 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]
- 80.Awano T, Ohmura T, Suginome M. J. Am. Chem. Soc. 2011;133:20738–20741. doi: 10.1021/ja210025q. [DOI] [PubMed] [Google Scholar]
- 81.Sandrock DL, Jean-Gérard L, Chen C.-y., Dreher SD, Molander GA. J. Am. Chem. Soc. 2010;132:17108–17110. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee JCH, McDonald R, Hall DG. Nature: Chemistry. 2011;3:894–899. doi: 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]
- 83.Kagan HB, Fiaud JC. Top. Stereochem. 1998;18:249–330. For a recent review of dynamic kinetic resolution see: Pellissier H. Tetrahedron. 2008;64:1563–1601.
- lxxxiv.Lundin PM, Esquivias J, Fu GC. Angew. Chem., Int. Ed. 2009;48:154–156. doi: 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxxv.Lou S, Fu GC. J. Am. Chem. Soc. 2010;132:1264–1266. doi: 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxxvi.Lou S, Fu GC. J. Am. Chem. Soc. 2010;132:5010–5011. doi: 10.1021/ja1017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxxvii.Fischer C, Fu GC. J. Am. Chem. Soc. 2005;127:4594–4595. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- lxxxviii.Lundin PM, Fu GC. J. Am. Chem. Soc. 2010;132:11027–11029. doi: 10.1021/ja105148g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- lxxxix.Dai X, Strotman NA, Fu GC. J. Am. Chem. Soc. 2008;130:3302–3303. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- xc.Saito B, Fu GC. J. Am. Chem. Soc. 2008;130:6694–6695. doi: 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- xci.Smith SW, Fu GC. J. Am. Chem. Soc. 2008;130:12645–12647. doi: 10.1021/ja805165y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcii.Arp FO, Fu GC. J. Am. Chem. Soc. 2005;127:10482–10483. doi: 10.1021/ja053751f. [DOI] [PubMed] [Google Scholar]
- xciii.Owston NA, Fu GC. J. Am. Chem. Soc. 2010;132:11908–11909. doi: 10.1021/ja105924f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xciv.Lu Z, Wilsily A, Fu GC. J. Am. Chem. Soc. 2011;133:8154–8157. doi: 10.1021/ja203560q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcv.Zultanski SL, Fu GC. J. Am. Chem. Soc. 2011;133:15362–15364. doi: 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcvi.Oelke AJ, Sun J, Fu GC. J. Am. Chem. Soc. 2012;134:2966–2969. doi: 10.1021/ja300031w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcvii.Wilsily A, Tramutola F, Owston NA, Fu GC. J. Am. Chem. Soc. 2012;134:5794–5797. doi: 10.1021/ja301612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcviii.Choi J, Fu GC. J. Am. Chem. Soc. 2012;134:9102–9105. doi: 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xcix.Smith G, Kochi JK. J. Organomet. Chem. 1980;198:199–214. [Google Scholar]
- c.Anderson TJ, Jones GD, Vicic DA. J. Am. Chem. Soc. 2004;126:8100–8101. doi: 10.1021/ja0483903. [DOI] [PubMed] [Google Scholar]
- ci.See ref. xciv; Note: non-asymmetric cross-coupling of alkyl chlorides was found to be dependent on substrate concentration: Lu Z, Fu GC. Angew. Chem., Int. Ed. 2010;49:6676–6678. doi: 10.1002/anie.201003272.
- cii.Caeiro J, Pérez Sestelo J, Sarandeses LA. Chem.—Eur. J. 2008;14:741–746. doi: 10.1002/chem.200701035. [DOI] [PubMed] [Google Scholar]
- ciii.Consiglio G, Botteghi C. Helv. Chim. Acta. 1973;56:460–462. [Google Scholar]
- civ.Kiso Y, Tamao K, Miyake N, Yamamoto K, Kumada M. Tetrahedron Lett. 1974;15:3–6. [Google Scholar]
- cv.a Hayashi M, Takaoki K, Hashimoto Y, Saigo K. Enantiomer. 1997;2:293–296. [Google Scholar]; b Schwink L, Knochel P. Chem.—Eur. J. 1998;4:950–968. [Google Scholar]; c Horibe H, Fukuda Y, Kondo K, Okuno H, Murakami Y, Aoyama T. Tetrahedron. 2004;60:10701–10709. [Google Scholar]
- cvi.Hayashi T, Konishi M, Fukushima M, Mise T, Kagotani M, Tajika M, Kumada M. J. Am. Chem. Soc. 1982;104:180–186. Historical note: Hayashi T. J. Organomet. Chem. 2002;653:41–45.
- cvii.Pellet-Rostaing S, Saluzzo C, Ter Halle R, Breuzard J, Vial L, Le Guyader F, Lemaire M. Tetrahedron: Asymmetry. 2001;12:1983–1985. [Google Scholar]
- cviii.Hayashi T, Konishi M, Fukushima M, Kanehira K, Hioki T, Kumada M. J. Org. Chem. 1983;48:2195–2202. [Google Scholar]
- cix.Vriesema BK, Lemaire M, Buter J, Kellogg RM. J. Org. Chem. 1986;51:5169–5177. [Google Scholar]