

Abstract
Successful outcome of pregnancy depends upon genetic, cellular, and hormonal interactions, which lead to implantation, placentation, embryonic, and fetal development, parturition and fetal adaptation to extrauterine life. The fetal endocrine system commences development early in gestation and plays a modulating role on the various physiological organ systems and prepares the fetus for life after birth. Our current article provides an overview of the current knowledge of several aspects of this vast field of fetal endocrinology and the role of endocrine system on transition to extrauterine life. We also provide an insight into fetal endocrine adaptations pertinent to various clinically important situations like placental insufficiency and maternal malnutrition.
Keywords: Endocrinology, fetal, hypothalamic, pituitary
INTRODUCTION
A gamut of transition factors and epigenetic events act in coordination with autocrine, paracrine, and endocrine network of hormones and growth factors leading to maternal–placental–fetal interactions and evolution of fetal endocrine system. Hypothalamus and pituitary provide the baseline controlling framework.
Anterior pituitary develops from Rathke's pouch by 5th week.[1] By 7th week hypothalamus, pituitary stalk, and posterior pituitary develop.[1] Hypothalamic pituitary portal vessels formation starts by 12th-17th week, which matures till 30th-35th weeks of gestation. Hypothalamic cells develop by 15th-18th weeks. Dopamine, thyrotropin releasing hormone (TRH), gonadotropin releasing hormone (GnRH), and somatostatin are present in hypothalamic tissue by 10th-14th weeks of gestation. Lactotropes, somatotropes, corticotropes, thyrotropes, and gonadotropes are discernible by 7th-16th weeks. Growth hormone (GH), prolactin (PRL), thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and adrenocorticotropic hormone (ACTH) are detectable between 10 and 17 weeks. The current article focuses fetal endocrine axes maturation and adaptation to extrauterine life. We have referred to development in animals, mostly in sheep and in rat.
HYPOTHALAMIC–PITUITARY GONADAL AXIS
GnRH stimulates LH and FSH release from gonadotropes. In sheep fetus, LH, FSH, and GnRH are present at mid-gestation or before. Prior to the maturation of the fetal pituitary, chorionic gonadotropin (CG) from the placenta stimulates fetal gonadal growth, differentiation, and secretory activity. CGs levels are greater in female fetuses and show pulsatile secretion during fetal period. Hypothalamus–pituitary–gonadal (HPG) axis activity peaks at 30-40% gestation in the sheep fetus, decreasing until birth.[2] In the rat fetus tissue concentrations of GnRH, LH, and FSH increase continuously until birth.
In the sheep fetus, gonadal testosterone and hypothalamic CG secretion peaks at about same time, approximately 30-40% gestation [Figure 1]. The mid-gestational peak in gonadal steroid hormone secretion stimulates gonadal growth and differentiation. Later in gestation, the placenta synthesizes estrogens and androgens, controlled by the fetal hypothalamus–pituitary–adrenal (HPA) axis.[3]
Figure 1.
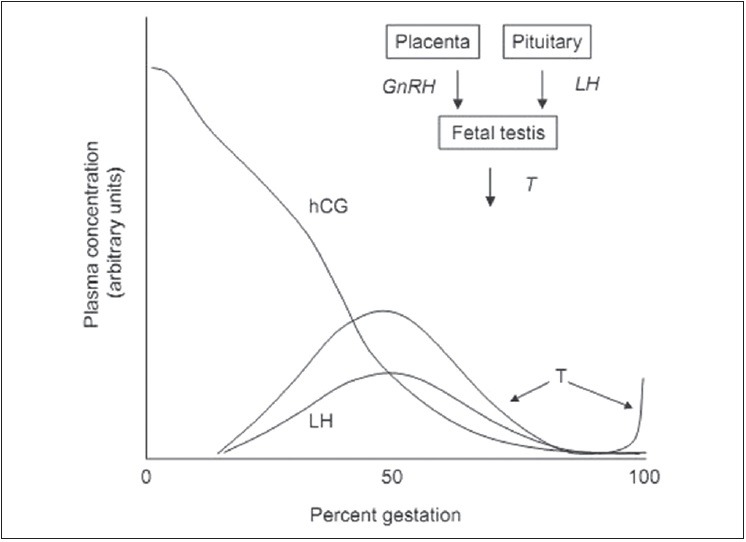
Schematic representation of the ontogeny of hCG, LH, and T in the plasma of a male fetus. The relationship of GnRH, LH and T is represented in the upper right corner of the fi gure. hCG: Human chorionic gonadotropin, LH: Luteinizing hormone, GnRH: Gonadotropin releasing hormone, T: Testosterone
Gonadal development
The mammalian gonad is derived from (a) primordial germ cells of the yolk sac wall, (b) stromal cells from primitive mesonephros.[4,5] By 4-5 weeks, the germ cells migrate from yolk sac and gonadal ridge derives from mesonephros. The germ cells incorporate into the gonadal ridge during the sixth week.[4] Gonadal embryogenesis is programmed by SRY, SF-1, SOX 1, DAX-1 genes.[6,7] CGs are not required for fetal gonadal development or sexual differentiation.[8]
Male gonadal differentiation begins at 7 weeks with organization of the gonadal blastema into interstitium and germ cell-containing testicular cords. Epithelium differentiates into tunica albuginea.[9] Primitive Sertoli cells and spermatogonia become visible within the cords. Leydig cells derived from interstitium by 8th week synthesize androgens. The fetal testes grow from approximately 20 mg at 14 weeks of gestation to 800 mg at birth; at 5-6 months they descend into inguinal canal with epididymis and ductus deferens.[9]
In females, differentiation of ovaries begins during 7th week. The gonadal blastema differentiates into interstitium and medullary cords containing oogonia. By 11-12 weeks, cortex contains oogonia surrounded; and medulla consists of connective tissue.[10] At 12 weeks, primitive granulosa cells begin to replicate and the oogonia in the deepest layers of the cortex enter their first meiotic division. Primordial follicles are observed at 18 weeks.[11] The number of oocytes progressively declines from 3-6 million at 5 months to 2 million at term.[11,12] By 5th-7th month stroma-derived thecal cells develop around mature primordial follicles. Each fetal ovary weighs about 15 mg at 14th gestational weeks and 300-350 mg at birth.[10] The number of surviving primary follicles at birth correlates with the duration of postpubertal ovulation. Interstitial steroid-producing cells are present after 12 weeks, and during third trimester theca cells with steroidogenic capacity surround the developing follicles.[12] Ovary produces less steroids, even in the presence of aromatase activity.[10,12]
Fetal estrogen and androgen biosynthesis and their effects
Fetal androgens and estrogens in late-gestation are majorly derived from placenta.[13] The placenta lacks cytochrome P450 (CYP17) and 17α-hydroxylase and 17,20-lyase activities [Figure 2]. Placenta synthesizes estrogen from precursors received from the fetal adrenal cortex. The adult zone or definitive zone of the fetal adrenal cortex lacks the enzyme 3β-hydroxysteroid dehydrogenase [Figure 2] and cannot, synthesize progesterone or 17α-hydroxyprogesterone. However, the fetal zone of the fetaladrenal cortex secretes dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) in response to ACTH. These steroidogenic intermediates used in biosynthesis of estrogens by placenta, bypassing CYP17 [Figures 2 and 3]. Estrogen in fetal bloodstream allows fetal growth and development. Estrogen increases uterine blood flow during pregnancy and at term increases myometrial contractility to initiate labor. In the male, leydig cells produce testosterone in response to human chorionic gonadotropin (hCG) and LH between 10 and 20 weeks.[9]
Figure 2.
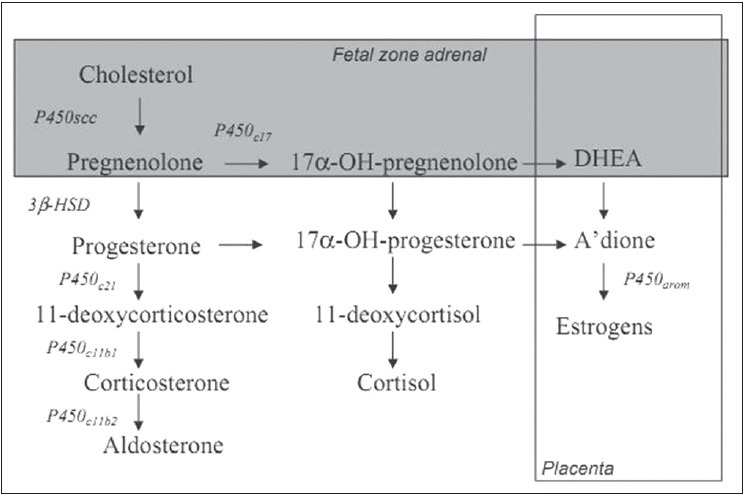
Steroid biosynthesis in the developing primate fetus. Steroidogenic enzymes are represented by the following abbreviations: P450c17 (17 and α-hydroxylase and 17,20-lyase activities); 3 and β-HSD (3 and β-hydroxysteroid dehydrogenase activity); P450c11b1 (11 and α-hydroxylase activity); P450c11b2 (aldosterone synthase activity); P450arom (aromatase activity). The fetal zone of the fetal adrenal cortex is capable of performing the reactions in the shaded area. The overlapping box (right) represents estrogen biosynthesis by the placenta. A-dione, androstenedione
Figure 3.
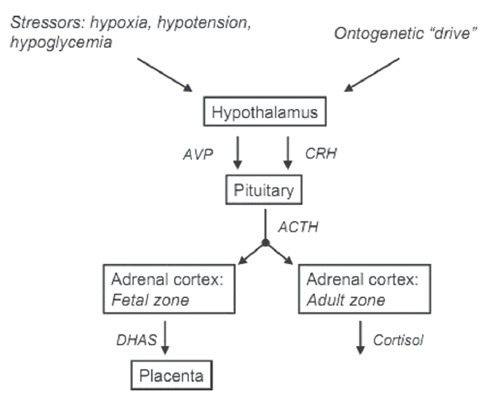
Relationship between hypothalamus, pituitary, and fetal and de and #64257; nitive zones of the fetal adrenal. Both zones of the fetal adrenal cortex are stimulated by ACTH secreted by the fetal anterior pituitary. The secretion of ACTH is stimulated acutely in response to stressors in utero, and chronically in a pattern that produces increased activity of the fetal hypothalamus–pituitary–adrenal axis independent of stressors at the end of gestation. CRH: Corticotropin releasing hormone, ACTH: Adrenocorticotropic hormone, DHAS: Dehydroepiandrosterone sulfate, AVP: Arginine vasopressin
Effect of testosterone and estrogen
Testosterone stimulates differentiation of primitive mesonephric ducts into ductus deferens, epididymides, seminal vesicles, and ejaculatory ducts. Androgen receptors appear in urogenital mesenchyme and epithelium at 8 and 12 weeks, respectively.[14] Dihydrotestosterone (DHT) is formed from testosterone by the 5α-reductase enzyme within the urogenital sinus and urogenital tubercle and acts on wolffian ducts. DHT stimulates male differentiation of the urogenital sinus and external genitalia, including differentiation of the prostate, growth of the genital tubercle to form a phallus, and fusion of the urogenital folds to form penile urethra. The sertoli cells produce antimüllerian hormone (AMH), which causes müllerian duct regression in male fetus after a 24-36 hour exposure.[15,16] AMH gene expression is activated by SRY gene.[15] Male phenotypic differentiation testosterone and AMH occurs between 8 and 14 weeks. In the female fetus, müllerian duct system differentiates in the absence of AMH, mesonephric ducts fail to develop in the absence of testosterone, and the undifferentiated urogenital sinus and external genitalia mature into female structures.
Estrogen acts through two nuclear receptors[17,18] expressed in 16-23 weeks. Estrogen receptor beta (ERβ) is predominant in testis, ovary, spleen, thymus, adrenal, brain, kidney, and skin. Estrogen receptor alpha (ERα) is prominent in uterus with relatively low levels in most other tissues.[17,18] Knockout of both ERα and ERβ genes has little impact on fetal development, but leads to hypoplastic uterus, fallopian tubes, vagina, and cervix postnatally, which are unresponsive to estrogen.[18] Gonadal hormones are involved in (a) structural development of rat brain and (b) control of CG production.[19,18,21]
HYPOTHALAMIC–PIUITARY–ADRENAL AXIS
ACTH is secreted by corticotropes in response to arginine vasopressin (AVP) and corticotropin releasing hormone (CRH).[22] Fetal HPA axis is controlled by paraventricular nuclei (PVN) in sheep fetuses.[23] The fetal HPA axis is activated progressively throughout latter gestation leading to increased biosynthesis of hypothalamic CRH, AVP, and proopiomelanocortin (POMC) in pituitary, increased sensitivity of steroidogenic tissue to ACTH, increased abundance of steroidogenic enzymes in adrenal cortex with increase in its size. This results in increased ACTH, cortisol levels in nonhuman fetal blood and DHEA, DHEAS and estrogens in human fetal blood. The molecular processing of POMC to ACTH is also stimulated by placental estrogen in latter developmental stages. In sheep the increased cortisol secretion is accompanied by an increased binding capacity for cortisol. Prior to term, there is an effective negative feedback mechanism by which cortisol inhibits fetal ACTH secretion. Estrogen selectively suppresses fetal zone growth during second half of pregnancy. At term, human placental estrogen leads to positive feedback cycle with progressive increase in fetal HPA activity resulting in increase in ACTH and cortisol in last 10 weeks of gestation. This helps in visceral and pulmonary maturity.[24] Placental release of ACTH and CRH into fetal blood stimulates fetal HPA axis and effects parturition. AVP and catecholamines also stimulate fetal ACTH secretion.[25]
Adrenal development
The adrenal glands develop cephalad to the bilaterally developing mesonephros by 3-4 weeks.[26,27] The fetal adrenal is composed of, fetal zone producing C19 androgens, transitional zone producing cortisol and outer definitive zone producing mineralocorticoids. By 9-12 weeks, fetal zone is steroidogenically active. At term, fetal adrenal gland weighs 8 grams (80%=fetal zone).[12,26,27] Fetal adrenal cortex is developed by genes including SF-1 (steroidogenic factor-1) and DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1), WT1 (Wilm's tumor 1), LIM1 (lin-11, Isl1 and mec-3 homologue) and growth factors like (FGF (fibroblast growth factor), (EGF (epithelial growth factor) and IGF-II (insulin like growth factor II), which are responsive to ACTH.[12] IGF-II augments ACTH stimulated expression of steroidogenic enzymes. Definitive zone produces cortisol from 30 weeks.[12]
Adrenal steroidogenesis and regulation
The steroidogenic acute regulatory protein (StAR) is the rate-limiting factor in adrenal steroidogenesis. The fetal adrenal expresses five steroidogenic apoenzymes: Two microsomal enzymes with 17-hydroxylase with 17,20-lyase (CYP17) and 21-hydroxylase (CYP21A2) activities, respectively, plus two mitochondrial cytochrome P450 enzymes providing cholesterol side-chain cleavage (CYP11A1) and C11/C18 hydroxylation of the parent steroid structure (CYP11B1/CYP11B2). A fifth enzyme in smooth endoplasmic reticulum, exhibits both 3ί-hydroxysteroid dehydrogenase (3bHSD) and 4, 5-isomerase activities. Definitive zone secretes cortisol (sheep, human) and corticosterone (rodent) in response to ACTH and aldosterone in response to angiotensin II and potassium, early in gestation. With low 3β HSD and high sulfotransferase activities, fetal zone produces DHEA, DHEAS, pregnenolone sulfate, several 53ί-hydroxysteroids, and limited amounts cortisol and aldosterone.[26,27] ACTH stimulates steroid production by activating StAR and increasing delivery of cholesterol to CYP 11A1; angiotensin II inhibits 3βHSD activity and promotes DHEA production in the fetal zone.[26] This provides DHEA substrate for placental estrogen production. The definitive zone contributes only a small fraction of total fetal adrenal steroid output. The substrate cholesterol is derived from circulating low-density lipoprotein (LDL) (70%) and from de novo adrenal synthesis. The fetal zone contains more LDL binding sites and has greater cholesterol synthesis rate resulting in greater steroidogenic activity. Both fetal adrenal cortisol and placental estradiol regulate hepatic synthesis of cholesterol in the fetus.
About two-thirds of fetal cortisol is derived from the fetal adrenal glands, and one-third is derived from placental transfer. A total of 80% of fetal cortisol is oxidized in fetal tissues or placenta by 11β HSD-II to cortisone or further metabolites. This isolates the fetus from maternal cortisol. As the fetus matures, the activity of 11β-HSD is increased, due to increased placental estrogen biosynthesis.[28] As term approaches, fetal liver and lung express 11-ketosteroid reductase activity that promotes local conversion of cortisone to cortisol.[26]
Adrenal hormones act through two nuclear receptors including glucocorticoidand mineralocorticoid receptors (GRs, MRs). GRs are present since mid-gestation in placenta, lung, brain, liver, and gut.[26,29,30] MRs are present from 12 to 16 weeks.[31] The human fetal adrenal gland secretes aldosterone, which is low in mid-gestation but increases 3-fold following caesarian section, and persists during the first year of extrauterine life.[26,32]
Adrenal medulla
The adrenal medulla becomes innervated by sympathetic preganglionic nerves at a 80% gestation (in sheep), and secretes catecholamines.[33] Prior to innervation, fetal adrenal is directly responsive to hypoxia. Biosynthesis of epinephrine is dependent upon the expression of phenylethanolamine-N-methyltransferase (PNMT), induced by cortisol. Hence adrenal medulla secretes epinephrine in late gestation.
The adrenal medulla responds to hypoxia and hypotension with increased secretion of catecholamines. This redistributes fetal ventricular output away from somatic tissues toward the umbilical–placental circulation. This also supports fetal glucose homeostasis; epinephrine secretion elevates plasma glucose following fetal distress.
HYPOTHALAMIC–PITUITARY–THYROID AXIS
In first trimester extrahypothalamic TRH stimulates pituitary TSH secretion, which mainly increases during second trimester. Progressive maturation of hypothalamic pituitary control and of thyroid gland responsiveness to TSH in late third trimester and early neonatal period leads to increased TSH and T4.[34,35] Pituitary TSH secretion responds to hypothyroxinemia and TRH early in third trimester.[36] The maternal and fetal HPT axes operate somewhat independently. The placental deiodinase converts T4 to rT3 and is relatively impermeable to T4 and T3. The placenta is also impermeable to TRH, TSH, and thyroid-binding globulins. This leads to a gradient of T4 and T3 from maternal to fetal plasma. Early in gestation placental transfer is the only source of fetal T4 and is essential for normal fetal brain development (between 12 and 20 weeks, before fetal thyroid hormonogenesis).[37]
Thyroid development
The buccopharyngeal cavity gives rise to (a) a midline thickening of pharyngeal floor (median anlage) and (b) paired caudal extensions of the fourth pharyngobranchial pouches (lateral anlagen).[36,38] The median anlage extends to fourth branchial arch by 25 days. By 50 days, the median and lateral anlagen fuse and migrate caudally to anterior neck. Colloid is visible by 70 days with demonstrable thyroglobulin synthesis and iodide accumulation. At 12 weeks, the fetal thyroid gland weighs 80 mg, which increases to 1-1.5 g at term. Genes involved in thyroid and parathyroid development are HEX, TTF1, TTF2 and PAX8.[36,39,40]
Thyroid hormone secretion
In humans, thyroglobulin biosynthesis starts by 25% gestation. The adult thyroid follicular cell can modify iodine transport or uptake with changes in dietary iodine intake[41,42] during 36-40 weeks, Fetal TSH and T4 are discernible early in second trimester. Type 3 deiodinase (D3) converts T4 to rT3 and is expressed in placenta, liver, and fetal skin leading to abundant fetal plasma rT3 circulating throughout the second and third trimesters. T3 concentrations increase in final stages of fetal development (30 weeks, suggesting late development of types 1 and 2 deiodinases (D1 and D2, respectively)) in liver, kidney, brain, and other tissues in association with low placental D3 activity[43] [Figure 4]. High levels of SULT (sulfotransferase) in fetal liver, lung, and brain by mid-gestation results in increased formation of iodothyronine sulfates.[34,44,45] The sulfated metabolites accumulate as a result of the low D1 activity in fetal tissues and because the sulfated iodothyronines are not substrates for D3.[41,44] Plasma T4-binding globulin and total T4 concentrations increase progressively from low levels at 16-18 weeks to maximal levels at 35-40 weeks. Free T4 levels also increase as a consequence of the increased T4 production.
Figure 4.

The ontogeny of thyrotropin, T4, T3, and rT3 in fetal plasma. The relationship of thyrotropin, T4, T3, and rT3 is represented in the upper left corner of the fi gure. rT3- Reverse T3
THYROID HORMONE ACTION
The circulating T3 levels at parturition (a) prepares fetal transition to extrauterine life and (b) stimulates maturation of vision and hearing postnatally in the mouse and toward the end of the second mid-trimester in the human fetus.[46] The period of brain dependency for thyroid hormone extends postnatally to 2-3 years of age, but the early weeks and months of life are most critical.
Thyroid hormone actions are mediated via two nuclear receptors. The genes for receptors are expressed on chromosome 17 and 3 for TRα and TR β.[47] There are four receptor isoforms–TRα1, TRα2, TRβ1, and TRβ2. TRα2 is the auto inhibitory isoform and can inhibit binding of other TRs. In human fetal brain, TRα1 and TRβ1 isoforms and receptor binding are present by 8-10 weeks; TRα1 isoforms increase 8- to 10-fold by 16-18 weeks.[36,48,49] Liver, heart, and lung receptor binding can be identified by 13-18 weeks.[36,49,50] Knockout of both the TRα and TRβ genes in mice results in elevated TSH levels, deafness, bradycardia, and decreased postnatal growth with delayed bone maturation.[36,47]
OTHER ANTERIOR PITUITARY HORMONES
Pituitary GH secretion starts by 8-10 weeks.[1] Fetal plasma GH levels in cord blood increases from 1 to 4 nmol/l at first trimester to 6 nmol/l at mid-gestation. Human somatotrophs respond predominantly to GHRH at 9-16 weeks; while response to the inhibitory somatostatin develops later in gestation[51] leading to progressive fall of plasma GH during second half of gestation to 1.5 nmol/l at term.[1] The response of plasma GH to somatostatin, GHRH, insulin and arginine are mature at term in humans; however, mature responses to sleep, glucose and L-dopa are present by 3 months of postnatal life. GH receptors are low in fetal liver and nutrition stimulates fetal IGF production.[52,53]
Fetal plasma prolactin levels are low until 25-30 weeks and increase to 11nmol/l at term.[1] Brain and hypothalamic control of PRL matures in late gestation and during the first months of extrauterine life.[1] Estrogen stimulates PRL release leading to high last trimester fetal plasma PRL levels. PRL receptors in fetal tissues during first trimester are implicated in fetal growth, skeletal maturation and adipose tissue maturation.[54]
POSTERIOR-PITUITARY
Neurohypophysis develops by 10-12 weeks.[55,56] AVP and oxytocin are synthesized by magnocellular neurons in supraoptic and paraventricular nuclei. AVP has three biological activities [Figure 5]. The vasopressor action on peripheral vessels is mediated by V1a. V1b and V2 receptors mediate Corticotropin-releasing and antidiuretic activity. It is less potent on fetal kidneys. AVP is an important fetal cardiovascular hormone.[57] During last trimester, fetal hypothalamic and pituitary responsiveness to both volume and osmolar stimuli for AVP secretion are well developed and AVP exerts antidiuretic effects on the fetal kidney.[55,56] AVP responds to hypoxia, hemorrhage, intrauterine bradycardia, meconium passage, more than to osmolar stimuli.[56,58,5,60] It maintains maternal circulatory homeostasis through its vasoconstrictor action, but has a limited effect on fetoplacental blood flow.[55,60] Fetal hypoxia is also a major stimulus for catecholamine release. Both fetal hypoxia and AVP stimulate anterior pituitary function including ACTH.[60] Aquaporin-1, 2, and 3 receptors in fetal and newborn kidney regulate free water clearance.[61,62] Maximal concentrating capacity by the fetal kidney is limited to about 600 mmol/L. AVP redistributes fetal ventricular output toward the umbilical–placental circulation, maximizing transfer of gases between maternal and fetal circulations.
Figure 5.
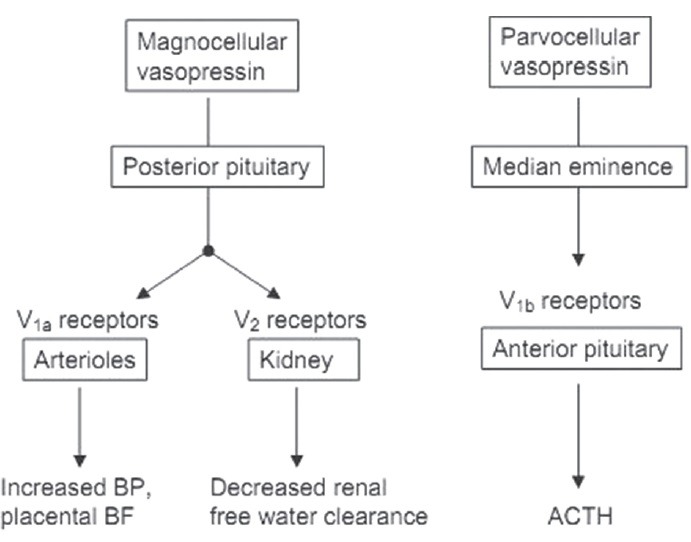
The synthesis, release, and action of AVP in the fetus. Magnocellular neurons in the paraventricular nucleus synthesize vasopressin, releasing the peptide into the bloodstream at the posterior pituitary. Parvocellular neurons in the hypothalamic nucleus synthesize vasopressin, which is released into the hypothalamo–hypophyseal portal blood at the median eminence. Portal blood vasopressin acts as a corticotropin-releasing factor at the corticotrope of the anterior pituitary. BF: Blood flow, BP: Blood pressure, ACTH: Adrenocorticotropic hormone
Oxytocin circulates in fetal plasma in higher concentrations, which increase as the fetus matures and during active labor. The placental barrier prevents fetal oxytocin from reaching the myometrium. Oxytocin stimulates the release of ACTH via V1b receptor.
INTERMEDIATE LOBE OF PITUITARY
Intermediate lobe cells begin to disappear near term and are virtually absent in the adult human pituitary.[63] The cells synthesize POMC, which after cleavage produces α-MSH and ß-endorphin and corticotropin and ß-lipotropin in anterior lobe in response to hypoxia.[64] α-MSH and corticotropin play a role in fetal adrenal activation, and α-MSH plays a role in fetal growth.[65,66] Neurointermediate lobe cells mature in late gestation, perhaps in response to cortisol in fetal plasma.[67]
ECTOPIC HORMONES
POMC secreted by the neuroendocrine cells of fetal lung might play a role in stress responsiveness, or in the timing of parturition, by altering adrenal sensitivity to circulating ACTH. The neuroendocrine cells also synthesize VIP (vasoactive intestinal peptide) and serotonin.
Kidney, liver, and testes from 16- to 20-week-old human fetuses produce hCG in vitro.[12] ACTH like immunoreactivity is present in neonatal rat pancreas and kidney.[68] Hypothalamic neuropeptides are also present in fetal gut tissues. TRH and somatostatin are produced in neonatal rat pancreas and gastrointestinal tissues.[69,70] In human neonatal pancreas and blood, TRH and somatostatin are derived mostly from extrahypothalamic sources.[71,72] Extrahypothalamic TRH controls fetal pituitary TSH secretion before maturation of hypothalamic TRH at term.[73]
OTHER ENDOCRINE SYSTEMS
Superior and inferior parathyroid glands develop respectively from 4th and 3rd pharyngeal pouches. Parafollicular C cells develop from 5th pouch between 5 and 12 weeks.[36,38] The genes implicated are HOX15, GCMB, GATA3, CRKL, and TBX1.[74,75] High fetal calcium concentrations (11-12 mg/dl) are maintained by active transport from maternal serum through an ATP-dependent calcium pump across syncytiotrophoblast.[75] The placental calcium pump is activated by a mid-molecule portion of PTHrP (67-86 amino acids) secreted by fetal parathyroid gland and placenta. Parathormone (PTH) or parathormone related peptide (PTHrP) fragments 1-34 activate PTH/PTHrP receptor leading to fetal skeletal calcium flux, calcium excretion through fetal kidney, renal 1, 25 (OH) 2 D production, calcium reabsorption from amniotic fluid. 1, 25 (OH) 2 D acts to enhance maternal–fetal calcium transport.[75] The placenta is impermeable to PTH, PTHrP and calcitonin, but is permeable to 25 (OH) D and 1, 25 (OH) 2 D.[75] Fetal hypercalcemia results in high blood levels of calcitonin in fetus, contributing to fetal bone mineral accretion and inhibition of bone resorption.[75] In addition, 25 (OH) D and 1, 25 (OH) 2 D play a role in fetal cartilage growth and bone mineral accretion.[76]
Fetal pancreas is identifiable by 4 weeks of gestation with α and β cells developing by 8-9 weeks. Insulin, glucagon, somatostatin, and pancreatic polypeptide are measurable by 8-10 weeks.[77] α cells are more numerous than β cells in early fetal pancreas and peak at mid-gestation; β cells increase throughout second half of gestation and at term α:β cells ratio is 1:1. Genes implicated in pancreatic development are PDX-1, HLXB9, ISL-1, or HES-1, whereas NGN3 or Beta 2 genes are responsible for endocrine development. Pancreatic β cells are functional by 14-24 weeks. Fetal rat pancreas releases insulin in response to leucine, arginbine, tolbutamide, or potassium, but respond minimally to glucose or pyruvate.[78,79,80] Similarly in monkey fetus, neither glucose nor arginine stimulates insulin release near term, but glucagon evokes insulin release. GH stimulates insulin gene expression and β cell hyperplasia and hypertrophy.[81] Fetal insulin unlike in adults, neither stimulates adenylate cyclase system, nor activates calcium channel.[80] Pancreatic glucagon concentrations are relatively high in fetal plasma and increase progressively with age.[78,79] The blunted capacity for insulin and glucagon secretion is due to deficient capacity of the fetal pancreatic islets to generate cyclic adenosine monophosphate (cAMP) or to destroy cAMP by phosphodieestrase.[78] Insulin and glucagon are not necessary for substrate metabolism in the fetus.[79] Glucose is obtained by placental transfer through facilitated diffusion. In addition, constant supply of glucose precludes the necessity for endogenous gluconeogenesis and gluconeogenetic enzyme activity is low in fetal liver. Similarly glycogen storage in fetus is modulated by fetal glucocorticoids and human placental lactogen (HPL), with insulin playing a role at term.[78,79] But the fetal hepatic glucagon receptors are reduced in number leading to relative resistance of fetal liver to the glycemic effect of glucagon.
ENDOCRINE REGULATION OF TRANSITION TO EXTRAUTERINE LIFE [FIGURE 6]
Figure 6.
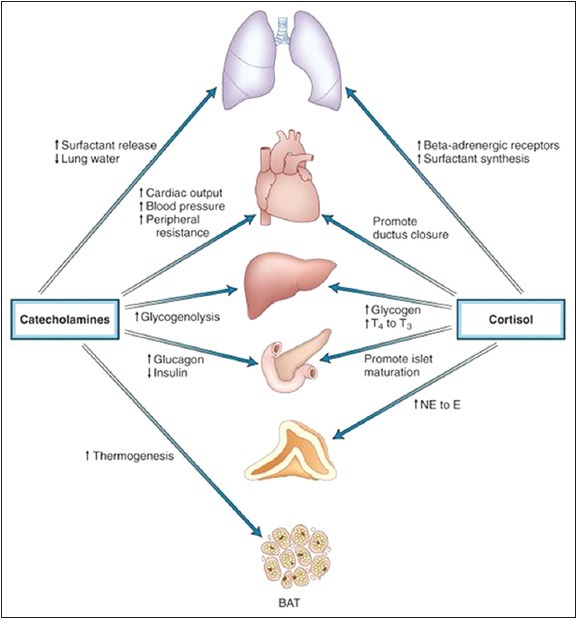
Actions of cortisol and catecholamines during fetal adaptation to the extrauterine environment. The prenatal cortisol surge acts to promote functional maturation of several organ systems as indicated. The neonatal catecholamine surge triggers or potentiates a number of the extrauterine cardiopulmonary and metabolic functional adaptations that are critical to extrauterine survival. See text for details. BAT: Brown adipose tissue, E: Epinephrine, NE: Norepinephrine, T3: Triiodothyronine, T4: Thyroxine
Adrenal cortex and autonomic nervous system are essential for extrauterine adaptation. In long term, there is adaptation to an environment of intermittent nutrient supply and transient substrate deficiency and maturation of the PTH-calcitonin system and the endocrine pancreas.
Cortisol
At term, estrogens stimulate placental 11β HSD leading to increased conversion of cortisol to cortisone. The resulting decrease in maternal-to-fetal cortisol transfer results in stimulation of fetal CRH and corticotropin secretion through the negative-feedback control loop. Placental CRH potentiates fetal adrenal activation resulting in increased cortisol production by the fetal adrenal.[82] The cortisol surge augments surfactant synthesis in lung tissue with increased lung liquid reabsorption; stimulates adrenomedullary PNMT with increased methylation of NE to epinephrine; increases conversion of T4 to T3 by hepatic D1; decreases sensitivity of the ductus arteriosus to prostaglandins, which facilitates ductus closure; induces maturation of several enzymes and transport processes of the small intestine; and stimulates maturation of hepatic enzymes.[83] The increased T3 levels stimulate ß-adrenergic receptor binding, potentiate surfactant synthesis in lung tissue, and increase the sensitivity of brown adipose tissue (BAT) to NE.
Delivery results in high neonatal cortisol levels despite lower plasma corticotropin concentrations. This is due to decreased inhibition of adrenal 3βHSD by estrogen and removal of placental CRH action on fetal pituitary corticotropin release. Plasma DHEAS and DHEA levels fall as the fetal adrenal atrophies.
Catecholamine surge
Parturition evokes catecholamine surge.[84] Plasma norepinephrine (NE) concentrations exceed epinephrine levels because of peripheral and adrenomedullary and paraaortic catecholamine release. These changes lead to increased blood pressure and cardiac inotropic effects; increased glucagon and decreased insulin secretion; increased thermogenesis with increased free fatty acid levels; and pulmonary adaptation by surfactant release.[84]
Others
BAT is the major site of thermogensis, seen maximally around kidney and adrenal glands, peaking at birth.[85] BAT mitochondria rich containing uncoupling protein-1 (UCP-1/thermogenin) that uncouples oxidation and phosphorylation of adenosine diphosphate, reduces ATP production, and consequently enhances thermogenesis.[85] D1 in BAT deiodinates T4 locally to T3, which stimulates thermogenin.[85] Catecholamine release is the stimulus for BAT thermogenesis in early neonatal period
The neonate shifts from a high-calcium environment regulated by PTHrP and calcitonin to a low-calcium environment controlled by PTH and vitamin D. The relatively obtunded PTH response and the high calcitonin levels lead to a 2-3 day period of transient neonatal hypocalcemia.[86] Inhibition of calcitonin secretion and stimulation of PTH secretion gradually result in increased serum calcium levels in the neonate. Low glomerular filtration and reduced renal responsiveness to PTH helps in calcium homeostasis in the newborn. These factors limit phosphate excretion and predispose the neonate to hyperphosphatemia, particularly if the diet includes high-phosphate milk such as unmodified cow's milk. PTH secretion and calcium homeostasis usually return to normal in 1-2 weeks in full-term infants and within 2-3 weeks in the small premature infant
Postbirth TSH increase is due to cooling of the neonate in the extrauterine environment.[34,36] The TSH surge peaks (70 mU/l) at 30 minutes leading to increased secretion of T4 and T3. Additionally increased hepatic conversion of T4 to T3 maintains the T3 level of 105-220 ng/dL. The reequilibration of thyrotropin levels to the normal extrauterine range is probably due to the readjustment of prevailing serum T3 levels and to maturation of feedback control of thyrotropin by thyroid hormones during the early weeks of life.[35,36] Production of rT3 by fetal and neonatal tissues abates by 3-4 weeks of age
Placental withdrawal of glucose supply leads to neonatal hypoglycemia,[78,79] which stimulates glucagon and catecholamine secretion while suppressing insulin secretion. This depletes hepatic glycogen stores. High glucagon/insulin ratio stimulates gluconeogenesis and normalized plasma glucose after 12-18 hours.[79] In the healthy term infant, glucose homeostasis is achieved within 5-7 days of life; in premature infants, 1-2 weeks may be required
Placental extraction leads to fall in fetal estrogen, progesterone, hCG, and HPL levels. The hypoestrogenic state removes major stimulus to fetal pituitary PRL release, and PRL levels decrease within several weeks. The relatively delayed fall may be due to lactotrope hyperplasia in the fetal pituitary or to delayed maturation of hypothalamic dopamine secretion. The gradual fall of GH levels during the early weeks of life is due to delayed maturation of hypothalamic-pituitary feedback control of GH release.[1] IGF-I and IGF-II levels fall to infantile values within a few days, presumably because of the removal of placental HPL and placental IGF production
In male infants, after a transient fall in testosterone levels as the hCG stimulus abates, pituitary LH secretion rebounds with secondary surge of estosterone.[1] In females, a transient, secondary surge in FSH may transiently elevate estrogen levels.
MANIPULATION OF FETAL ENDOCRINE DEVELOPMENT DUE TO ENVIRONMENTAL CHALLENGES
Exposure to nutritional and environmental challenges like hypoglycemia and hypoxemia leads to resetting of fetal growth trajectory and endocrine function. These developmental adaptations result in infertility, hypertension, obesity and insulin insensitivity in adults.[87] Fetus with either a disproportionately large or small placenta leads to altered placenta: Fetal size ratio resulting in increased mortality.
Response to placental insufficiency (hypoglycemia/hypoxemia) [Figure 7]
Figure 7.
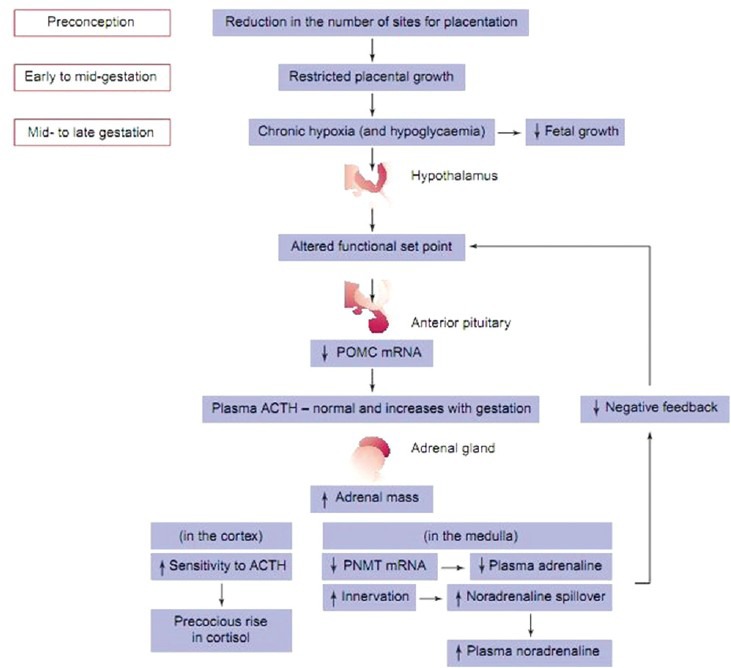
Developmental effect of placental restriction and resulting hypoxia on chronic adaptations in the fetal hypothalamic–pituitary–adrenal axis in the regulation of cortisol and catecholamine secretion in mid- to late-gestation. ACTH: Adrenocorticotrophicvhormone, PNMT: Phenylethanolamine-N-vethyltransferase, POMC: Pro-opiomelanocortin
Prolactin and its receptor in adipose tissue have a critical role in fetal development. When maternal nutrition is enhanced, abundance of prolactin receptor is associated with higher UCP-1 concentration.[85] This facilitates the fetal metabolic response at birth and minimizes risk of hypothermia when volume: Surface area is greater than in singleton births. Placental insufficiency leads to decrease in prolactin receptor in brown adipose tissue and may impact on thermoregulation after birth[88]
Fetal plasma concentrations of anabolic hormones (insulin, IGF-I, prolactin and thyroid hormones) near term are normally decreased (with the exception of IGF-II), in accordance with the degree of fetal hypoxaemia or hypoglycaemia.[89] Plasma concentrations of catabolic hormones like cortisol and catecholamines are higher. The capacity of the fetal adrenal medulla to synthesize catecholamines is potentially impaired from at least mid-gestation in placentally restricted fetuses in view of suppression of adrenaline-synthesizing enzyme PNMT.[90] Even though plasma noradrenaline concentrations are significantly increased in placentally restricted fetuses and there is an inverse relationship between prevailing arterial pO2 and plasma noradrenaline. Enhanced sympathetic innervations of specific fetal tissues in late gestation results in overspill of catecholamines.[91]
Response to malnutrition [Figure 8]
Figure 8.
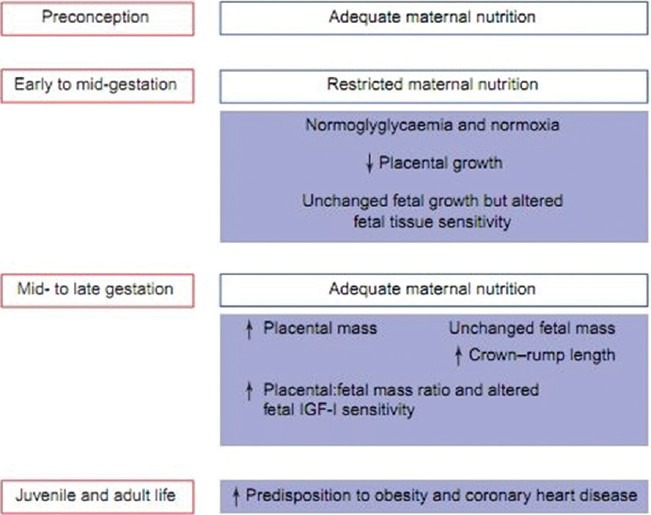
Effect of maternal nutrient restriction in early gestation on the relationship between placental and fetal growth throughout gestation with respect to adaptations in the fetal metabolic and hormonal environment that could contribute to an increased predisposition to adult disease in the resulting offspring. IGF-I: Insulin-like growth factor I
Dietary restriction during early gestation had the greatest effect on the ratio of placental size: Birth weight and resulted in the lowest perceived health. Infants born after dietary restriction in early gestation were longer and heavier having larger placenta. These individuals were at much greater risk of obesity and coronary heart disease, but not hypertension in adulthood.[92] A period of maternal under nutrition of >1 month in mid- or late-gestation is associated with an enlarged placenta at term[93]
There is increase in IGF-II content in skeletal muscle at 80 days of gestation with subsequent decline in IGF-II between mid- and late-gestation. In contrast, IGF-I and IGF-II mRNA as well as growth hormone receptor messenger ribonucleic acid (mRNA) are enhanced in the livers of previously nutrient-restricted fetuses when the mother is subsequently re-fed at term.[94] These adaptations may result in changes in hepatic glucose production in postnatal life
Fetal responses to nutritional deprivation are mediated through changes in maternal or fetal cortisol production and exposure. Excess, or inappropriate, fetal exposure to glucocorticoids reprograms fetal development.[95]
CONCLUSION
Various genes, hormones, and growth factors act as cellular communicating cascade leading to maternal–placental–fetal interactions and fetal maturation. Fetal endocrine system plays a significant role in helping the fetus adjust to extrauterine environment. Use of functional genomics and the ability to determine a large range of molecular responses within fetal organs and tissues after hormonal stimulation should greatly facilitate understanding of the consequences of fetal endocrine adaptation to perturbations of the intrauterine environment.
ACKNOWLEDGEMENTS
All the authors would extend their heartfelt thanks to, Mrs. Sruti Jammula, M Pharm, Dr Jagadeesh Tangudu, M Tech, MS, PhD, and Mrs. Sowmya Jammula, M Tech for their immense and selfless contribution toward manuscript preparation, language editing, and final approval of text.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Grumbach MM, Gluckman PD. The human fetal hypothalamus and pituitary gland: The maturation of neuroendocrine mechanisms controlling secretion of fetal pituitary growth hormone, prolactin, gonadotropins, adrenocorticotropin-related peptides, and thyrotropin. In: Tulchinsky D, Little AB, editors. Maternal Fetal Endocrinology. 2nd ed. Philadelphia: WB Saunders; 1994. pp. 193–261. [Google Scholar]
- 2.Wu Y, He Z, Zhang L, Jiang H, Zhang W. Ontogeny of immunoreactive Lh and Fsh cells in relation to early ovarian differentiation and development in protogynous hermaphroditic ricefield Eel Monopterus albus. Biol Reprod. 2012;86:93. doi: 10.1095/biolreprod.111.095646. [DOI] [PubMed] [Google Scholar]
- 3.Pepe GJ, Albrecht ED. Regulation of the primate fetal adrenal cortex. Endocr Rev. 1990;11:151–76. doi: 10.1210/edrv-11-1-151. [DOI] [PubMed] [Google Scholar]
- 4.Erickson RP, Blecher SR. Genetics of sex determination and differentiation. In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology. 3rd ed. Philadelphia: WB Saunders; 2004. pp. 1935–41. [Google Scholar]
- 5.Lee MM. Molecular genetic control of sex differentiation. In: Pescovitz OH, Eugster EA, editors. Pediatric Endocrinology. Philadelphia: Lippincott Williams and Wilkins; 2004. pp. 231–42. [Google Scholar]
- 6.Harley VR, Clarkson MJ, Argentaro A. The molecular action and regulation of the testis-determining factors, SRY (sex-determining region on the Y chromosome) and SOX9 [SRY-related high-mobility group (HMG) Box9] Endocr Rev. 2003;24:466–87. doi: 10.1210/er.2002-0025. [DOI] [PubMed] [Google Scholar]
- 7.Park SY, Jameson JL. Minireview: Transcriptional regulation of gonadal development and differentiation. Endocrinology. 2005;146:1035–42. doi: 10.1210/en.2004-1454. [DOI] [PubMed] [Google Scholar]
- 8.Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol. 2001;15:172–83. doi: 10.1210/mend.15.1.0582. [DOI] [PubMed] [Google Scholar]
- 9.Aslan AR, Kogan BA, Gondos B. Testicular development. In: Polin RA, Fox WW, Abmans SH, editors. Fetal and Neonatal Physiology. 3rd ed. Philadelphia: WB Saunders; 2004. pp. 1950–5. [Google Scholar]
- 10.Byskov AG, Westergaard LG. Differentiation of the ovary. In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology. 3rd ed. Philadelphila: WB Saunders; 2004. pp. 1941–9. [Google Scholar]
- 11.Fulton N, Martins da Silva SJ, Bayne RA, Anderson RA. Germ cell proliferation and apoptosis in the developing human ovary. J Clin Endocrinol Metab. 2005;90:4664–70. doi: 10.1210/jc.2005-0219. [DOI] [PubMed] [Google Scholar]
- 12.Mesiano S, Jaffe RB. Neuroendocrine-metabolic regulation of pregnancy. In: Strauss JF 3rd, Barbieri RL, editors. Reproductive Endocrinology. 5th ed. Philadelphia: WB Saunders; 2004. pp. 327–66. [Google Scholar]
- 13.Diczfalusy E. Endocrine functions of the human fetoplacental unit. 1964. Am J Obstet Gynecol. 2005;193:2024. doi: 10.1016/j.ajog.2005.02.117. [DOI] [PubMed] [Google Scholar]
- 14.Sajjad Y, Quenby S, Nickson P, Lewis-Jones DI, Vince G. Immunohistochemical localization of androgen receptors in the urogenital tracts of human embryos. Reproduction. 2004;128:331–9. doi: 10.1530/rep.1.00227. [DOI] [PubMed] [Google Scholar]
- 15.Josso N, Belville C, Dicard JY. Mutations in AMH and its receptors. Endocrinology. 2003;13:247–51. [Google Scholar]
- 16.Rouiller-Fabre V, Carmona S, Merhi RA, Cate R, Habert R, Vigier B. Effect of anti-Mullerian hormone on Sertoli and Leydig cell functions in fetal and immature rats. Endocrinology. 1998;139:1213–20. doi: 10.1210/endo.139.3.5785. [DOI] [PubMed] [Google Scholar]
- 17.Brandenberger AW, Tee MK, Lee JY, Chao V, Jaffe RB. Tissue distribution of estrogen receptors alpha (ER-alpha) and beta (ER-beta) mRNA in the midgestational human fetus. J Clin Endocrinol Metab. 1997;82:3509–12. doi: 10.1210/jcem.82.10.4400. [DOI] [PubMed] [Google Scholar]
- 18.Couse JF, Korach KS. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Falgueras A, Pinos H, Collado P, Pasaro E, Fernandez R, Jordan CL, et al. The role of the androgen receptor in CNS masculinization. Brain Res. 2005;1035:13–23. doi: 10.1016/j.brainres.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 20.Naftolin F, Brawer JR. The effect of estrogens on hypothalamic structure and function. Am J Obstet Gynecol. 1978;132:758–65. doi: 10.1016/s0002-9378(78)80010-6. [DOI] [PubMed] [Google Scholar]
- 21.Colciago A, Negri-Cesi P, Pravettoni A, Mornati O, Casati L, Celotti F. Prenatal Aroclor 1254 exposure and brain sexual differentiation: Effect on the expression of testosterone metabolizing enzymes and androgen receptors in the hypothalamus of male and female rats. Reprod Toxicol. 2006;22:738–45. doi: 10.1016/j.reprotox.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Wood CE. Estrogen/hypothalamus-pituitary-adrenal axis interactions in the fetus: The interplay between placenta and fetal brain. J Soc Gynecol Investig. 2005;12:67–76. doi: 10.1016/j.jsgi.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 23.Myers DA, McDonald TJ, Nathanielsz PW. Effect of bilateral lesions of the ovine fetal hypothalamic paraventricular nuclei at 118-122 days of gestation on subsequent adrenocortical steroidogenic enzyme gene expression. Endocrinology. 1992;131:305–10. doi: 10.1210/endo.131.1.1612010. [DOI] [PubMed] [Google Scholar]
- 24.Snegovskikh V, Park JS, Norwitz ER. Endocrinology of parturition. Endocrinol Metab Clin North Am. 2006;35:173–91. doi: 10.1016/j.ecl.2005.09.012. viii. [DOI] [PubMed] [Google Scholar]
- 25.Rivier C, Vale W. Neuroendocrine interactions between corticotrophin releasing factor and vasopressin on adrenocorticotropic hormone secretion in the rat. In: Schrier WW, editor. Vasopressin. New York: Raven; 1985. pp. 181–8. [Google Scholar]
- 26.Winter JS. Fetal and neonatal adrenocortical physiology. In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology. Philadelphia: WB Saunders; 2004. pp. 1915–25. [Google Scholar]
- 27.Geller DH, Miller WL. Molecular development of the adrenal gland. In: Pescovitz OH, Eugster EA, editors. Pediatric Endocrinology. Philadelphia: Lippincott Williams and Wilkins; 2004. pp. 548–67. [Google Scholar]
- 28.Pepe GJ, Albrecht ED. Central integrative role of oestrogen in the regulation of placental steroidogenic maturation and the development of the fetal pituitary-adrenocortical axis in the baboon. Hum Reprod Update. 1998;4:406–19. doi: 10.1093/humupd/4.4.406. [DOI] [PubMed] [Google Scholar]
- 29.Yang K, Jones SA, Challis JR. Changes in glucocorticoid receptor number in the hypothalamus and pituitary of the sheep fetus with gestational age and after adrenocorticotropin treatment. Endocrinology. 1990;126:11–7. doi: 10.1210/endo-126-1-11. [DOI] [PubMed] [Google Scholar]
- 30.Noguchi KK, Lau K, Smith DJ, Swiney BS, Farber NB. Glucocorticoid receptor stimulation and the regulation of neonatal cerebellar neural progenitor cell apoptosis. Neurobiol Dis. 2011;43:356–63. doi: 10.1016/j.nbd.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, et al. Mineralocorticoid receptor knockout mice: Pathophysiology of Na+metabolism. Proc Natl Acad Sci U S A. 1998;95:9424–9. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beitins IZ, Graham GG, Kowarski A, Migeon CJ. Adrenal function in normal infants and in marasmus and kwashiorkor: Plasma aldosterone concentration and aldosterone secretion rate. J Pediatr. 1974;84:444–51. doi: 10.1016/s0022-3476(74)80737-7. [DOI] [PubMed] [Google Scholar]
- 33.Sklar CA, Mueller PL, Gluckman PD, Kaplan SL, Rudolph AM, Grumbach MM. Hormone ontogeny in the ovine fetus. VII. Circulating luteinizing hormone and follicle-stimulating hormone in mid- and late gestation. Endocrinology. 1981;108:874–80. doi: 10.1210/endo-108-3-874. [DOI] [PubMed] [Google Scholar]
- 34.Burrow GN, Fisher DA, Larsen PR. Maternal and fetal thyroid function. N Engl J Med. 1994;331:1072–8. doi: 10.1056/NEJM199410203311608. [DOI] [PubMed] [Google Scholar]
- 35.Fisher DA, Nelson JC, Carlton EI, Wilcox RB. Maturation of human hypothalamic-pituitary-thyroid function and control. Thyroid. 2000;10:229–34. doi: 10.1089/thy.2000.10.229. [DOI] [PubMed] [Google Scholar]
- 36.Brown RS, Huang SA, Fisher DA. The maturation of thyroid function in the perinatal period and during childhood. In: Braverman LE, Utiger RD, editors. The Thyroid. 9th ed. Philadelphia: JB Lippincott; 2005. pp. 1013–28. [Google Scholar]
- 37.Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Role of thyroid hormone during early brain development. Eur J Endocrinol. 2004;151(Suppl 3):U25–37. doi: 10.1530/eje.0.151u025. [DOI] [PubMed] [Google Scholar]
- 38.Santisteban P. Development and anatomy of the hypothalamic-pituitary axis. In: Braverman LE, Utiger RD, editors. The Thyroid. 9th ed. Philadelphia: JB Lippincott; 2005. pp. 8–25. [Google Scholar]
- 39.Parlato R, Rosica A, Rodriguez-Mallon A, Affuso A, Postiglione MP, Arra C, et al. An integrated regulatory network controlling survival and migration in thyroid organogenesis. Dev Biol. 2004;276:464–75. doi: 10.1016/j.ydbio.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 40.Trueba SS, Augé J, Mattei G, Etchevers H, Martinovic J, Czernichow P, et al. PAX8, TITF1, and FOXE1 gene expression patterns during human development: New insights into human thyroid development and thyroid dysgenesis-associated malformations. J Clin Endocrinol Metab. 2005;90:455–62. doi: 10.1210/jc.2004-1358. [DOI] [PubMed] [Google Scholar]
- 41.Eng PH, Cardona GR, Fang SL, Previti M, Alex S, Carrasco N, et al. Escape from the acute Wolff-Chaikoff effect is associated with a decrease in thyroid sodium/iodide symporter messenger ribonucleic acid and protein. Endocrinology. 1999;140:3404–10. doi: 10.1210/endo.140.8.6893. [DOI] [PubMed] [Google Scholar]
- 42.Sherwin JR. Development of regulatory mechanisms in the thyroid: Failure of iodide to suppress iodide transport activity. Proc Soc Exp Biol Med. 1982;169:458–62. doi: 10.3181/00379727-169-41375. [DOI] [PubMed] [Google Scholar]
- 43.Hume R, Simpson J, Delahunty C, van Toor H, Wu SY, Williams FL, et al. Human fetal and cord serum thyroid hormones: Developmental trends and interrelationships. J Clin Endocrinol Metab. 2004;89:4097–103. doi: 10.1210/jc.2004-0573. [DOI] [PubMed] [Google Scholar]
- 44.Polk DH, Reviczky A, Wu SY, Huang WS, Fisher DA. Metabolism of sulfoconjugated thyroid hormone derivatives in developing sheep. Am J Physiol. 1994;266:E892–6. doi: 10.1152/ajpendo.1994.266.6.E892. [DOI] [PubMed] [Google Scholar]
- 45.Richard K, Hume R, Kaptein E, Stanley EL, Visser TJ, Coughtrie MW. Sulfation of thyroid hormone and dopamine during human development: Ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J Clin Endocrinol Metab. 2001;86:2734–42. doi: 10.1210/jcem.86.6.7569. [DOI] [PubMed] [Google Scholar]
- 46.St Germain DL, Hernandez A, Schneider MJ, Galton VA. Insights into the role of deiodinases from studies of genetically modified animals. Thyroid. 2005;15:905–16. doi: 10.1089/thy.2005.15.905. [DOI] [PubMed] [Google Scholar]
- 47.Yen P. Genomic and nongenomic actions of thyroid hormones. In: Braverman LE, Utiger RD, editors. The Thyroid. 9th ed. Philadelphia: Lippincott Williams and Wilkins; 2005. pp. 135–50. [Google Scholar]
- 48.Kilby MD, Gittoes N, McCabe C, Verhaeg J, Franklyn JA. Expression of thyroid receptor isoforms in the human fetal central nervous system and the effects of intrauterine growth restriction. Clin Endocrinol (Oxf) 2000;53:469–77. doi: 10.1046/j.1365-2265.2000.01074.x. [DOI] [PubMed] [Google Scholar]
- 49.Iskaros J, Pickard M, Evans I, Sinha A, Hardiman P, Ekins R. Thyroid hormone receptor gene expression in first trimester human fetal brain. J Clin Endocrinol Metab. 2000;85:2620–3. doi: 10.1210/jcem.85.7.6766. [DOI] [PubMed] [Google Scholar]
- 50.Rajatapiti P, Kester MH, de Krijger RR, Rottier R, Visser TJ, Tibboel D. Expression of glucocorticoid, retinoid, and thyroid hormone receptors during human lung development. J Clin Endocrinol Metab. 2005;90:4309–14. doi: 10.1210/jc.2005-0556. [DOI] [PubMed] [Google Scholar]
- 51.Anderson LL, Jeftinija S, Scanes CG. Growth hormone secretion: Molecular and cellular mechanisms and in vivo approaches. Exp Biol Med (Maywood) 2004;229:291–302. doi: 10.1177/153537020422900403. [DOI] [PubMed] [Google Scholar]
- 52.DeLeon DD, Cohen P, Katz LE. Growth factor regulation of fetal growth. In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology. 3rd ed. Philadelphia: WB Saunders; 2004. pp. 1880–90. [Google Scholar]
- 53.Kind KL, Owens JA, Robinson JS, Quinn KJ, Grant PA, Walton PE, et al. Effect of restriction of placental growth on expression of IGFs in fetal sheep: Relationship to fetal growth, circulating IGFs and binding proteins. J Endocrinol. 1995;146:23–34. doi: 10.1677/joe.0.1460023. [DOI] [PubMed] [Google Scholar]
- 54.Clément-Lacroix P, Ormandy C, Lepescheux L, Ammann P, Damotte D, Goffin V, et al. Osteoblasts are a new target for prolactin: Analysis of bone formation in prolactin receptor knockout mice. Endocrinology. 1999;140:96–105. doi: 10.1210/endo.140.1.6436. [DOI] [PubMed] [Google Scholar]
- 55.Leake RD, Fisher DA. Ontogeny of vasopressin in man. In: Czernichow P, Robinson AG, editors. Diabetes Insipidus in Man, Frontiers in Hormone Research. Vol. 13. Basel: S Karger; 1985. pp. 42–51. [Google Scholar]
- 56.Leake RD. The fetal-maternal neurohypophysial system. In: Polin RA, Fox WW, editors. Fetal and Neonatal Physiology. 2nd ed. Philadelphia: WB Saunders; 1998. pp. 2442–6. [Google Scholar]
- 57.Wood CE, Tong H. Central nervous system regulation of reflex responses to hypotension during fetal life. Am J Physiol. 1999;277:R1541–52. doi: 10.1152/ajpregu.1999.277.6.R1541. [DOI] [PubMed] [Google Scholar]
- 58.Zhao X, Nijland MJ, Ervin MG, Ross MG. Regulation of hypothalamic arginine vasopressin messenger ribonucleic acid and pituitary arginine vasopressin content in fetal sheep: Effects of acute tonicity alterations and fetal maturation. Am J Obstet Gynecol. 1998;179:899–905. doi: 10.1016/s0002-9378(98)70186-3. [DOI] [PubMed] [Google Scholar]
- 59.DeVane GW, Porter JC. An apparent stress-induced release or arginine vasopressin by human neonates. J Clin Endocrinol Metab. 1980;51:1412–6. doi: 10.1210/jcem-51-6-1412. [DOI] [PubMed] [Google Scholar]
- 60.Matthews SG, Challis JR. Regulation of CRH and AVP mRNA in the developing ovine hypothalamus: Effects of stress and glucocorticoids. Am J Physiol. 1995;268:E1096–107. doi: 10.1152/ajpendo.1995.268.6.E1096. [DOI] [PubMed] [Google Scholar]
- 61.Devuyst O, Burrow CR, Smith BL, Agre P, Knepper MA, Wilson PD. Expression of aquaporins-1 and -2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am J Physiol. 1996;271:F169–83. doi: 10.1152/ajprenal.1996.271.1.F169. [DOI] [PubMed] [Google Scholar]
- 62.Baum MA, Ruddy MK, Hosselet CA, Harris HW. The perinatal expression of aquaporin-2 and aquaporin-3 in developing kidney. Pediatr Res. 1998;43:783–90. doi: 10.1203/00006450-199806000-00011. [DOI] [PubMed] [Google Scholar]
- 63.Visser M, Swaab DF. Life span changes in the presence of alpha-melanocyte-stimulating-hormone-containing cells in the human pituitary. J Dev Physiol. 1979;1:161–78. [PubMed] [Google Scholar]
- 64.Perry RA, Mulvogue HM, McMillen IC, Robinson PM. Immunohistochemical localization of ACTH in the adult and fetal sheep pituitary. J Dev Physiol. 1985;7:397–404. [PubMed] [Google Scholar]
- 65.Glickman JA, Carson GD, Challis JR. Differential effects of synthetic adrenocorticotropin1-24 and alpha-melanocyte-stimulating hormone on adrenal function in human and sheep fetuses. Endocrinology. 1979;104:34–9. doi: 10.1210/endo-104-1-34. [DOI] [PubMed] [Google Scholar]
- 66.Swaab DF, Martin JT. Functions of alpha-melanotropin and other opiomelanocortin peptides in labour, intrauterine growth and brain development. Ciba Found Symp. 1981;81:196–217. doi: 10.1002/9780470720646.ch12. [DOI] [PubMed] [Google Scholar]
- 67.Fora MA, Butler TG, Rose JC, Schwartz J. Adrenocorticotropin secretion by fetal sheep anterior and intermediate lobe pituitary cells in vitro: Effects of gestation and adrenalectomy. Endocrinology. 1996;137:3394–400. doi: 10.1210/endo.137.8.8754766. [DOI] [PubMed] [Google Scholar]
- 68.Mesiano S, Jaffe RB. Neuroendocrine-metabolic regulation of pregnancy. In: Strauss JF III, Barbieri RL, editors. Reproductive Endocrinology. 5th ed. Philadelphia: WB Saunders; 2004. pp. 327–66. [Google Scholar]
- 69.Wu P, Jackson IM. Identification, characterization and localization of thyrotropin-releasing hormone precursor peptides in perinatal rat pancreas. Regul Pept. 1988;22:347–60. doi: 10.1016/0167-0115(88)90111-5. [DOI] [PubMed] [Google Scholar]
- 70.Koshimizu T. The development of pancreatic and gastrointestinal somatostatin-like immunoreactivity and its relationship to feeding in neonatal rats. Endocrinology. 1983;112:911–6. doi: 10.1210/endo-112-3-911. [DOI] [PubMed] [Google Scholar]
- 71.Rahier J, Wallon J, Henquin JC. Abundance of somatostatin cells in the human neonatal pancreas. Diabetologia. 1980;18:251–4. doi: 10.1007/BF00251925. [DOI] [PubMed] [Google Scholar]
- 72.Leduque P, Aratan-Spire S, Czernichow P, Dubois PM. Ontogenesis of thyrotropin-releasing hormone in the human fetal pancreas. A combined radioimmunological and immunocytochemical study. J Clin Invest. 1986;78:1028–34. doi: 10.1172/JCI112657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Polk DH, Reviczky A, Lam RW, Fisher DA. Thyrotropin-releasing hormone in ovine fetus: Ontogeny and effect of thyroid hormone. Am J Physiol. 1991;260:E53–8. doi: 10.1152/ajpendo.1991.260.1.E53. [DOI] [PubMed] [Google Scholar]
- 74.Hochberg Ze’ev, Tiosano D. Disorders of mineral metabolism. In: Pescovitz OH, Eugster EA, editors. Pediatric Endocrinology. Philadelphia: Lippincott Williams and Wilkins; 2004. pp. 614–40. [Google Scholar]
- 75.Prada JA. Calcium-regulating hormones. In: Polin RA, Fox WW, editors. Fetal and Neonatal Physiology. 2nd ed. Philadelphia: WB Saunders; 1998. pp. 2287–96. [Google Scholar]
- 76.Ross R, Halbert K, Tsang RC. Determination of the production and metabolic clearance rates of 1,25-dihydroxyvitamin D3 in the pregnant sheep and its chronically catheterized fetus by primed infusion technique. Pediatr Res. 1989;26:633–8. doi: 10.1203/00006450-198912000-00024. [DOI] [PubMed] [Google Scholar]
- 77.Edlund H. Pancreatic organogenesis--developmental mechanisms and implications for therapy. Nat Rev Genet. 2002;3:524–32. doi: 10.1038/nrg841. [DOI] [PubMed] [Google Scholar]
- 78.Sperling MA. Carbohydrate metabolism: Insulin and glucagons. In: Tulchinsky D, Little AB, editors. Maternal-Fetal Endocrinology. 2nd ed. Philadelphia: WB Saunders; 1994. pp. 380–400. [Google Scholar]
- 79.Girard J. Control of fetal and neonatal glucose metabolism by pancreatic hormones. Baillieres Clin Endocrinol Metab. 1989;3:817–36. doi: 10.1016/s0950-351x(89)80055-2. [DOI] [PubMed] [Google Scholar]
- 80.Ammon HP, Glocker C, Waldner RG, Wahl MA. Insulin release from pancreatic islets of fetal rats mediated by leucine β-BCH, tolbutamide, glibenclamide, arginine, potassium chloride, and theophylline does not require stimulation of Ca2+net uptake. Cell Calcium. 1989;10:441–50. doi: 10.1016/0143-4160(89)90035-3. [DOI] [PubMed] [Google Scholar]
- 81.Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, et al. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287:E405–13. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 82.Pepe GJ, Albrecht ED. Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocr Rev. 1995;16:608–48. doi: 10.1210/edrv-16-5-608. [DOI] [PubMed] [Google Scholar]
- 83.Jobe AH, Newnham JP, Moss TJ, Ikegami M. Differential effects of maternal betamethasone and cortisol on lung maturation and growth in fetal sheep. Am J Obstet Gynecol. 2003;188:22–8. doi: 10.1067/mob.2003.61. [DOI] [PubMed] [Google Scholar]
- 84.Padbury JF. Functional maturation of the adrenal medulla and peripheral sympathetic nervous system. Baillieres Clin Endocrinol Metab. 1989;3:689–705. doi: 10.1016/s0950-351x(89)80049-7. [DOI] [PubMed] [Google Scholar]
- 85.Lee JY, Takahashi N, Yasubuchi M, Kim YI, Hashizaki H, Kim MJ, et al. Triiodothyronine induces UCP-1 expression and mitochondrial biogenesis in human adipocytes. Am J Physiol Cell Physiol. 2012;302:C463–72. doi: 10.1152/ajpcell.00010.2011. [DOI] [PubMed] [Google Scholar]
- 86.Fthenakis GC, Arsenos G, Brozos C, Fragkou IA, Giadinis ND, Giannenas I, et al. Health management of ewes during pregnancy. Anim Reprod Sci. 2012;130:198–212. doi: 10.1016/j.anireprosci.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 87.Symonds ME, Budge H, Stephenson T, McMillen IC. Fetal endocrinology and development--manipulation and adaptation to long-term nutritional and environmental challenges. Reproduction. 2001;121:853–62. doi: 10.1530/rep.0.1210853. [DOI] [PubMed] [Google Scholar]
- 88.Budge H, Dandrea J, Ingleton P, Stephenson T, Symonds ME. Effect of fetal number on prolactin receptor abundance in perirenal adipose tissue of neonatal lambs. Early Hum Dev. 2000;58:72. doi: 10.1016/s0378-3782(00)00059-1. [DOI] [PubMed] [Google Scholar]
- 89.Heltemes A, Gingery A, Soldner EL, Bozadjieva N, Jahr KN, Johnson BK, et al. Chronic placental ischemia alters amniotic fluid milieu and results in impaired glucose tolerance, insulin resistance and hyperleptinemia in young rats. Exp Biol Med (Maywood) 2010;235:892–9. doi: 10.1258/ebm.2010.009357. [DOI] [PubMed] [Google Scholar]
- 90.Coulter CL, McMillen IC, Robinson JS, Owens JA. Placental restriction alters adrenal medullary development in the midgestation sheep fetus. Pediatr Res. 1998;44:656–62. doi: 10.1203/00006450-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 91.Edwards LJ, McMillen IC. Impact of maternal undernutrition during the periconceptional period, fetal number, and fetal sex on the development of the hypothalamo-pituitary adrenal axis in sheep during late gestation. Biol Reprod. 2002;66:1562–9. doi: 10.1095/biolreprod66.5.1562. [DOI] [PubMed] [Google Scholar]
- 92.Luo ZC, Nuyt AM, Delvin E, Audibert F, Girard I, Shatenstein B, et al. Maternal and fetal IGF-I and IGF-II levels, fetal growth, and gestational diabetes. J Clin Endocrinol Metab. 2012;97:1720–8. doi: 10.1210/jc.2011-3296. [DOI] [PubMed] [Google Scholar]
- 93.Yajnik CS, Deshmukh US. Fetal programming: Maternal nutrition and role of one-carbon metabolism. Rev Endocr Metab Disord. 2012;13:121–7. doi: 10.1007/s11154-012-9214-8. [DOI] [PubMed] [Google Scholar]
- 94.Törölä H, Lehtihalmes M, Yliherva A, Olsén P. Feeding skill milestones of preterm infants born with extremely low birth weight (ELBW) Infant Behav Dev. 2012;35:187–94. doi: 10.1016/j.infbeh.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 95.Field T, Hernandez-Reif M, Diego M, Figueiredo B, Schanberg S, Kuhn C. Prenatal cortisol, prematurity and low birthweight. Infant Behav Dev. 2006;29:268–75. doi: 10.1016/j.infbeh.2005.12.010. [DOI] [PubMed] [Google Scholar]