

Abstract
Acromegaly is characterized by chronic, excess secretion of growth hormone (GH) from a pituitary adenoma, and elevated hepatic insulin-like growth factor 1 (IGF-1) levels. Significant progress has been made in the development of medical therapies to achieve biochemical and symptomatic control in acromegaly. In this review we discuss the three currently available medical therapies, which include somatostatin analogs, dopamine agonists and pegvisomant. We describe a step-wise approach in which a somatostatin analog is followed by the addition of a dopamine agonist, and then if required the addition of or replacement by pegvisomant. New somatostatin agonists such as pasireotide, and the introduction of new orally-acting somatostatin agonists, should increase the therapeutic choices available in the near future.
Keywords: Acromegaly, cabergoline, lanreotide, medical therapy, octreotide, pasireotide, pegvisomant, somatostatin analogs
INTRODUCTION
Most cases of acromegaly are due to chronic, excess secretion of growth hormone (GH) from a pituitary adenoma. GH induces the synthesis of hepatic insulin-like growth factor 1(IGF-1); elevated GH and IGF-1 levels cause severe metabolic dysfunction and result in significant morbidity and mortality. Mortality associated with acromegaly is at least two-fold higher than the general population.[1,2] Mortality rates in acromegaly are reduced to baseline if GH and (probably) IGF-1 levels are sufficiently controlled.[3] However, the criteria for the biochemical cure of acromegaly are a contentious area; the current definition includes GH levels <0.4 μg/L after oral glucose load, and IGF-1 levels within the normal age - and gender-adjusted range, although mortality data have tended to rely on random GH levels of <2.5 μg/l or, more recently, <1 μg/L.[4] Additional therapeutic goals in acromegaly include reducing tumor size, preserving pituitary function, and preventing disease recurrence. The main treatment modalities are pituitary surgery, medical therapy, and radiotherapy. Transsphenoidal surgery is the first-line treatment in patients whose tumor is likely to be completely resected and particularly those where the adenoma is compressing neurological and visual structures.[5] Remission rate are high in those with microadenomas.[6] However, approximately 70% patients with acromegaly have an underlying macroadenoma; the majority of these patients will not be cured by surgery alone, and will require adjuvant medical therapy or radiotherapy.[6] Thus, approximately 50% of patients will not be controlled by primary surgery. The roles of surgery and radiotherapy are beyond the scope of this review. In this review, we discuss the three currently available classes of medical therapy, which respectively target pituitary somatostatin receptors, pituitary dopamine receptors, and peripheral GH receptors.
SOMATOSTATIN ANALOGS
Somatostatin analogs are the first choice medical therapy in acromegaly and have been in clinical use since the 1980s. Somatostatin action is mediated by G-protein-coupled membrane receptors, five of which have been identified (sst1-5). The majority of GH-secreting adenomas express sst2 and sst5, and stimulation of these receptors by a somatostatin analog suppresses GH secretion by the adenoma and hepatic IGF-1 production. Octreotide and lanreotide are synthetic analogs of somatostatin and selectively bind to sst2 and lower affinity for sst5, with a longer half-life than the endogenous hormone. Depot formulations of these analogs are usually administered every 4 weeks, at least initially: Octreotide Long-Acting Repeatable (LAR) (Sandostatin LAR) and Lanreotide Autogel (Somatuline Depot). Somatostatin analogs are usually well-tolerated and adverse events are largely mild and transient.[7]
Traditionally, somatostatin analogs have been used as an adjuvant after incomplete surgical resection or to control excess GH secretion until radiotherapy becomes effective. Classical studies have reported that somatostatin analogs result in “control” of GH and IGF-1 levels in 48-67% patients with acromegaly.[8] However, it should be noted that many of these studies had selected patients for somatostatin analog sensitivity, and control rates in unselected patients fall to 20-30% based on both safe GH levels and normalized IGF-1 levels.[9] This leaves a proportion of patients in whom GH and IGF-1 levels remain uncontrolled with this class of medications, and one option for these patients is to try higher-dose somatostatin analog therapy. In 11 patients with uncontrolled acromegaly on standard doses of somatostatin analogs, high dose Octreotide LAR (up to 60 mg/28 days compared with the usual dose 30 mg/28 days) controlled GH levels in 10 patients (91%) and normalized IGF-1 levels in 4 patients (36%).[10]
Ramírez et al. recently reported that a few patients (2.4%) who are well controlled on small doses of somatostatin analogs were able to maintain long-term remission after ceasing therapy.[11] Only 12 patients met the study inclusion criteria, which included biochemical normalization on a stable dose of Octreotide LAR for at least 1 year, with an injection interval of at least 8 weeks, no history of radiotherapy, no dopamine agonist use in the preceding 6 months, and a small tumor remnant (<8 mm). Five of these patients (42%) remained biochemically cured after somatostatin analog therapy was withdrawn at 12 months follow-up.
Wide-spread interest in the use of somatostatin analogs as primary therapy for acromegaly was stimulated by Newman et al. in 1998.[12] They reported equal efficacy of subcutaneous Octreotide when used as primary therapy or as an adjuvant therapy following surgery to control GH and IGF-1 levels in patients with large or invasive tumors without visual or neurological compromise. Similar outcomes have been reported with the depot formulations, which also appear to reduce pituitary tumor size in most patients. A prospective, long-term study (up to 9 years) in 62 patients with de novo acromegaly showed that approximately 70% patients treated with Octreotide LAR as first-line therapy achieved GH levels <2.5 μg/L and/or normal IGF-1 levels, and tumor volume decreased by over 25% in 82% patients.[13] Another prospective study of 104 patients with newly diagnosed acromegaly compared Octreotide LAR and surgery and reported a comparable reduction in tumor volume in both groups.[14] Primary therapy with somatostatin analogs is a very high cost approach. Suitable candidates for first-line therapy with somatostatin analogs include those with no risk of visual impairment from the pituitary adenoma, patients who are unlikely to achieve biochemical cure by surgery, patients at high surgical risk and patients who decline surgical intervention.
Pasireotide (SOM230, Novartis, Basel) is a novel multi-receptor targeted somatostatin analog with high-affinity for sst1, sst2, sst3, and particularly sst5. Pasireotide has been proposed to have a stronger inhibitory effect on GH secretion than Octreotide due to its receptor binding profile. A phase II study has shown that pasireotide can effectively control GH and IGF-1 levels in patients with de novo and persistent acromegaly, and was more effective than subcutaneous Octreotide.[15] After 3 months of pasireotide therapy (200-600 μg twice daily), 27% of 51 patients achieved biochemical cure and 39% had significant tumor shrinkage (>20%). Drug-related adverse events were common, particularly gastrointestinal side-effects and worsening dysglycemia. Results from a large, randomized, double-blind phase III study, which compared Octreotide LAR and pasireotide LAR in patients with acromegaly were presented at the 2012 Joint International Congress of Endocrinology and the European Congress of Endocrinology meeting.[16] Significantly more patients with acromegaly were biochemically cured after 12 months treatment with pasireotide LAR than Octreotide LAR (31.3% vs. 19.2%, P = 0.007). Data from the 6-month extension study in which patients who did not achieve full biochemical control were offered the other treatment showed that 21% of the 81 patients who switched to pasireotide LAR achieved biochemical control of their disease compared with 2.6% of 38 patients who switched to Octreotide LAR. It is of interest that the control of GH levels with pasireotide was similar to octreotide, only the IGF-1 levels were significantly lower with pasireotide. It is therefore possible that the differential effects of the analogs is more dependent on pasireotide selectively inhibiting insulin release, which is a co-determinant with GH of IGF-1 levels. The side effect profile of pasireotide LAR includes hyperglycemia and diabetes mellitus, which has been reported previously.[17] These results indicate that pasireotide offers a promising therapeutic option for patients with acromegaly who are resistant or refractory to currently available somatostatin analogs.
DOPAMINE AGONISTS
Dopamine agonists were the first effective medical therapy for acromegaly and have been available since the 1970s. This class of oral medications is generally well-tolerated and relatively inexpensive. A meta-analysis of 15 studies (none of which was randomized or placebo-controlled) of cabergoline therapy in acromegaly showed that 51 of 149 patients (34%) achieved normal IGF-1 levels.[18] In contrast, the first prospective study of cabergoline in acromegaly recently reported that IGF-1 levels normalized in only 11% of patients with acromegaly after an 18 week trial of cabergoline therapy alone (maximum dose 3.5 mg/week) with insignificant changes in IGF-1 levels across the group.[19] The use of dopamine agonists alone in acromegaly has been largely superseded by somatostatin analogs. However, combined cabergoline and somatostatin analog therapy may improve the therapeutic response in patients who do not achieve adequate biochemical control with maximal doses of somatostatin analogs. The meta-analysis reported that IGF-1 levels normalized in 40 of 77 patients (52%) after cabergoline was added to on-going somatostatin analog treatment across 5 studies.[18] Furthermore, 17 of 49 (35%) patients experienced a reduction in tumor size.[18] These data suggest that the biochemical control of acromegaly (based on IGF-1 levels) is achieved in over half of patients when cabergoline is added to somatostatin therapy that has failed to normalize IGF-1 levels alone. The addition of cabergoline remained helpful even when patients had normal prolactin levels and appeared to correlate with tumor dopamine receptor 2 expression. The outcome was better in patients with lower pre-treatment IGF-1 levels. Randomized control trials are needed to confirm the beneficial effects of combined somatostatin analogs and dopamine agonists, although clinical experience suggests that it is an effective approach in individual patients. There is an ongoing concern about the (possible) effect of cabergoline on cardiac valves. Bromocriptine was used in the past and may still be used where cabergoline is unavailable or cost-prohibitive. However, high daily doses of bromocriptine are usually necessary (10-30 mg daily) and the control rate is probably < 10%.
PEGVISOMANT
Pegvisomant is a pegylated analogue of GH, which acts as a GH receptor antagonist. In the original trial, pegvisomant was highly effective when the dose was appropriately titrated: IGF-1 levels were normalized in up to 97% patients with persistent active acromegaly after pegvisomant treatment.[20] However, it is a highly expensive therapy, which has limited its widespread use. Pegvisomant is typically used in patients with acromegaly who have not responded to surgery, radiotherapy, and other medical therapies.[5] Following the original highly positive trial results, the ACROSTUDY is a global, non-interventional, surveillance study, which has recently provided data on the use of pegvisomant in routine clinical practice. A report on over 1200 patients who were enrolled on to the ACROSTUDY database up to December 2009 indicated that pegvisomant is an effective and safe therapy for patients with acromegaly who are not cured by other means: 63% of patients had normal IGF-1 levels after 5 years of pegvisomant treatment (mean dose 18 mg/day).[21] This efficacy was lower than expected and may reflect inadequate dosing or problems with compliance. In general, there is a dose-response relationship between the basal IGF-1 level and the dose of pegvisomant required to normalize the level. Treatment-related serious adverse events were reported in only 2% patients and most commonly involved recurrent pituitary tumor, abnormal liver function tests, and dysglycemia. Thirty follow-up MRI scans were reported with an increase in pituitary tumor size, although, reassuringly, only 13 were thought to be clinically significant. The majority of patients in the ACROSTUDY administered pegvisomant daily. In contrast, one previous study reported that weekly dosing with pegvisomant was as effective as daily injections in controlling IGF-1 levels and was preferred by patients.[22] In the ACROSTUDY, approximately one-third of patients also used another medical treatment, usually somatostatin analogs. A prospective UK multi-center study concluded that pegvisomant therapy alone or in combination with somatostatin analogs was equally efficacious and tolerated in patients with acromegaly who were not biochemically cured by somatostatin analog therapy alone.[23] Combined somatostatin analog and pegvisomant therapy normalized IGF-1 levels in almost two-thirds of patients. They also noted increased incidence of transaminitis when using the combination therapy. Combination therapy did not offer significant cost saving as the dose of pegvisomant was only 5 mg/day lower in combination with somatostatin analogs than used as a monotherapy (20 mg/day vs. 15 mg/day). A previous study showed that monthly somatostatin analogs combined with weekly pegvisomant injections was highly effective: 18 of 19 (95%) patients normalized IGF-1 levels with a median pegvisomant dose of 60 mg/week.[24]
Another prospective UK multi-center study assessed the efficacy of low-dose pegvisomant (10 mg/day) combined with cabergoline in 24 patients with active acromegaly, of which 19 completed the study.[19] A total of 13 patients (68%) achieved normal IGF-1 levels after 12 weeks of combined therapy. Interestingly there was a significant rise in IGF-1 levels after cabergoline therapy was withdrawn and only 5 patients (26%) had normal IGF-1 levels after 12 weeks of low-dose pegvisomant monotherapy. This study suggests that combination therapy with pegvisomant and dopamine agonists may facilitate lower pegvisomant doses, which could be a more cost-effective way to use this medication.
CONCLUSIONS
The control of GH and IGF-1 levels is an important aspect of the management of acromegaly to reduce associated morbidity and mortality. Medical therapies play a key role in achieving biochemical and symptomatic control in acromegaly [Figure 1]. Although consensus guidelines are available for acromegaly, a survey of experts within the field highlighted different approaches to management, particularly in patients with macroadenomas without visual compromise where opinion was divided between primary therapy with somatostatin analogs versus neurosurgery.[25] Further unresolved questions include the use of combination medical therapies and the cost-effectiveness of various therapies compared with surgery. Significant progress has been made in the medical management of acromegaly. The currently available medical therapies which include somatostatin analogs, dopamine agonists and pegvisomant are able to achieve biochemical cure in the vast majority of patients; however, the widespread use of these therapies is restrained by cost. Future developments, such as the use of orally-acting somatostatin analogs which are currently under clinical trial, should further increase the therapeutic possibilities.
Figure 1.
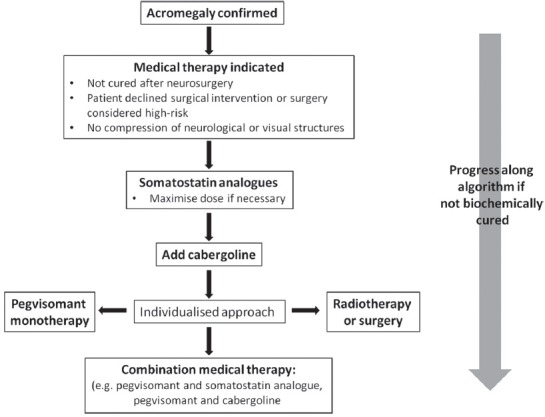
Algorithm to guide use of currently-available medical therapies in the management of acromegaly
Footnotes
Source of Support: Nil
Conflict of Interest: ABG has received consultancy fees and research funding from Novartis, Ipsen and Pfizer
REFERENCES
- 1.Holdaway IM, Rajasoorya RC, Gamble GD. Factors influencing mortality in acromegaly. J Clin Endocrinol Metab. 2004;89:667–74. doi: 10.1210/jc.2003-031199. [DOI] [PubMed] [Google Scholar]
- 2.Kauppinen-Mäkelin R, Sane T, Reunanen A, Välimäki MJ, Niskanen L, Markkanen H, et al. A nationwide survey of mortality in acromegaly. J Clin Endocrinol Metab. 2005;90:4081–6. doi: 10.1210/jc.2004-1381. [DOI] [PubMed] [Google Scholar]
- 3.Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP. Mortality in acromegaly: A metaanalysis. J Clin Endocrinol Metab. 2008;93:61–7. doi: 10.1210/jc.2007-1191. [DOI] [PubMed] [Google Scholar]
- 4.Giustina A, Chanson P, Bronstein MD, Klibanski A, Lamberts S, Casanueva FF, et al. A consensus on criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010;95:3141–8. doi: 10.1210/jc.2009-2670. [DOI] [PubMed] [Google Scholar]
- 5.Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, et al. Guidelines for acromegaly management: An update. J Clin Endocrinol Metab. 2009;94:1509–17. doi: 10.1210/jc.2008-2421. [DOI] [PubMed] [Google Scholar]
- 6.Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol. 2005;152:379–87. doi: 10.1530/eje.1.01863. [DOI] [PubMed] [Google Scholar]
- 7.Freda PU. Somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2002;87:3013–8. doi: 10.1210/jcem.87.7.8665. [DOI] [PubMed] [Google Scholar]
- 8.Freda PU, Katznelson L, van der Lely AJ, Reyes CM, Zhao S, Rabinowitz D. Long-acting somatostatin analog therapy of acromegaly: A meta-analysis. J Clin Endocrinol Metab. 2005;90:4465–73. doi: 10.1210/jc.2005-0260. [DOI] [PubMed] [Google Scholar]
- 9.Mercado M, Borges F, Bouterfa H, Chang TC, Chervin A, Farrall AJ, et al. A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf) 2007;66:859–68. doi: 10.1111/j.1365-2265.2007.02825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giustina A, Bonadonna S, Bugari G, Colao A, Cozzi R, Cannavo S, et al. High-dose intramuscular octreotide in patients with acromegaly inadequately controlled on conventional somatostatin analogue therapy: A randomised controlled trial. Eur J Endocrinol. 2009;161:331–8. doi: 10.1530/EJE-09-0372. [DOI] [PubMed] [Google Scholar]
- 11.Ramírez C, Vargas G, González B, Grossman A, Rábago J, Sosa E, et al. Discontinuation of octreotide LAR after long term, successful treatment of patients with acromegaly: Is it worth trying? Eur J Endocrinol. 2012;166:21–6. doi: 10.1530/EJE-11-0738. [DOI] [PubMed] [Google Scholar]
- 12.Newman CB, Melmed S, George A, Torigian D, Duhaney M, Snyder P, et al. Octreotide as primary therapy for acromegaly. J Clin Endocrinol Metab. 1998;83:3034–40. doi: 10.1210/jcem.83.9.5109. [DOI] [PubMed] [Google Scholar]
- 13.Cozzi R, Montini M, Attanasio R, Albizzi M, Lasio G, Lodrini S, et al. Primary treatment of acromegaly with octreotide LAR: A long-term (up to nine years) prospective study of its efficacy in the control of disease activity and tumor shrinkage. J Clin Endocrinol Metab. 2006;91:1397–403. doi: 10.1210/jc.2005-2347. [DOI] [PubMed] [Google Scholar]
- 14.Colao A, Cappabianca P, Caron P, De Menis E, Farrall AJ, Gadelha MR, et al. Octreotide LAR vs. surgery in newly diagnosed patients with acromegaly: A randomized, open-label, multicentre study. Clin Endocrinol (Oxf) 2009;70:757–68. doi: 10.1111/j.1365-2265.2008.03441.x. [DOI] [PubMed] [Google Scholar]
- 15.Petersenn S, Schopohl J, Barkan A, Mohideen P, Colao A, Abs R, et al. Pasireotide (SOM230) demonstrates efficacy and safety in patients with acromegaly: A randomized, multicenter, phase II trial. J Clin Endocrinol Metab. 2010;95:2781–9. doi: 10.1210/jc.2009-2272. [DOI] [PubMed] [Google Scholar]
- 16.Colao A, Bronstein M, Freda P, Gu F, Shen C, Gadelha M, et al. Pasireotide LAR is significantly more effective than octreotide LAR at inducing biochemical control in patients with acromegaly: Results of a 12-month randomized, double-blind, multicenter, Phase III study. Endocr Abstr. 2012;29:OC1. [Google Scholar]
- 17.Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-month phase 3 study of pasireotide in Cushing's disease. N Engl J Med. 2012;366:914–24. doi: 10.1056/NEJMoa1105743. [DOI] [PubMed] [Google Scholar]
- 18.Sandret L, Maison P, Chanson P. Place of cabergoline in acromegaly: A meta-analysis. J Clin Endocrinol Metab. 2011;96:1327–35. doi: 10.1210/jc.2010-2443. [DOI] [PubMed] [Google Scholar]
- 19.Higham CE, Atkinson AB, Aylwin S, Bidlingmaier M, Drake WM, Lewis A, et al. Effective combination treatment with cabergoline and low-dose pegvisomant in active acromegaly: A prospective clinical trial. J Clin Endocrinol Metab. 2012;97:1187–93. doi: 10.1210/jc.2011-2603. [DOI] [PubMed] [Google Scholar]
- 20.van der Lely AJ, Hutson RK, Trainer PJ, Besser GM, Barkan AL, Katznelson L, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001;358:1754–9. doi: 10.1016/s0140-6736(01)06844-1. [DOI] [PubMed] [Google Scholar]
- 21.van der Lely AJ, Biller BM, Brue T, Buchfelder M, Ghigo E, Gomez R, et al. Long-term safety of pegvisomant in patients with acromegaly: Comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97:1589–97. doi: 10.1210/jc.2011-2508. [DOI] [PubMed] [Google Scholar]
- 22.Higham CE, Thomas JD, Bidlingmaier M, Drake WM, Trainer PJ. Successful use of weekly pegvisomant administration in patients with acromegaly. Eur J Endocrinol. 2009;161:21–5. doi: 10.1530/EJE-08-0990. [DOI] [PubMed] [Google Scholar]
- 23.Trainer PJ, Ezzat S, D’souza GA, Layton G, Strasburger CJ. A randomized, controlled, multicentre trial comparing pegvisomant alone with combination therapy of pegvisomant and long-acting octreotide in patients with acromegaly. Clin Endocrinol (Oxf) 2009;71:549–57. doi: 10.1111/j.1365-2265.2009.03620.x. [DOI] [PubMed] [Google Scholar]
- 24.Feenstra J, de Herder WW, ten Have SM, van den Beld AW, Feelders RA, Janssen JA, et al. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005;365:1644–6. doi: 10.1016/S0140-6736(05)63011-5. [DOI] [PubMed] [Google Scholar]
- 25.Giustina A, Bronstein MD, Casanueva FF, Chanson P, Ghigo E, Ho KK, et al. Current management practices for acromegaly: An international survey. Pituitary. 2011;14:125–33. doi: 10.1007/s11102-010-0269-9. [DOI] [PubMed] [Google Scholar]