

Abstract
Phosphorus is a common anion. It plays an important role in energy generation. Renal phosphate handling is regulated by three organs parathyroid, kidney and bone through feedback loops. These counter regulatory loops also regulate intestinal absorption and thus maintain serum phosphorus concentration in physiologic range. The parathyroid hormone, vitamin D, Fibrogenic growth factor 23 (FGF23) and klotho coreceptor are the key regulators of phosphorus balance in body.
Keywords: Klotho, phosphorus, parathyroid hormone, vitamin D
INTRODUCTION
Phosphorus is a common anion, distributed ubiquitously throughout the body. Approximately 80%-85% of the ~600 gm of total body phosphorus is present in the skeleton. The remaining is distributed widely in the form of organic phosphate compounds that play fundamental roles in many aspects of cellular metabolism. The maintenance of normal phosphate homeostasis is critical for diverse physiologic processes including cell signaling, nucleic acid synthesis, energy homeostasis, formation of lipid bilayers, and bone formation. The concentration of phosphorus influences the activity of many metabolic pathways such as ammoniagenesis, glycolysis, gluconeogenesis, parathyroid hormone (PTH) secretion, and phosphate reabsorption, as well as the formation of 1,25-dihydroxycholecalciferol[1,25(OH) 2D3] from 25-hydroxycholecalciferol [25(OH) D3].
PHOSPHORUS IN BLOOD
Plasma phosphate is present in ionized, complexed, and protein-bound forms. The normal concentration of phosphorus in plasma is 3-4.5 mg/dL and it is present in plasma primarily as HPO4-2 and H2PO4-1.[1] If phosphate were totally filterable through artificial and glomerular membranes, its concentration in the ultrafiltrate would be 1.18 times that of plasma, however, measured ultrafilterable (UF) phosphate to plasma phosphate ratios have been found to range from 0.89-0.96, indicating that about 25% of plasma phosphate is bound to protein. Of the UF phosphate, approximately 60% is ionized and 40% is complexed to the major plasma cations, mainly Ca2+, Mg2+, and Na+. The fraction of total phosphate that is UF declines with hypercalcemia, probably due to the formation of calcium-phosphate-proteinate complexes.
PHOSPHORUS IN CELL
Intracellular phosphate is primarily sequestered in intracellular organelles, or incorporated into organic compounds like adenosine phosphates, creatine phosphate and in erythrocytes, 2,3-diphosphoglycerate. The cytosolic free inorganic phosphate concentration is only about 1 μM. Nevertheless, this is above its electrochemical equilibrium value as predicted from the membrane potential, suggesting that there must be active transport of phosphate into cells. The regulation of intracellular phosphate levels is closely linked to cellular metabolic activity. Inhibition of phosphate uptake impairs cellular metabolic function, whereas increasing extracellular phosphate concentration stimulates mitochondrial respiration. Conversely, bathing cells in glucose reduces phosphate uptake and in conditions of limited phosphate availability reduces mitochondrial respiration, oxidative phosphorylation, and ATP content, a phenomenon called the Crabtree effect.
ABSORPTION AND REABSORPTION OF PHOSPHATE IN INTESTINE AND KIDNEY
In mammals, absorption and reabsorption of phosphate take place primarily in the intestine and kidney, respectively. The daily dietary intake of phosphate is 800 mg-1500 mg. Dietary deficiency of phosphorus is rare as phosphate is found in many foods, including dairy products, meat, and cereal grains. Approximately 65% of ingested phosphate is absorbed, primarily by the duodenum and jejunum [Figure 1] and it varies proportionately with dietary intake. Under the influence of vitamin D, phosphorus transport takes place in proximal segments of the small intestine and appears to involve both passive and active components. The movement of phosphorus from the intestinal lumen to the blood requires (a) transport across the luminal brush-border membrane of the intestine; (b) transport through the cytoplasm; and (c) transport across the basolateral plasma membrane of the epithelium. The rate-limiting step and the main driving force of absorption is the luminal membrane step.[2] Polyvalent cations present in diet such as Ca2+, Mg2+, and Al3+ bind to intestinal luminal phosphate and decrease its absorption. That is why phosphate binder in the patients with renal failure is prescribed with meals. Secreted digestive juices contain about 3 mg/kg/day of phosphate. Once absorbed, phosphate in the extracellular fluid may exchange with the pool in bone, with 200 mg of phosphate typically entering and leaving the skeleton daily as it is continuously remodeled. Ultimately, the kidneys are responsible for the excretion of a substantial excess of phosphate, about 700-900 mg per day. During periods of growth, a greater proportion of phosphate is retained for bone deposition, but this still constitutes a small percentage of dietary intakes. Renal phosphate excretion is the principal mechanism by which the body regulates extracellular phosphate balance [Figure 1].
Figure 1.
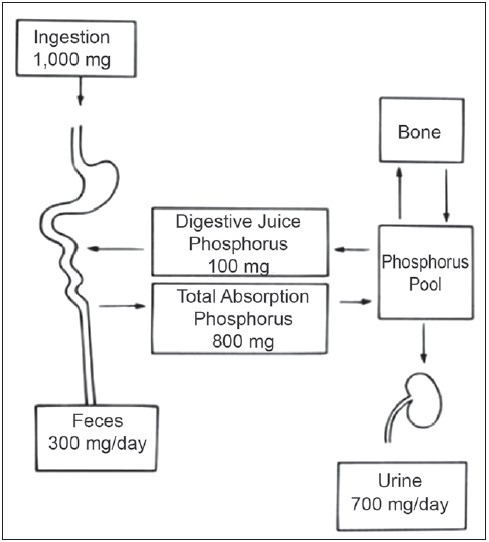
Phosphate balance at level of intestine and kidney
RENAL REABSORPTION OF PHOSPHORUS
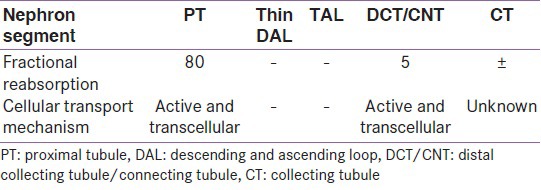
Most of the inorganic phosphorus (Pi) in serum (90%-95%) is UF at the level of the glomerulus. Approximately 7 g of phosphorus is filtered daily by the kidney, at physiologic levels of serum phosphorus, of which 80%-90% is reabsorbed by the renal tubules and the remainder is excreted in the urine (approximately 700 mg) equal to intestinal absorption.[3] So at steady-state, adults are in a state of balance between intake and excretion of phosphorus [Figure 1]. Evidences have demonstrated that 60%-70% of the filtered phosphorus is reabsorbed in the proximal tubule and there is also evidence that a significant amount of filtered phosphorus is reabsorbed in distal segments of the nephron [Table 1].[4] When serum phosphorus level increases and the filtered load of phosphorus increases, the capacity to reabsorb phosphorus also increases. However, a maximum rate of transport (Tm) for phosphorus reabsorption is obtained usually at serum phosphorus concentrations of 6 mg/dL. There is a direct correlation between Tm phosphorus values and glomerular filtration rate (GFR) even when the GFR is varied over a broad range. Micro puncture studies suggest two different mechanisms responsible for phosphorus reabsorption in the proximal tubule. In the first third of the proximal tubule, in which only 10%-15% of the filtered sodium and fluid is reabsorbed, the ratio of tubular fluid (TF) phosphorus to plasma ultra-filterable (UF) phosphorus falls to values of approximately 0.6. This indicates that the first third of the proximal tubule accounts for approximately 50% of the total amount of phosphorus reabsorbed in this segment of the nephron. In the last two-thirds of the proximal tubule, the reabsorption of phosphorus parallels the movement of salt and water. In the remaining 70% of the pars convoluta, the TF: UF phosphorous ratio remains at a value of 0.6-0.7, whereas fluid reabsorption increases to approximately 60%-70% of the filtered load. Thus, in the last two-thirds of proximal tubule, the TF: UF phosphorus reabsorption ratio is directly proportional to sodium and fluid reabsorption. A significant amount of phosphorus, perhaps on the order of 20%-30%, is reabsorbed beyond the portion of the proximal tubule that is accessible to micropuncture. There is little phosphorus transport within the loop of Henle, with most transport distal to micropuncture accessibility occurring in the distal convoluted tubule. In this location, Pastoriza-Munoz et al.[4] found that approximately 15% of filtered phosphorus is reabsorbed under baseline conditions in animals subjected to parathyroidectomy, but that value falls to about 6% after administration of large doses of PTH. The collecting duct is a potential site for distal nephron reabsorption of phosphorus.[5,6,7] Transport in this nephron segment may explain the discrepancy between the amount of phosphorus delivered to the late distal tubule in micropuncture studies and the considerably smaller amount of phosphorus that appears in the final urine of the same kidney. Phosphorus transport in the cortical collecting tubule is independent of regulation by PTH. This is in agreement with the absence of PTH-dependent adenylate cyclase in the cortical collecting tubule.
Table 1.
Segmental handling of phosphate along with renal tubule
MOLECULAR AND CELLULAR MECHANISM OF PHOSPHATE REGULATION
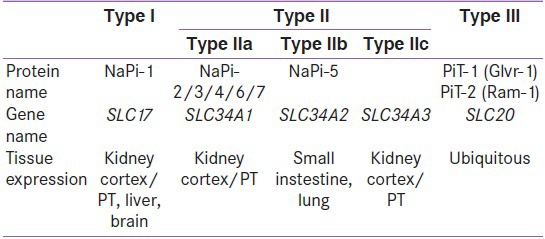
Because the movement of Pi into the cell does not occur by simple diffusion, 3 families of NA-coupled Pi co transporters (NAPT) mediate the transport of Pi across the cell membrane[8] [Figure 2]. The NA-coupled Pi co-transporters that are important in Pi uptake in vertebrates belongs to two large families, the NAPT type II and the NAPT type III families [Table 2]. The major role of NAPT type II has been clearly demonstrated in Npt2-/-(knockout) mice. Compared to wild-type mice, the Npt2-/-mice display a higher urinary excretion of Pi and lower NAPT co-transport activity (mainly mediated by NAPT-IIa and additionally by NAPT-IIc) in the brush border membranes (BBM) of renal proximal tubules.[9] This suggests that the regulation of Pi reabsorption is achieved mainly by controlling the apical expression of NAPT-IIa in BBM of renal proximal tubules.[10,11] Consistently, phosphaturic factors reduce the expression of NAPT-IIa, whereas factors that increase renal Pi reabsorption increase NAPT-IIa in BBM. Therefore, control mechanisms that regulate apical expression and membrane retrieval of NAPT-IIa are essential to our understanding of how Pi homeostasis is achieved.
Figure 2.
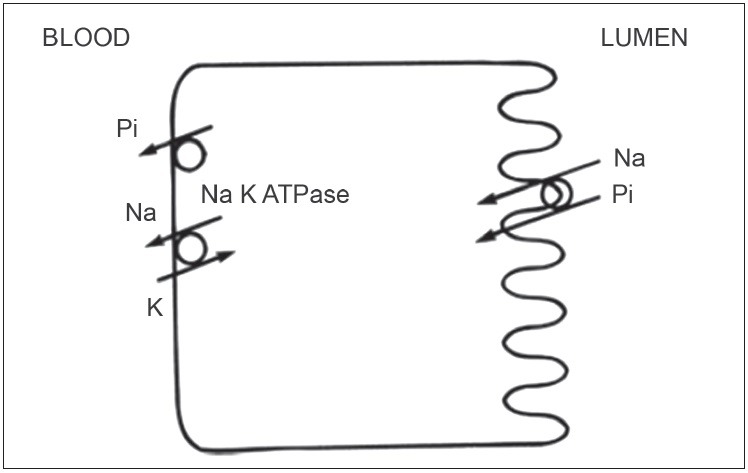
Molecular and cellular mechanism of phosphate handling at tubular cell membrane
Table 2.
The three families of Na-Pi co-tranasporters
REGULATION OF PHOSPHATE ABSORPTION IN RENAL TUBULE
NAPT co-transport activity and organ specific Pi absorptive processes are regulated by PTH and 1,25(OH) 2D, which interact in a coordinate fashion to regulate Pi homeostasis. In the kidney, various factors, most importantly PTH, influence the efficiency of renal Pi reabsorption.[12] Animals fed with a low-Pi diet have decreased serum Pi concentrations that are associated with a reciprocal increase in circulating Ca++ concentrations. The increase in serum Ca++ inhibits PTH release, which in turn reduces the renal excretion of Pi into the urine. In addition, a low-Pi diet and reductions in serum Pi are associated with increased 1,25(OH) vit D synthesis,[13] which consequently restores serum Pi concentrations to normal state by increasing intestinal (mediated via NAPT-IIb) and renal Pi recoveries. It is, however, difficult to discriminate between direct versus indirect effects of 1,25(OH) 2D on renal Pi reabsorption, as in vivo, the vitamin D status is closely associated with alterations in plasma calcium and PTH concentrations. Conversely, when animals are fed a high-Pi diet, serum Ca++ concentrations decrease, and PTH release is increased. Recently, Martin et al. suggested that changes in PTH secretion in response to dietary Pi occur rapidly (within 10 min) and independently of changes in serum Pi or Ca++ concentrations,[13] suggesting that a signal emanating from the intestine may affect PTH secretion.[14] An elevation in serum Pi after a high-Pi meal also reduces 1,25(OH) 2D synthesis and intestinal Pi absorption.
Other than PTH and 1,25(OH) 2D, several phosphaturic peptides such as fibroblast growth factor 23 (FGF23), secreted frizzled related protein 4, matrix extracellular phosphoglycoprotein, and FGF7 have been demonstrated to inhibit the activity of NAPT-IIa co-transporters in renal epithelial cells.[12,15,16,17,18,19,20] Furthermore, current studies have strongly suggested the importance of the network of proteins interacting with NAPT-IIa co-transporters in apical membrane expression of NAPT-IIa co-transporters and the fate of endocytosed NAPT-IIa co-transporters.[21]
RELATIONSHIP OF PTH, FGF23, KLOTHO, AND PHOSPHATE REGULATION
The regulation of Pi is complex and involves both acute and chronic processes. Alteration in PTH-vitamin D axis do not completely explains the pathogenesis of several hypophosphatemic disorders and normal Pi physiology and therefore, the role of phosphatonin, a circulating factor that is responsible for renal Pi reabsorption and altered vitamin D regulation has been proposed [Figure 3].
Figure 3.
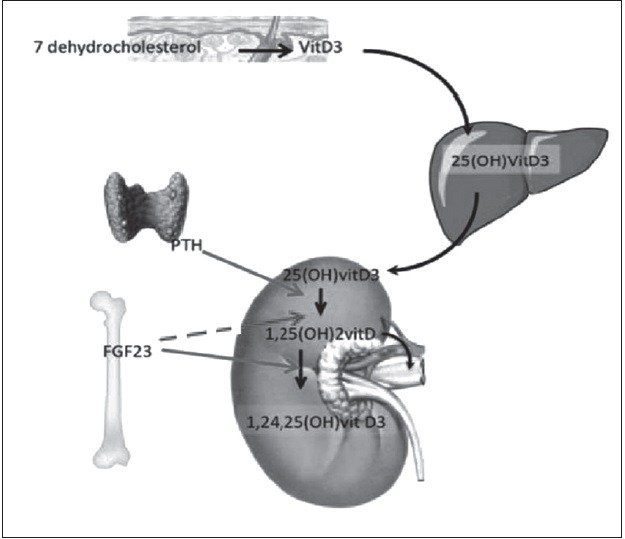
Relationship of vitamin D, PTH, FGF-23 and phosphate handling-vitamin D3 is produced in skin from a UVB-mediated conversion of 7-dehydrocholesterol. In liver, vitamin D is converted to 25(OH) vitD3, which is released in blood. In the kidney, 25(OH) vitD3 is converted to 1-dihydroxyvitamin D [1,25(OH) 2vitD]. Two hormones regulate this step: PTH and FGF23, a hormone that is synthesized by osteocytes and osteoblasts. In kidney, PTH stimulates whereas FGF23 represses 25(OH) D-1α-hydroxylation. In kidney, FGF23 increases expression of 24-hydroxylase an enzyme that inactivates calcitriol
Recent evidences on FGF23 showed that this hormone regulates serum phosphate concentration and calcitriol metabolism. FGF23 is present in the plasma of healthy individual, as an intact peptide in its active form. Intact FGF23 is cleaved within cells, in disorders that alter FGF23 glycosylation and results in the release of fragments.[22,23,24] It has been observed that those patients who develop hyperphosphatemia and tumoral calcinosis had low intact circulating FGF23 concentration and increased plasma FGF23 fragment concentration. The enzyme that cleaves FGF23 in body is unknown.
FGF23 is a circulating 32k-Da peptide secreted by osteocytes, osteoblast and osteoclast in response to hyperphosphatemia and vitamin D.[25,26,27] It decreases renal phosphate reabsorption by lowering NPT2a and NPT2c expression, and it diminishes calcitriol synthesis by inhibiting 1α hydroxylase and stimulating its catabolizing enzyme 24, 25 hydroxylase. The reduced calcitriol decreases intestinal NPT2b expression and phosphate reabsorption[28] as it is not observed in mice with disrupted VDR gene.[29] FGF23 binds to and activates a composite receptor formed by the conjunction of FGF receptor 1 (FGFR1), FGFR3 and or FGFR4 with klotho.
Over expression of FGF23 mRNA in bone cells, leads too many bone disorders.[27,30] FGF23 mRNA is also present, albeit at lower levels, in the liver and in the kidney.[25] One of the principal roles of FGF23 is the control of phosphate balance. Infusion of recombinant FGF23 and over expression of FGF23 gene results in marked increase in urinary phosphate excretion and severe hypophosphatemia.[31,32,33] First of all, FGF23 has been reported in a patient with tumor induced osteomalacia (TIO). Cai et al.[34] described a case who had TIO and in whom the biochemical phenotype of hypophosphatemia, renal Pi wasting and reduced serum 1α25(OH) 2D3 and osteomalacia disappeared after removal of tumor. Conditioned medium from cultured tumor cells specifically inhibited NATPi transport in renal epithelia and implantation of tumor cell in nude mice recapitulated the syndrome. Subsequently, its role has been observed in X-linked hypophosphatemic rickets and autosomal dominant hypophosphatemic rickets.[35,36,37]
Klotho was discovered by Kuro-O et al.[38] in 1997. Klotho is a protein containing 1012–amino acid present as a cell surface molecule, bound to the plasma membrane by a short one-span transmembrane domain. Its intracellular tail is less than 15 amino acids long. Klotho expression is restricted to a small number of tissues like in kidneys, parathyroid glands, brain, and skeletal muscle.[38,39,40] In the kidney, Klotho is mainly expressed in the distal tubule, whereas FGF23 exerts its action on the proximal tubule. The mechanism by which FGF23 modifies proximal tubule functions is unknown. Klotho is also present in plasma and urine. The major soluble form of Klotho originates from the shedding of the full-length protein. A smaller circulating form comes from RNA splicing at exon 3 in Klotho gene. It has been reported that ADAM 10 and 17 can cleave and release Klotho from the cell surface; however, it is unknown whether this is a physiologic role of these enzymes.[41] The physiologic role of the circulating forms of Klotho remains to be established.
Klotho acts as a co-factor that is mandatory for FGF23 action. Klotho gene was discovered after the accidental disruption, in mice of a gene that is encoding a protein, led to a phenotype very similar to that seen in FGF23 knockout mice but without an increase in serum FGF23.[42,43] Studies performed in cultured cells showed that FGF23 binds to FGF receptors with less affinity but that expression of Klotho markedly increases the affinity. In the presence of FGF23, Klotho binds to FGF receptors has been observed in co-immunoprecipitation studies. Then Klotho seemed important for the stimulation of the FGF receptor–signaling pathway, which involves extracellular signal–regulated kinase 1/2 and FGF receptor substrate phosphorylation, by FGF23.[42,43] These data suggest that Klotho is a co-receptor for FGF23. All of the effects of FGF23 are mediated by Klotho and that the principal function of Klotho is to permit FGF23 action, this is suggested by the fact that phenotype of mice with a double disruption of FGF23 and Klotho gene is similar to that of FGF23 or Klotho knockout mice.[43,44]
KLOTHO, FGF23 AND ITS ACTION ON PARATHYROID
Although klotho is highly expressed in the parathyroid glands, the role of the protein in parathyroid tissue is uncertain. Multiple functions and effects of klotho in the parathyroid glands have been proposed [Figure 4]. Biological activity of klotho plays role in mineral homeostasis. Lack of klotho in mice resembles the clinical picture of human aging, and, at the same time, plasma calcium and phosphate levels are elevated and plasma levels of 1,25(OH) 2D are increased.[45] It has already been discussed that Klotho is an obligatory co-receptor for fibroblast growth factor 23 (FGF23), converting the canonical FGF receptors FGFR 1, 3, and 4 to FGF23-specific receptors. FGFR1 is highly expressed in the parathyroid glands, with the IIIC splice form being predominant, whereas expression of FGF receptors 3 and 4 is minimal.[46] The parathyroid gland is a target tissue for the action of FGF23, with FGF23 having an inhibitory effect on parathyroid hormone (PTH) secretion and mRNA expression. Furthermore, FGF23 has been shown to inhibit low calcium–induced parathyroid cell proliferation.[47,48] Klotho/FGF23 might also influence the autocrine vitamin D system in parathyroid cells. It has been proposed that FGF23 stimulates 1-α hydroxylase and increases the local production of calcitriol (opposite of the effect in the kidneys) and thereby potentially inhibits PTH gene activity.[49] Along this line, administration of FGF23 in vivo to rats resulted in up regulation of parathyroid VDR protein.[48] Conversely, Klotho has been found to participate in trafficking of the Na +/K+-ATPase to the parathyroid cell membrane, thereby increasing PTH secretion;[50] thus, mechanisms that relate klotho to increased as well as to decreased PTH secretion have been proposed. This uncertainty has persisted in studies of human parathyroid tissue. In both primary hyperparathyroidism (HPT) and secondary HPT, klotho has been shown to be severely suppressed in adenomas and in nodular hyperplasia;[51,52] however, in other studies of human uremic parathyroid glands, the expression of klotho was variable with significantly decreased or increased expression in different parathyroid nodules.[53]
Figure 4.
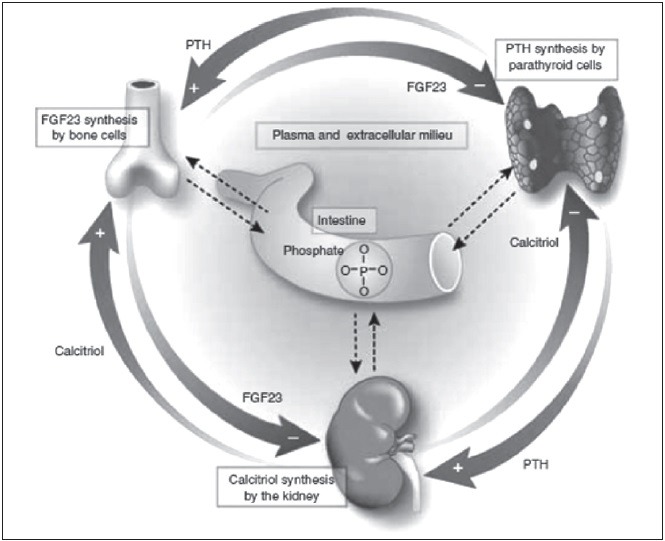
Three feedback loops showing the regulation of phosphate balance in body. Parathyroid gland produce PTH stimulates phosphate excretion and calcitriol synthesis in kidney; this lowers phosphorus and calcitriol inhibits PTH. PTH stimulates FGF23 and phosphate release following an increase in bone remodeling. FGF23 inhibits PTH but phosphate stimulates PTH production. FGF23 at kidney level stimulates urinary phosphate excretion and inhibits calcitriol, tending to reduce serum phosphorus. Renal-PTH stimulated calcitriol production stimulates FGF23 production by bone cells Lopez et al.54
SUMMARY OF FEEDBACK LOOPS REGULATING PHOSPHORUS BALANCE IN BODY
Lopez et al.[54] have studied the direct and indirect effect of PTH on circulating level of FGF23 in vivo. They demonstrated that PTH is necessary for FGF23 secretion. High phosphate does not stimulate FGF23 in a state of low PTH. It appears that PTH is likely to be more important than phosphate in regulation of FGF23 secretion. The decrease in calcitriol that ensures parathyroidectomy plays an important role in the inhibitory effect of low PTH on FGF23 production. Recently, Torres et al.[55] proposed the three feedback loops precisely regulating serum phosphorus level has been proposed in a commentary on study of Lopez et al.[54] Serum phosphorus is tightly regulated by three organs parathyroid, kidney and bone through three feedback loops. Parathyroid gland produce PTH which on kidney stimulates phosphate excretion and calcitriol synthesis; then in turn low phosphorus and calcitriol directly inhibit PTH production. On the bone PTH stimulates FGF23 and phosphate release following an increase in bone remodelling. FGF23 inhibits PTH secretion but phosphate will tend to stimulate PTH production. FGF23 at kidney level stimulates urinary phosphate excretion and inhibits calcitriol production, tending to reduce serum phosphorus level by these two mechanisms. On the other hand the renal-PTH stimulated calcitriol production stimulates FGF23 production by bone cells. These three counter regulatory loops maintain tightly controlled intestinal absorption and serum phosphorus concentration. Torres et al. has also postulated that there should be an intestinal phosphate sensor which alters one of the organs in case of phosphate overload or phosphate deficiency.[55]
FGF-23 IN CHRONIC KIDNEY DISEASE AND THE DEVELOPMENT OF BONE MINERAL DISORDER
Serum intact FGF-23 concentrations rise with a decline in GFR in CKD patients.[56,57,58] Renal Pi excretion decreases with reductions in GFR and the attendant retention of Pi is postulated to be the proximate cause of elevated FGF-23 in patients with CKD. However, reduced FGF-23 clearance could also be a possible factor to play a role as well.[59,60,61,62] The increase in FGF-23 is not sufficient to correct the hyperphosphatemia in advanced CKD. The increase in serum FGF-23 levels is associated with a decline in 1α,25(OH) 2D concentrations.[63] Diminished 1α,25(OH) 2D3 concentrations lead to increased PTH synthesis and contribute to the secondary hyperparathyroidism that is seen in CKD. Because FGF-23 inhibits 25(OH) D 1α-hydroxylase activity and 1α,25(OH) 2D3 synthesis, it is very likely that elevated FGF-23 plays an indirect role in the pathogenesis of secondary hyperparathyroidism in CKD patients and FGF-23 concentrations positively correlate with PTH concentrations. It has also been suggested that elevated serum FGF-23 concentrations are predictive of the development of refractory secondary hyperparathyroidism in dialysis patients.[64] It is interesting that FGF-23 levels continue to be elevated despite reductions in PTH levels after calcitriol therapy.[64] In another study, FGF-23 levels decreased after surgical parathyroidectomy in patients with secondary hyperparathyroidism.[65] Inorganic phosphorus (Pi) retention increases both PTH and FGF-23 concentrations and that there may not be a direct interaction between these two peptides. Whether FGF-23 directly influences bone formation or resorption in the context of CKD remains to be determined.
The calcium sensing receptor (CaSR) present on parathyroid gland senses low calcium level in blood which occurs consequent upon high phosphate and low calcitriol level and stimulates parathyroid gland to increase the secretion, synthesis and proliferation of parathyroid cells leading to secondary hyperparathyroidism.
FGF-23 IN PRIMARY HYPERPARATHYROIDISM, HUMORAL HYPERCALCEMIA OF MALIGNANCY, AND MALIGNANCIES
The changes in FGF-23 in primary hyperparathyroidism are not much studied. In contrast to primary hyperparathyroidism, in which serum calcium concentrations are elevated, serum Pi is reduced, and serum 1α,25(OH) 2D concentrations are elevated, patients with humoral hypercalcemia of malignancy exhibit hypercalcemia, hypophosphatemia, and inappropriately low serum 1α,25(OH) 2D concentrations. Serum FGF-23 is elevated 5-10 fold in humoral hypercalcemia of malignancy, whereas it is normal in primary hyperparathyroidism. It is possible that the low serum 1α,25(OH) 2D concentrations that are seen in humoral hypercalcemia of malignancy could be due to increases in FGF-23 concentrations.[66] Intact FGF-23 is elevated in metastatic ovarian cancer, in which serum Pi levels are normal.[67] FGF-23 may not be elevated sufficiently in this disease, suggesting that there is a threshold beyond which FGF-23 becomes phosphaturic.
FGF-23 IN NEPHROLITHIASIS
It has been observed that stone formers with increased urinary Pi excretion and hypophosphatemia had higher serum FGF-23 concentrations when compared with stone formers without Pi wasting and normal individuals.[68] Normal serum 1α,25(OH) 2D concentrations in the hypophosphatemic stone formers with hypercalciuria were unusual because most hypophosphatemic stone formers have elevated or high-normal serum 1α,25(OH) 2D concentrations. The role of FGF-23 in nephrolithiasis may be explored in future.
FGF-23 AND ECTOPIC CALCIFICATION
It has also observed that FGF-23–null mice have considerable mineralization in nonskeletal tissues, including the heart and the kidney.[69] Although these mice also have hyperphosphatemia and elevated 1α,25(OH) 2D3 concentrations that could influence calcification. The possible role of FGF-23 in vascular calcification may be area of future research.
CONCLUSION
Renal phosphate handling is regulated by three organs parathyroid, kidney and bone through three feedback loops. These three counter regulatory loops maintain tightly controlled intestinal absorption and serum phosphorus concentration. The parathyroid hormone, vitamin D, FGF23 and klotho coreceptor are the key regulator of phosphorus balance in body.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Fuchs R, Peterlik M. Intestinal phosphate transport. In: Massry SG, Ritz E, Jahn H, editors. Phosphate and Minerals in Health and Disease. New York: Plenum Press; 1980. p. 381. [Google Scholar]
- 2.Levine BS, Kleeman CR. Hypophosphatemia and hyperphosphatemia: Clinical and pathophysiologic aspects. In: Maxwell MH, Kleeman CR, editors. Clinical Disorders of Fluid and Electrolyte Metabolism. New York: McGraw Hill; 1994. p. 1040. [Google Scholar]
- 3.Knox FG, Osswald H, Marchand GR, Spielman WS, Haas JA, Berndt T, et al. Phosphate transport along the nephron. Am J Physiol. 1977;233:F261–8. doi: 10.1152/ajprenal.1977.233.4.F261. [DOI] [PubMed] [Google Scholar]
- 4.Pastoriza-Muñoz E, Colindres RE, Lassiter WE, Lechene C. Effect of parathyroid hormone on phosphate reabsorption in rat distal convolution. Am J Physiol. 1978;235:F321–30. doi: 10.1152/ajprenal.1978.235.4.F321. [DOI] [PubMed] [Google Scholar]
- 5.Peraino RA, Suki WN. Phosphate transport by isolated rabbit cortical collecting tubule. Am J Physiol. 1980;238:F358–62. doi: 10.1152/ajprenal.1980.238.5.F358. [DOI] [PubMed] [Google Scholar]
- 6.Shareghi GR, Agus ZS. Phosphate transport in the light segment of the rabbit cortical collecting tubule. Am J Physiol. 1982;242:F379–84. doi: 10.1152/ajprenal.1982.242.4.F379. [DOI] [PubMed] [Google Scholar]
- 7.Chabardès D, Imbert M, Clique A, Montégut M, Morel F. PTH sensitive adenyl cyclase activity in different segments of the rabbit nephron. Pflugers Arch. 1975;354:229–39. doi: 10.1007/BF00584646. [DOI] [PubMed] [Google Scholar]
- 8.Werner A, Kinne RK. Evolution of the Na-P (i) cotransport systems. Am J Physiol Regul Integr Comp Physiol. 2001;280:R301–12. doi: 10.1152/ajpregu.2001.280.2.R301. [DOI] [PubMed] [Google Scholar]
- 9.Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998;95:5372–7. doi: 10.1073/pnas.95.9.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murer H, Hernando N, Forster I, Biber J. Regulation of Na/Pi transporter in the proximal tubule. Annu Rev Physiol. 2003;65:531–42. doi: 10.1146/annurev.physiol.65.042902.092424. [DOI] [PubMed] [Google Scholar]
- 11.Hernando N, Biber J, Forster I, Murer H. Recent advances in renal phosphate transport. Ther Apher Dial. 2005;9:323–7. doi: 10.1111/j.1744-9987.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 12.Berndt T, Kumar R. Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol. 2007;69:341–59. doi: 10.1146/annurev.physiol.69.040705.141729. [DOI] [PubMed] [Google Scholar]
- 13.Condamine L, Menaa C, Vrtovsnik F, Friedlander G, Garabédian M. Local action of phosphate depletion and insulin-like growth factor 1 on in vitro production of 1,25-dihydroxyvitamin D by cultured mammalian kidney cells. J Clin Invest. 1994;94:1673–9. doi: 10.1172/JCI117512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin DR, Ritter CS, Slatopolsky E, Brown AJ. Acute regulation of parathyroid hormone by dietary phosphate. Am J Physiol Endocrinol Metab. 2005;289:e729–34. doi: 10.1152/ajpendo.00065.2005. [DOI] [PubMed] [Google Scholar]
- 15.Landsman A, Lichtstein D, Bacaner M, Ilani A. Dietary phosphate-dependent growth is not mediated by changes in plasma phosphate concentration. Br J Nutr. 2001;86:217–23. [PubMed] [Google Scholar]
- 16.ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–8. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 17.Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, et al. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67:54–68. doi: 10.1006/geno.2000.6235. [DOI] [PubMed] [Google Scholar]
- 18.Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–94. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, et al. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002;17:1102–10. doi: 10.1359/jbmr.2002.17.6.1102. [DOI] [PubMed] [Google Scholar]
- 20.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: An inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab. 2005;90:1012–20. doi: 10.1210/jc.2004-0357. [DOI] [PubMed] [Google Scholar]
- 21.Hernando N, Gisler SM, Pribanic S, Déliot N, Capuano P, Wagner CA, et al. NaPi-IIa and interacting partners. J Physiol. 2005;567:21–6. doi: 10.1113/jphysiol.2005.087049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–81. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 23.Benet-Pagès A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–90. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 24.Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, et al. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006;91:4037–42. doi: 10.1210/jc.2006-0305. [DOI] [PubMed] [Google Scholar]
- 25.Mirams M, Robinson BG, Mason RS, Nelson AE. Bone as a source of FGF23: Regulation by phosphate? Bone. 2004;35:1192–9. doi: 10.1016/j.bone.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 26.Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–32. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:e38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 28.Inoue Y, Segawa H, Kaneko I, Yamanaka S, Kusano K, Kawakami E, et al. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J. 2005;390:325–31. doi: 10.1042/BJ20041799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, et al. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha, 25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278:2206–11. doi: 10.1074/jbc.M207872200. [DOI] [PubMed] [Google Scholar]
- 30.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–92. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–82. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- 32.Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–94. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 33.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–35. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 34.Cai Q, Hodgson SF, Kao PC, Lennon VA, Klee GG, Zinsmiester AR, et al. Brief report: Inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med. 1994;330:1645–9. doi: 10.1056/NEJM199406093302304. [DOI] [PubMed] [Google Scholar]
- 35.Rowe PS, Ong AC, Cockerill FJ, Goulding JN, Hewison M. Candidate 56 and 58 kDa protein (s) responsible for mediating the renal defects in oncogenic hypophosphatemic osteomalacia. Bone. 1996;18:159–69. doi: 10.1016/8756-3282(95)00458-0. [DOI] [PubMed] [Google Scholar]
- 36.Meyer RA, Jr, Tenenhouse HS, Meyer MH, Klugerman AH. The renal phosphate transport defect in normal mice parabiosed to X-linked hypophosphatemic mice persists after parathyroidectomy. J Bone Miner Res. 1989;4:523–32. doi: 10.1002/jbmr.5650040411. [DOI] [PubMed] [Google Scholar]
- 37.Meyer RA, Jr, Meyer MH, Gray RW. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res. 1989;4:493–500. doi: 10.1002/jbmr.5650040407. [DOI] [PubMed] [Google Scholar]
- 38.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 39.Li SA, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct. 2004;29:91–9. doi: 10.1247/csf.29.91. [DOI] [PubMed] [Google Scholar]
- 40.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–8. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007;104:19796–801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 43.Segawa H, Yamanaka S, Ohno Y, Onitsuka A, Shiozawa K, Aranami F, et al. Correlation between hyperphosphatemia and type II Na-Pi cotransporter activity in klotho mice. Am J Physiol Renal Physiol. 2007;292:F769–79. doi: 10.1152/ajprenal.00248.2006. [DOI] [PubMed] [Google Scholar]
- 44.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–3. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewin E, Olgaard K. Klotho, an important new factor for the activity of Ca2+channels, connecting calcium homeostasis, ageing and uraemia. Nephrol Dial Transplant. 2006;21:1770–2. doi: 10.1093/ndt/gfl178. [DOI] [PubMed] [Google Scholar]
- 46.Hofman-Bang J, Martuseviciene G, Santini MA, Olgaard K, Lewin E. Increased parathyroid expression of klotho in uremic rats. Kidney Int. 2010;78:1119–27. doi: 10.1038/ki.2010.215. [DOI] [PubMed] [Google Scholar]
- 47.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–8. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canalejo R, Canalejo A, Martinez-Moreno JM, Rodriguez-Ortiz ME, Estepa JC, Mendoza FJ, et al. FGF23 fails to inhibit uremic parathyroid glands. J Am Soc Nephrol. 2010;21:1125–35. doi: 10.1681/ASN.2009040427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lafage-Proust MH. Does the downregulation of the FGF23 signaling pathway in hyperplastic parathyroid glands contribute to refractory secondary hyperparathyroidism in CKD patients? Kidney Int. 2010;77:390–2. doi: 10.1038/ki.2009.512. [DOI] [PubMed] [Google Scholar]
- 50.Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–8. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- 51.Komaba H, Goto S, Fujii H, Hamada Y, Kobayashi A, Shibuya K, et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int. 2010;77:232–8. doi: 10.1038/ki.2009.414. [DOI] [PubMed] [Google Scholar]
- 52.Kumata C, Mizobuchi M, Ogata H, Koiwa F, Nakazawa A, Kondo F, et al. Involvement of alpha-klotho and fibroblast growth factor receptor in the development of secondary hyperparathyroidism. Am J Nephrol. 2010;31:230–8. doi: 10.1159/000274483. [DOI] [PubMed] [Google Scholar]
- 53.Ohkido I, Yokoyama K, Imura A, Utsunomiya Y, Hosoya T, Nabeshima Y. Persistent alpha-Klotho (a-Kl) expression in the parathyroid glands of patients with secondary hyperparathyroidism. Nephrol Dial Transplant. 2010;25:1007–8. doi: 10.1093/ndt/gfp743. author reply 1008-9. [DOI] [PubMed] [Google Scholar]
- 54.López I, Rodríguez-Ortiz ME, Almadén Y, Guerrero F, de Oca AM, Pineda C, et al. Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int. 2011;80:475–82. doi: 10.1038/ki.2011.107. [DOI] [PubMed] [Google Scholar]
- 55.Torres PA, De Brauwere DP. Three feedback loops precisely regulating serum phosphate concentration. Kidney Int. 2011;80:443–5. doi: 10.1038/ki.2011.146. [DOI] [PubMed] [Google Scholar]
- 56.Nakanishi S, Kazama JJ, Nii-Kono T, Omori K, Yamashita T, Fukumoto S, et al. Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients. Kidney Int. 2005;67:1171–8. doi: 10.1111/j.1523-1755.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- 57.Shigematsu T, Kazama JJ, Yamashita T, Fukumoto S, Hosoya T, Gejyo F, et al. Possible involvement of circulating fibroblast growth factor 23 in the development of secondary hyperparathyroidism associated with renal insufficiency. Am J Kidney Dis. 2004;44:250–6. doi: 10.1053/j.ajkd.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 58.Pande S, Ritter CS, Rothstein M, Wiesen K, Vassiliadis J, Kumar R, et al. FGF-23 and sFRP-4 in chronic kidney disease and post-renal transplantation. Nephron Physiol. 2006;104:P23–32. doi: 10.1159/000093277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slatopolsky E, Gradowska L, Kashemsant C, Keltner R, Manley C, Bricker NS. The control of phosphate excretion in uremia. J Clin Invest. 1966;45:672–7. doi: 10.1172/JCI105382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slatopolsky E, Robson AM, Elkan I, Bricker NS. Control of phosphate excretion in uremic man. J Clin Invest. 1968;47:1865–74. doi: 10.1172/JCI105877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bricker NS, Klahr S, Lubowitz H, Slatopolsky E. The pathophysiology of renal insufficiency. On the functional transformations in the residual nephrons with advancing disease. Pediatr Clin North Am. 1971;18:595–611. doi: 10.1016/s0031-3955(16)32568-8. [DOI] [PubMed] [Google Scholar]
- 62.Slatopolsky E, Bricker NS. The role of phosphorus restriction in the prevention of secondary hyperparathyroidism in chronic renal disease. Kidney Int. 1973;4:141–5. doi: 10.1038/ki.1973.92. [DOI] [PubMed] [Google Scholar]
- 63.Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol. 2005;289:F1088–95. doi: 10.1152/ajprenal.00474.2004. [DOI] [PubMed] [Google Scholar]
- 64.Kazama JJ, Sato F, Omori K, Hama H, Yamamoto S, Maruyama H, et al. Pretreatment serum FGF-23 levels predict the efficacy of calcitriol therapy in dialysis patients. Kidney Int. 2005;67:1120–5. doi: 10.1111/j.1523-1755.2005.00178.x. [DOI] [PubMed] [Google Scholar]
- 65.Sato T, Tominaga Y, Ueki T, Goto N, Matsuoka S, Katayama A, et al. Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis. 2004;44:481–7. [PubMed] [Google Scholar]
- 66.Singh RJ, Kumar R. Fibroblast growth factor 23 concentrations in humoral hypercalcemia of malignancy and hyperparathyroidism. Mayo Clin Proc. 2003;78:826–9. doi: 10.4065/78.7.826. [DOI] [PubMed] [Google Scholar]
- 67.Tebben PJ, Kalli KR, Cliby WA, Hartmann LC, Grande JP, Singh RJ, et al. Elevated fibroblast growth factor 23 in women with malignant ovarian tumors. Mayo Clin Proc. 2005;80:745–51. doi: 10.1016/S0025-6196(11)61528-0. [DOI] [PubMed] [Google Scholar]
- 68.Rendina D, Mossetti G, De Filippo G, Cioffi M, Strazzullo P. Fibroblast growth factor 23 is increased in calcium nephrolithiasis with hypophosphatemia and renal phosphate leak. J Clin Endocrinol Metab. 2006;91:959–63. doi: 10.1210/jc.2005-1606. [DOI] [PubMed] [Google Scholar]
- 69.Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]