

Abstract
Our laboratory investigates systemic autoimmune disease in the context of mouse models of systemic lupus erythematosus (SLE). SLE is associated with high titers of serum autoantibodies of the IgG class that are predominantly directed against nuclear antigens, with pathological manifestations that are considered by many to be characteristic of an immune-complex mediated disease. In this review, we focus on the known and potential roles of somatic mutagenesis in SLE. We will argue that antinuclear antibodies (ANA) arise predominantly from nonautoreactive B cells that are transformed into autoreactive cells by the process of somatic hypermutation (SHM), which is normally associated with affinity maturation during the germinal center reaction. We will also discuss the role of SHM in creating antigenic peptides in the V region of the B cell receptor (BCR) and its potential to open an avenue of unregulated T cell help to autoreactive B cells. Finally, we will end this review with new experimental evidence suggesting that spontaneous somatic mutagenesis of genes that regulate B cell survival and activation is a rate-limiting causative factor in the development of ANA.
Keywords: Somatic mutation, Autoimmunity, Anti-nuclear Antibodies, Lupus, Haplodeficiency
1. Introduction
SLE is the prototypical systemic autoimmune disease, characterized by a breach of self-tolerance in B cells that is dependent upon T cells, as assessed in spontaneous mouse models of this disease. Antibodies directed against nucleosomes and associated components such as dsDNA and histones are most consistently observed, and those directed to ribonuclear proteins are common (1). Systemic autoimmunity is associated with multiple genetic loci, which is most obvious from the disease susceptibility of specific inbred strains of mice and affirmed through genome-wide association studies in humans (2, 3). Although this modicum of predictability has proved valuable to investigators of SLE, it is clear that there is a substantial stochastic component to disease development. And while the last two decades have witnessed enormous strides in progress revealing mechanisms that censor self-reactive lymphocytes under physiological circumstances, the events that precipitate loss of self-tolerance in SLE, and autoimmune disease in general, remain largely unknown. Studies in experimental mouse models of SLE have elucidated some of the pathology, immune cells and molecules associated with the development of ANA. However they have yet to explain a feature shared amongst models: isogenic mice of the same sex, housed in a common clean facility and even within the same cage, have different kinetics of ANA onset (4). These delayed, but variable, kinetics indicate that a stochastic event is required prior to the activation and expansion of autoimmune lymphocytes, and that this event does not require an exogenous pathogen.
In this review we will highlight the roles of somatic mutation in SLE. We will discuss evidence that SHM is the primary mechanism that generates anti-nuclear B cells in SLE, and that somatically mutated peptides from the V region of the B cell receptor (BCR) may be an avenue of CD4 T cell help to autoreactive B cells. We will also provide experimental evidence that somatic mutagenesis leading to the loss of function in immunoregulatory genes may be the stochastic rate-limiting step responsible for autoimmunity.
2. Anti-nuclear B cells
2.1 Tolerance in anti-nuclear B cells
In humans, anti-nuclear B cells represent ~60% of immature B cells in the bone marrow, however they constitute less than 10% of B cells in the circulation (5). This implies that, while a high frequency of V(D)J rearrangements produce an autoreactive BCR, B cells with such receptors are ultimately purged from the mature B cell pool. In studies dating from the early 1990’s, it has been shown in various BCR transgene models that autoreactive B cells are censored by three major mechanisms: receptor editing, clonal deletion and anergy (6–14). In the case of anti-nuclear B cells, several groups have shown with the 3H9 BCR Tg model and its derivatives that all three mechanisms of tolerance may apply (15). Receptor editing and clonal deletion are the most efficient self-tolerance mechanisms, as anti-nuclear specificities are physically erased by changing the BCR (receptor editing) or by eliminating the B cell (clonal deletion). For high-avidity clones, receptor editing appears to be the primary mode of central self-tolerance, with clonal deletion serving as a backup (9). In the case of peripheral self-antigens, clonal deletion appears to be the predominant mode (16, 17). In contrast, B cells that chronically engage self-antigens with low avidity or low valency may enter the mature B cell pool in an unresponsive state, known as anergy (10, 18). Due to its reversible state (19, 20), many investigators consider anergic B cells to be potentially dangerous. We do not adhere to this view for lack of evidence that such low affinity clones develop into high-avidity ANA secreting cells characteristic of SLE (14), and because recent unpublished studies from this laboratory suggest that anergic B cells play a regulatory role as enforcers of self-tolerance. All of these inferred self-tolerance mechanisms, of course, must be qualified by the artificial nature of the genetic systems required to produce traceable populations of autoreactive B cells.
2.2 Origin of anti-nuclear B cells
A fundamental question in autoimmunity is how anti-nuclear B cells arise in a disease such as SLE. A simple explanation is that, in lupus-prone genetic backgrounds, central tolerance in B cells is defective such that B cells born with an anti-nuclear BCR can enter into the periphery. Once in the periphery, anti-nuclear B cells can be recruited into an immune response like conventional antigen-specific follicular B cells. We refer to this model as the germline-founder hypothesis (Fig. 1A). Much of the evidence supporting this hypothesis comes from work involving mice that are genetically predisposed to spontaneous systemic autoimmunity and that carry, in addition, Ig heavy chain transgenes derived from B cell hybridomas producing ANA. These include D42, 3H9 and its derivative 3H956R, which has an extra affinity-boosting arginine residue at position 56 in CDR2 of the 3H9 heavy chain (7, 13, 21–25). However, even in these exaggerated scenarios involving high-avidity BCR, self-tolerance appears to be largely intact, and only modest differences in kinetics of ANA development distinguish the Tg animals from the nonTg controls. Moreover, based on hybridoma sampling, most of the Tg-encoded ANA in site-directed models appears to be IgM, and recent studies have indicated that IgM ANA can have an ameliorating effect on pathology (26–30). These observations question the view that the high-avidity IgG ANA of SLE are derived from autoreactive antecedents generated by V(D)J recombination in the bone marrow. Our recent studies in autoimmune predisposed B6.Nba2 Aicda−/− mice also challenge this view (unpublished). A majority of serum IgM autoantibodies arising in such mice bind cytoplasmic rather than nuclear antigens. Few mice produce high-titer antibodies to the nucleosome, which is the most predominant IgG ANA specificity in mouse models of SLE. And among those B cell clones that do produce anti-nucleosome Ab, an unusually large fraction use the distal Jk5 gene segments, which is atypical of ANA from lupus-prone mice (31).
Figure 1. Origin of anti-nuclear B cells in SLE.

(A) Germline-founder hypothesis: an anti-nuclear B cell, generated upon VDJ recombination in the bone marrow, evades tolerance checkpoints and enters the peripheral circulation. Upon recognition of a self-antigen, this autoreactive cells enters a germinal center (GC) and initiates an autoimmune response. (B) Mutation-founder hypothesis: a B cell with a normal, nonautoreactive receptor is recruited into a GC by an immunogen. Somatic hypermutation (SHM) generates an anti-nuclear B cell, which is the antecedent of an autoimmune lineage.
As anergy is a reversible state, several authors have proposed that anti-nuclear B cells observed in SLE could be derived from low-avidity anergic precursors generated in the bone marrow by V(D)J recombination. For example, Yachimovich-Cohen et al., demonstrated that B cells carrying a site-directed D42 heavy chain Tg and a Vκ8Jκ5 light-chain Tg were anergic. When these transgenes were crossed into a lupus-prone strain, the anergic state was apparently lost, as assessed in vitro by proliferation and upregulation of IgM in response to LPS, and in vivo by the spontaneous activation of B cells producing IgM ANA encoded by this pair of Tg (as assessed by hybridoma sampling). However this result was not confirmed in a subsequent study. Moreover, only 1 of 22 sampled hybridomas that produced IgG ANA expressed both the heavy and light-chain Tg (7). Collectively, these and the preceding observations challenge the view that anti-nuclear clones generated by V(D)J recombination in the bone marrow, whether of high- or low-avidity, are the precursors to the IgG anti-nuclear clones observed in spontaneous SLE.
An alternative to the germline-founder hypothesis is that IgG anti-nuclear B cells of lupus originate from normal precursors that are transformed into autoreactive cells via the process of somatic hypermutation (SHM) (Fig. 1B). Such autoreactive B cells would have a distinct advantage over those generated in the BM because the former would not have to escape early self-tolerance checkpoints that precede B cell activation and SHM in germinal centers. We refer to this as the mutation-founder hypothesis.
Distinguishing between germline-founder and mutation-founder hypotheses has proved to be difficult even in BCR Tg models, where interpretations are obscure because ANA-producing B cells often expressed edited receptors. It is unclear whether receptor editing in such cases failed to extinguish autoreactive specificities. A plausible alternative is that receptor editing successfully extinguished autoreactivity and thereby allowed the edited cells to participate in immune responses, only to acquire somatic mutations rendering them autoreactive once again. Such cells would only have to escape the final self-tolerance checkpoints that precede terminal differentiation.
To define the role of somatic mutagenesis in generating ANA, several groups have attempted to revert somatic mutations in anti-nuclear clones to germline sequence, with the expectation that this would eliminate autoreactivity if the mutation-founder idea were correct. These studies have produced mixed results (32, 33). The most conclusive study was by performed by Wellman et al. (34), who found that reverting somatic mutations ablated anti-nuclear reactivity in two clones derived from SLE patients. In all of these studies, however, it was never possible to unambiguously identify and revert all somatic mutations. This is largely because of undefined sequences in CDR3, which is created in part through addition of untemplated nucleotides by terminal deoxynucleotidyl transferase (TdT) during V(D)J recombination in the bone marrow (35). Somatic mutations that subsequently land at these sites cannot be identified due to the unknown starting sequence.
To conduct a definitive test of the mutation-founder hypothesis, Guo et al. developed a spontaneous lupus model in which all somatic mutations, including those in CDR3, could be identified for reversion analysis (36). The model is predicated on the B6.Nba2 strain, which carries a distal segment of chromosome 1 (Nba2 interval) from the autoimmune-prone NZB strain and develops ANA with properties and kinetics that are essentially indistinguishable from those observed in classical models of SLE (37). This segment of chromosome 1 is also syntenic with one in humans that is associated with SLE (38). A homozygous deficiency in the Tdt gene and heterozygous deficiencies in the Igh and kappa loci were bred into mice of the B6.Nba2 genetic background (B6.Nba2; Tdt−/−; JH+/−;Jk+/−) to facilitate identification of somatic mutations in CDR3 and to assist interpretations regarding receptor editing.
Thirty-three IgG ANA-secreting hybridomas belonging to 14 clones (lineages) were obtained from spontaneously autoimmune mice in this model. It was possible to identify every somatic mutation in heavy and light chain V region genes expressed by 10 of these clones. When all of these somatic mutations were reverted to germline sequence, anti-nuclear activity was completely lost in 9 of 10 clones, and it was reduced by ~95% in the 10th clone. In addition, the 9 revertant antibodies without detectable anti-nuclear reactivity showed no evidence of autoreactivity against any tissue antigen, as assessed by immunofluorescence performed on whole frozen sections of neonatal mice. Moreover, the 14 clones showed little evidence of receptor editing. None expressed dual kappa and lambda light chains, and 10 of them expressed kappa V genes that had joined to proximal Jk1 or Jk2 gene segments. Had the cells emerged from the BM with autoreactive receptors, we would have expected them to edit their receptors as evidenced by frequent use of distal Jk segments or expression of Vλ genes. But this was not observed. Importantly, a predominant use of proximal Jk1 and Jk2 gene segments was reported from a survey of published ANA from several mouse models of SLE (31). Taken together with this observation, our results form a strong case for the view that most IgG ANA clones in spontaneous murine SLE are generated via the process of SHM.
A caveat of course is that the frequency of anti-nuclear B cells within a TdT-deficient repertoire of newly generated B cells may be substantially less than it is in a TdT+ repertoire. A deficiency in TdT does ameliorate nephritis in two mouse models of SLE, and it reduces ANA in MRLlpr/lpr mice (39, 40). However, it is not clear whether reduced pathology and ANA are due a TdT deficiency in B cells or T cells. Feeney et al.,(40) reported that B220+ double-negative T cells, which arise independently of B cells (41), are decreased in TdT deficient MRLlpr/lpr mice. Moreover, Conde et al.,(42) have shown that ANA titers were not affected by a TdT deficiency in the (NZB x NZW)F1 mouse model, which is the closest correlate of human SLE and the predecessor of the B6.Nba2 model that we analyzed (42). Our most recent unpublished studies in B6.Nba2 Aicda−/− mice, in which VHCDR3 diversity is intact, also support the view that most of the prototypical IgG ANA in SLE that bind complexes of dsDNA and histones are created by SHM.
A high frequency of mutations producing arginine codons was observed among the panel of 33 ANA hybridomas from the TdT-deficient B6.Nba2 mice. Arg residues are well known for their ability to interact with nucleic acids (43–45). In agreement with this fact and the mutation-founder idea, at least one Arg mutation was shared by all clone members for 5 of the 6 multimember lineages. In addition, a single Arg mutation was largely responsible for generating the anti-nuclear specificity of one of the clones (the only one tested). Curiously, 20 out of 30 such Arg mutations occurred at just two germline serine codons: AGC and AGT. AGC is the most intrinsically preferred target of SHM. Moreover, AGC and AGT can mutate to an Arg codon by any one of 3 distinct single base changes (46). These are the only codons with this property. Finally, these codons are not underrepresented in germline V region genes, as initially suggested from observations by Radic and Weigert (47). On the contrary, they are overrepresented (~2X) in all mouse and human V region genes with respect to either random codon use or expected use, as calculated from species-specific codon preference tables (36). Taken together, these observations strongly suggest that SHM routinely generates B cells with anti-nuclear BCR. Presumably these are efficiently censored, otherwise high-titer ANA would be the rule rather than the exception.
3. T cell help to ANA-producing B cells
ANA observed in spontaneous models of SLE bear many features characteristic of clonal selection in T cell-dependent immunity (48, 49). They are IgG, somatically mutated, of high avidity and frequently derived from expanded lineages of B cells, as revealed through hybridoma sampling studies. A substantial body of work indicates that if T cell help in SLE-prone mice is impeded either genetically or physically with cytotoxic or blocking antibodies, ANA are severely reduced or absent (50–54). While evidence for T cell help in ANA development is considerable, the nature, stage and specificity of such help are largely unknown. Two major models for CD4 T cell help to anti-nuclear B cells in SLE have emerged. One postulates that such helper cells recognize peptides derived from nucleosomal antigens acquired by B cells. Evidence for this comes from observed proliferative and cytokine responses to nucleosomes and histone peptides by CD4 T cells of lupus-prone strains of mice and from the induction of ANA by cloned lines of such T cells (55, 56). These T cell responses were not restricted by MHC II and appeared to be largely determined by the TCR Vα region (57). However, in autoimmune-prone (NZB x SWR)F1 mice carrying transgenes encoding TCRα or TCRαβ chains expressed by one of these clones, ANA responses were diminished relative to nonTg controls, and the transgenic T cells that escaped central tolerance were skewed towards a Treg phenotype (58).
An alternative but not mutually-exclusive hypothesis regarding T cell help emerged from the initial work of Hahn and colleagues, who observed that T cells from SLE-prone (NZB X NZW)F1 mice proliferated in response to peptides synthesized to match sequences specified by V regions of ANA (59). ANA titers and other lupus manifestations were increased when (NZB x NZW)F1 mice were immunized with several such antigenic peptides (60). Because peptides from the BCR are processed and self-presented in MHC II by activated B cells upon internalization of antigen (61–64) these observations raised the possibility that anti-nuclear B cells receive T cell help via this BCR receptor presentation avenue. It is noteworthy that in related studies, CD4 T cells from cerebral spinal fluid (CSF) of autoimmune patients with multiple sclerosis were found to react with peptides from V regions of autologous CSF antibodies (65, 66).
Studies from our laboratory have shown that T cells attain a state of tolerance to germline-encoded Ig V region sequences, even in autoimmune mice (67–69). In addition, it is clear that somatic mutations can create MHC II-restricted epitopes for CD4 T cells (70, 71). We therefore speculated that somatic mutations in ANA might generate neo-antigenic determinants mediating T cell help to anti-nuclear B cells in SLE. Indirect support for this idea was obtained from analyses of somatic mutations in lineages of ANA-producing hybridomas generated from a spontaneously autoimmune mouse. A somatic mutation in the VH that marked the initiation of a large lineage created an immunodominant MHC II-restricted determinant but had no influence on ANA affinity (72). Several members of a second lineage from the same animal that used a highly related Vκ gene that apparently carried the same somatic mutation (72).
Prospective experimental support for the receptor presentation hypothesis was obtained from two different models, both involving a complementary pair of Tg mice. In each case, one mouse carried an Ig light chain Tg with a defined MHC II-restricted epitope in the V region, and the other contained CD4 T cells expressing TCRαβ Tg encoding a receptor that reacted with the corresponding V region epitope. Both epitopes were originally generated via SHM. When such Tg T cells were transferred into the complementary Ig Tg adoptive recipients, IgG ANA and SLE-associated pathologies were induced (73, 74). Although the receptor presentation avenue of help to ANA B cells has yet to be conclusively demonstrated, there is evidence supporting its plausibility and a central role for SHM in creating immunogenic peptides in the BCR that direct T cell help. A key difference between this receptor presentation model and nucleosome peptide presentation model is that T helper cells of the former do not have to escape central tolerance to a ubiquitous self-antigen (Fig. 2).
Figure 2. Origin of T cell help for anti-nuclear B cells.

Source of T cell help to antinuclear B cells in SLE. (A) Defect in central and/or peripheral T cell tolerance in a lupus-prone background allows autoreactive CD4 T cells reactive with peptides from histones and/or germline-encoded BCR-V regions to escape self-tolerance and help anti-nuclear B cells. (B) Self-tolerance in CD4 T cells is intact with respect to peptides from histones and germline-encoded BCR-V regions; however CD4 T cells that recognize somatically generated BCR-V region-derived peptides provide a source of help to autoreactive B cells. Autoimmune prone-genetic backgrounds might have a defective regulation of this type of T-B interaction, resulting in the production of anti-nuclear antibodies by an autoimmune B cell.
4. Somatic mutagenesis and autoimmunity
Although our results argue that SHM of Ig V region genes is responsible for creating a majority of the anti-nuclear B cells in SLE, this alone cannot explain the spontaneous development of ANA in SLE. As codon analyses of mouse and human Ig V region genes indicates that SHM likely creates B cells with anti-nuclear specificities routinely during immune responses, other rate limiting steps must stand between generation of self-reactive clones and autoimmunity (36).
Genetic association studies in humans and mouse SLE models have revealed the complex, multigenic nature of SLE (3, 75–77). This is in agreement with reverse-genetic studies demonstrating that ANA development is promoted by loss of many different single, immunoregulatory molecules involved in a diversity of processes including transcription, programmed cell death, phagocytosis, and receptor signaling (78–80). Genetics alone, however, cannot account for the key and universal observation among SLE-prone strains that ANA appear with delayed kinetics, typically not appearing until the age of 5 months or more. Moreover, there is considerable variation in the kinetics of ANA development among isogenic lines of mice of the same sex, housed in a common clean facility and even within the same cage. Collectively, these observations suggest that a rate-limiting stochastic event is behind the activation and expansion of autoreactive lymphocyte clones and that this event does not require a specific exogenous pathogen (4).
While it is commonly held that mutation of germline DNA is the foundation of evolution and mutation of somatic genes is the most proximal cause of cancer, there is little discussion, much less experimentation, regarding a potential role for sporadic somatic mutation in autoimmune disease. This is somewhat surprising in view of a monograph on the subject published by F.M. Burnet in 1972 (81) and the fact that several proliferative diseases are known to be caused by somatic mutation. Included among these are patients with various proliferative syndromes, caused in part by somatic mutations in Fas (Cd95, Apo-1) NRAS or KRAS (82–85). The idea that somatic mutation might be the stochastic event catalyzing autoimmunity was recently proposed again in a review by C.C. Goodnow, although to our knowledge, no one has developed an experimental model to test this hypothesis (4).
4.1 The haplodeficient mouse model
To test the hypothesis that somatic mutagenesis might be the stochastic event behind the development of ANA, we aged a cohort of B6 mice that were heterozygous for the lpr mutant allele of the gene for Fas and examined sera for ANA. Impaired expression of Fas in murine T cells, B cells or myeloid cells compromises self-tolerance, as manifested in part by development of IgG ANA (86–92). We reasoned that if somatic mutagenesis were the cause of autoimmunity, then inheriting a single defective copy of a gene regulating immune homeostasis would greatly increase the odds of developing autoimmunity. The rationale for this idea originates from a large body of work demonstrating that inheriting a single defective allele of a tumor suppressor gene dramatically increases the chances of developing cancer, due in part to loss of heterozygosity (93–96). In fact, although lpr is generally considered to be a recessive allele, there is a report of heterozygous lpr/+ mice developing ANA (97).
In this experiment, some of our B6lpr/+ mice developed IgG ANA in an age-dependent manner. Among a cohort of 10 week-old female mice, only 1 of 16 mice developed anti-chromatin IgG (data not shown), but by 6 months of age, this frequency increased to ~50% (Fig. 3A). Sera with anti-chromatin IgG also stained nuclei of HEp-2 cells with a homogeneous pattern (Fig. 3B). Similar results were obtained from a second cohort of B6lpr/+ mice. Among age (8 months) and sex matched B6 control mice, none produced detectable ANA. And among a second cohort of 11 B6 mice that were 12 months old, we have only seen one with detectable serum IgG ANA.
Figure 3. Stochastic ANA and hypergammaglobulinemia in B6lpr/+ mice.
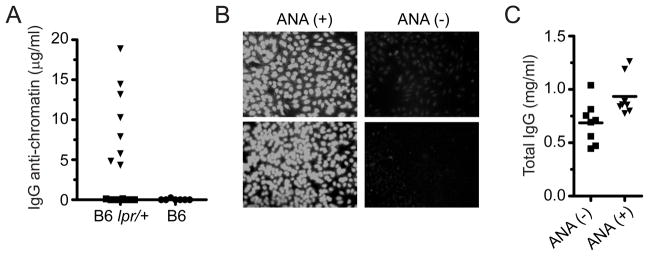
(A) Total IgG anti-chromatin in B6lpr/+ (n=16) and wildtype B6 mice (n=7) at 6 months of age. (B) Representative IgG HEp-2 stain of B6lpr/+ anti-chromatin positive [ANA(+)] or negative [ANA(-)] observed in A. (C) Total IgGκ in sera of B6lpr/+ mice. Mice were segregated on the basis of the presence (▼) or absence (■) of IgG anti-chromatin antibodies. Anti-chromatin assays: 96 well trays were coated with 10 μg/ml of calf-chromatin or bovine serum albumin (BSA). Bound IgG Abs were detected with goat anti-mouse IgG heavy-chain specific antibody (SouthernBiotech). Specific IgG anti-chromatin Abs were calculated by subtracting counts to BSA-coated wells. Concentrations were calculated using a standard curve generated with the 3H9/Vκ4 mAb. Total IgG Abs were assayed in a similar manner, except that trays were coated with goat anti-mouse IgG (heavy chain-specific) at 1 μg/ml and bound antibodies were detected with biotin-goat anti-mouse κ.
The development of ANA in only some of the haplodeficient mice in our SPF facility may be taken as a measure of evidence for the somatic mutation hypothesis of autoimmunity. To evaluate this more closely, we performed quantitative binding studies of ANA. Since high levels of IgG can produce a false positive signal in a solid phase immunoassay for anti-chromatin antibody, we quantified total serum IgG in B6lpr/+ mice. As shown in Fig. 3C, mice with IgG anti-chromatin had approximately 1.4 times more total serum IgG than those without detectable anti-chromatin. This modest increase in total IgG was not nearly enough to produce false positive signals equivalent to those observed for the positive sera in the chromatin-binding assay. In this assay, where the sensitivity of detection for 3 standard deviations above background was 600 ng/ml for a 1/100 dilution of serum, the mean concentration of IgG anti-chromatin observed was ~9.5 μg/ml, and the lowest concentration was ~4.2 μg/ml. Moreover, sera of B6lpr/+ nonproducers were indistinguishable from those of wildtype B6 mice. This clear distinction between anti-chromatin producers and nonproducers implicates a stochastic event(s) in autoantibody development within a subset of B6lpr/+ mice. In agreement with this, the total concentration of anti-chromatin antibody in ANA+ animals was low, as if derived from one B cell clone.
4.2 ANA not due to allelic exclusion of Fas in B6lpr/+ lymphocytes
We considered the possibility that Fas might be subject to allelic exclusion, such that the population of cells responsible for ANA in B6lpr/+mice was devoid of Fas. In view of reports that Fas deficiency in either T cells or B cells can promote autoimmunity, we examined both cell types for evidence of allelic exclusion. However, flow cytometric analyses of Fas levels on thymocytes, activated B cells and activated T cells from B6, B6lpr/+ and B6lpr/lpr mice, indicated biallelic expression in all cases. Therefore, allelic exclusion of Fas cannot account for the spontaneous development of ANA in B6lpr/+ mice (Fig. 4)
Figure 4. Allelic exclusion of Fas in lymphocytes.
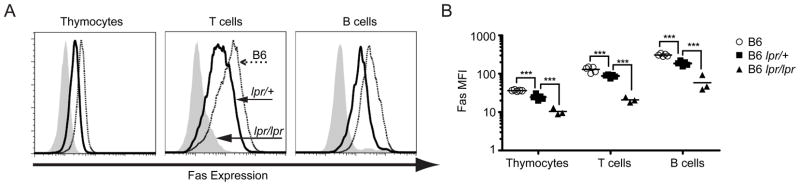
(A) Left panel, ex vivo expression of Fas on thymocytes. Middle panel, Fas expression on CD4 T cells after 48 hours of in vitro stimulation with PMA/ionomycin. Right panel, expression of Fas on B cells after in vitro stimulation with anti-μ and anti-CD40. Solid histograms, thick line and dotted line indicate Fas expression by cells from B6lpr/lpr, B6lpr/+ and wildtype B6 mice respectively. (B) Mean fluorescence intensity (MFI) for stains involving multiple animals (*** p<0.01).
4.3 Normal affinity maturation in haplodeficient mice
In addition to culling autoreactive B cells from the repertoire, Fas has been reported to promote affinity maturation in B cells (98, 99). This was confirmed by results of an experiment illustrated in Fig. 5A, showing that the rate of affinity maturation was impaired in B6lpr/lpr mice. If a haplodeficiency in Fas impaired the functional outcome of Fas signaling, then we would have expected affinity maturation to be compromised in B6lpr/+ mice. However, we found that the kinetics, quantities and the affinities of the anti-NP IgG responses of B6lpr/+ mice were indistinguishable from those of wildtype B6 mice (Fig. 5B–C). Thus, a haplodeficiency in Fas had no discernible effect on the selection of B cells during a T cell-dependent immune response. This result provides additional support for the idea that autoimmunity in B6lpr/+ mice is due to a stochastic event, as opposed to a graded gene-dose effect compromising the outcome of the Fas signaling pathway.
Figure 5. Equivalent NP antibody responses by wildtype B6 and B6lpr/+ mice.
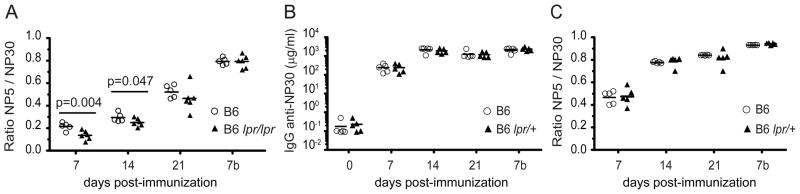
Mice were immunized with 100 μg of NP-chicken gamma globulin (CGG) in alum i.p. at day 0. Sera from indicated time points were tested for IgG anti-(NP-BSA). Relative affinity was calculated using the ratio of binding to NP4-BSA versus NP30-BSA. Values represented by each point were obtained from binding curves generated with serial 2-fold dilutions of sera. Secondary injection was performed i.p. on day 22 with 100 μg of NP-CGG in PBS and is indicated in graph by a red arrow. Day 7b refers to sera obtained 7 days after the secondary injection. Total NP Abs were calculated using the B1–8 mAb as a standard. (A) Impaired affinity maturation in B6lpr/lpr compared to B6 mouse, immunized with NP-CGG. (B–C) Normal kinetics, quantities and affinity maturation in serum from B6lpr/+ immunized with NP-CGG.
4.4 Development of ANA in mice with other haplodeficiencies
It was conceivable that autoimmunity in B6lpr/+ mice was an idiosyncrasy of the lpr mutation or, more generally, of lymphocyte death receptors or lymphoproliferation. Therefore, we extended our analyses to other previously described SLE models in which genetic deficiencies are known to promote disease. In addition, as a more rigorous test of the mutation/autoimmunity hypothesis, we chose two genes, Cd22 and Pten, whose expression was primarily restricted to, or could be conditionally restricted to, the B cell lineage.
CD22 is a surface receptor found on mature B cells that dampens signals through the BCR by recruiting SHIP-1 to the BCR synapse via the CD22 cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM) domain. A homozygous deficiency in Cd22 promotes B cell hyperactivity and development of ANA (80). PTEN is also a signaling attenuator as well as a tumor suppressor. It dampens signaling by dephosphorylating end products of PI3 kinase. PTEN regulates a diversity of cellular processes such as proliferation, cell growth, survival and motility. Similar to CD22, a PTEN deficiency in B cells leads to development of ANA in an age-dependent manner(79). In addition, Pten+/− mice develop ANA (100).
We crossed CD19-Cre mice to Ptenflox/flox mice to produce F1 mice in which a heterozygous Pten deficiency was largely restricted to cells of the B lineage. Cohorts of female Cd22+/− and Ptenflox/+ Cd19-Cre mice were aged and their sera tested for the presence of IgG ANA. As shown in Fig. 6A, many of these mice (~40%) developed IgG ANA by 7 months of age. ANA developed in 4 of 10 Cd22+/− mice and in 9 of 21 Ptenflox/+ Cd19-Cre mice. In contrast, in the cohort of B6 mice that were 12 months old, only 1 of 11 developed ANA. As in the case of the B6lpr/+ mice, there was a clear and absolute distinction between ANA producers and nonproducers (Fig. 6A). In agreement with our previous findings, these results are consistent with a stochastic event leading to the development of ANA. Notably, ANA developed even in animals in which the haplodeficiency was largely restricted to the B cell lineage.
Figure 6. Stochastic ANA in Cd22 and Pten-conditional heterozygous deficient mice.
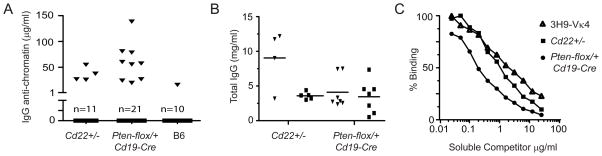
(A–B) IgG anti-chromatin antibodies and total IgG in sera of Cd22+/−, Pten flox/+ Cd19-Cre and B6 mice. Mice were segregated into anti-chromatin positive (▼) and anti-chromatin negative (■) groups as defined by a solid phase immunoassay. Assays were performed as described in Fig. 3. (C) Sera of ANA positive mice were incubated in calf chromatin-coated wells in the presence of competing soluble chromatin at the indicated concentrations. Bound IgG anti-chromatin Abs were detected using a goat anti-mouse IgG (SouthernBiotech). The high-affinity, anti-nuclear mAb 3H9/Vκ4 was included for comparison.
Although total IgG was higher in both ANA-producing groups (Fig. 6B), the increase was modest and could not account for the ANA signals detected in heterozygous mice, indicating that ANA were products of clonal selection. As an additional test of clonal selection in ANA development, we compared the relative affinities of the anti-chromatin antibodies in a competition assay with soluble chromatin. Figure 6C shows that soluble chromatin inhibited the binding of ANA from Cd22+/− and Ptenflox/+ CD19-Cre mice to immobilized chromatin at concentrations that were less than, or equivalent to, those required to compete with a high-affinity monoclonal ANA (3H9/Vκ4) derived from an autoimmune MRLlpr/lpr mouse (101). These results indicate that the serum IgG ANA in haplodeficient mice bound chromatin with affinities that were comparable to those of known high-affinity autoantibody products of somatic hypermutation derived from established mouse models of SLE.
These results also reinforce the interpretation that ANA in haplodeficient mice were derived from stochastic events involving one, or a few, B cell clones per mouse. It has been reported that antibody elicited during anti-hapten immune responses, in milligram/ml quantities, is derived from approximately 20 B cell clones (102). Using 3H9/Vκ4 mAb as a standard, we estimated ANA to be in the range of ~5–100 μg/ml (Fig. 3 and 6). However, the binding signal observed in an immunoassay is determined by total Ab concentration and affinity. Because our antibodies were of high-affinity relative to the standard, particularly those from the haplodeficient Pten mice, we can infer that the actual concentrations of ANA were less than or equal to our estimates. This supports the idea that ~one B cell clone participated in ANA production in a given animal. Thus, by all tested criteria, the observed ANA in haplodeficient mice appear to be specific products of clonal selection involving rare B cells.
Collectively, the preceding results demonstrate that autoantibodies develop in mice that are haplodeficient in genes associated with lymphocyte self-tolerance. ANA arose in mice that were heterozygous-deficient in every gene that we tested: a lymphocyte death receptor, an inositide phosphatase, and a cell surface attenuator of BCR signaling. This was true, even in cases in which the haplodeficiency was largely restricted to the B cell lineage. The all-or-nothing nature of ANA appearance in these mice housed in an SPF facility, a kinetic delay in ANA appearance, and a role for clonal selection in ANA development argue together for a causative stochastic event in autoimmunity.
5. Final Considerations
We believe that it is unlikely that a pathogen or a single genetic defect can be held accountable for the stochastic development of autoimmunity. In fact, the notion that immunity precedes autoimmunity is supported by the findings that virtually all of the ANA in both spontaneous non-Tg mouse models and human SLE patients are generated from normal B cells via the process of somatic hypermutation (34, 36). The antecedent B cell clones were probably driven into a T cell-dependent immune response, perhaps by a bacterium, a virus or an endogenous retrovirus, before acquiring somatic mutations that generated anti-nuclear specificities. We have reported evidence that such autoantibodies are generated frequently by somatic mutations in V region genes that convert AGC and AGT serine codons into arginine codons (36). Somatic mutations can also create neo antigenic epitopes for CD4 T cells in BCR-V regions of anti-nuclear B cells, providing an avenue of help to B cells by T cells that were not subjected to thymic deletion (72). Yet, clones producing these mutant autoantibodies are usually censored or autoimmunity would be the rule and not the exception. Thus, while it is plausible that infection may be an instigator of ANA development, we think it is unlikely to be the root cause.
We think the most plausible explanation for the stochastic nature of autoimmunity is a requirement for somatic mutagenesis in genes that are vital to immunoregulation. In the haplodeficient mice we examined, such mutations would likely occur in the wildtype allele and/or in other genes encoding products contributing a signaling pathway shared with the product of the wildtype allele. We favor this idea, in part, because of studies involving tumor suppressor genes, in which the inheritance of one deficient allele greatly increases the likelihood of cancer (93–96). In these studies, cancer was often associated with loss of heterozygosity due to somatic mutation in the wildtype allele or loss of the wildtype allele by genetic recombination. Recent studies by various sequencing consortia have revealed that cancer cells carry a substantial mutational load and that mutations in specific genes are associated with particular cancerous tissue types (103–109). Moreover, results of these studies indicate that somatic mutations in multiple driver genes are probably required to unleash a fully transformed phenotype. This is consistent with earlier kinetic studies indicating that multiple rate-limiting steps are involved in tumorigenesis (110). A kinetic delay in autoimmune disease is also observed in genetically susceptible mice. This is true even in reverse-genetic models in which animals inherit 2 null alleles of immunoregulatory genes. These observations imply that mutations in several genes may be required prior to the onset of an autoimmune response (Fig. 7).
Figure 7. Stochastic autoimmunity due to somatic mutations in immunoregulatory genes.
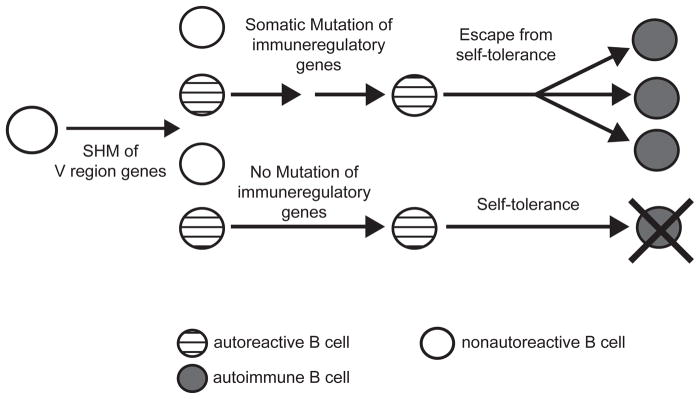
A nonautoreactive B cell gives rise to anti-nuclear B cells in the GC. Most such B cells are censored by self-tolerance mechanisms unless they acquire additional critical somatic mutations that alter or ablate the function of immunoregulatory genes involved in self-tolerance. Somatic mutations in immuneregulatory genes could arise in an AID-dependent and/or independent manner.
While the most straightforward interpretation of the haplodeficient mouse model is that somatic mutation played a causative role in ANA development, we are mindful that in order to formally prove this, the driver mutations must be identified. This will necessitate isolating ANA-specific B cells, which has not been possible to date due to technical considerations that include low frequencies of these cells and the complexity of the autoantigen. Nonetheless, we believe that the haplodeficient mouse model may ultimately prove to be a powerful device to assess the potential role of somatic mutagenesis in autoimmunity.
Acknowledgments
We would like to thank Judith Spiegel for proofreading the manuscript. This work was funded by grants from the National Institutes of Health: R01AI033613, R01AI073945, R03AI088408, and F30DK091102.
Footnotes
The authors declare no conflict of interest.
References
- 1.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303–306. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 2.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Advances in immunology. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 3.Flesher DL, Sun X, Behrens TW, Graham RR, Criswell LA. Recent advances in the genetics of systemic lupus erythematosus. Expert Rev Clin Immunol. 2010;6:461–479. doi: 10.1586/eci.10.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 5.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 6.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. The Journal of experimental medicine. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yachimovich-Cohen N, Fischel R, Bachar N, Yarkoni Y, Eilat D. Autoimmune NZB/NZW F1 mice utilize B cell receptor editing for generating high-affinity anti-dsDNA autoantibodies from low-affinity precursors. European Journal of Immunology. 2003;33:2469–2478. doi: 10.1002/eji.200324025. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Louzoun Y, Weigert M. Editing anti-DNA B cells by Vlambdax. The Journal of experimental medicine. 2004;199:337–346. doi: 10.1084/jem.20031712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nature Immunology. 2004;5:645–650. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 10.Benschop RJ, Aviszus K, Zhang X, Manser T, Cambier JC, Wysocki LJ. Activation and anergy in bone marrow B cells of a novel immunoglobulin transgenic mouse that is both hapten specific and autoreactive. Immunity. 2001;14:33–43. doi: 10.1016/s1074-7613(01)00087-5. [DOI] [PubMed] [Google Scholar]
- 11.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. The Journal of experimental medicine. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Jiang Y, Prak EL, Radic M, Weigert M. Editors and editing of anti-DNA receptors. Immunity. 2001;15:947–957. doi: 10.1016/s1074-7613(01)00251-5. [DOI] [PubMed] [Google Scholar]
- 13.Chen C, Li H, Tian Q, Beardall M, Xu Y, Casanova N, Weigert M. Selection of anti-double-stranded DNA B cells in autoimmune MRL-lpr/lpr mice. Journal of immunology. 2006;176:5183–5190. doi: 10.4049/jimmunol.176.9.5183. [DOI] [PubMed] [Google Scholar]
- 14.Kat I, Makdasi E, Fischel R, Eilat D. B-cell anergy is maintained in anti-DNA transgenic NZB/NZW mice. International Immunology. 2010;22:101–111. doi: 10.1093/intimm/dxp120. [DOI] [PubMed] [Google Scholar]
- 15.Fields ML, Erikson J. The regulation of lupus-associated autoantibodies: immunoglobulin transgenic models. Current Opinion in Immunology. 2003;15:709–717. doi: 10.1016/j.coi.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 16.Ota T, Ota M, Duong BH, Gavin AL, Nemazee D. Liver-expressed Igkappa superantigen induces tolerance of polyclonal B cells by clonal deletion not kappa to lambda receptor editing. The Journal of experimental medicine. 2011;208:617–629. doi: 10.1084/jem.20102265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kench JA, Russell DM, Nemazee D. Efficient peripheral clonal elimination of B lymphocytes in MRL/lpr mice bearing autoantibody transgenes. The Journal of experimental medicine. 1998;188:909–917. doi: 10.1084/jem.188.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 19.Rathmell JC, Cooke MP, Ho WY, Grein J, Townsend SE, Davis MM, Goodnow CC. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181–184. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 20.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nature Immunology. 2005;6:1160–1167. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
- 21.Steeves MA, Marion TN. Tolerance to DNA in (NZB x NZW)F1 mice that inherit an anti-DNA V(H) as a conventional micro H chain transgene but not as a V(H) knock-in transgene. Journal of immunology. 2004;172:6568–6577. doi: 10.4049/jimmunol.172.11.6568. [DOI] [PubMed] [Google Scholar]
- 22.Sekiguchi DR, Jainandunsing SM, Fields ML, Maldonado MA, Madaio MP, Erikson J, Weigert M, Eisenberg RA. Chronic graft-versus-host in Ig knockin transgenic mice abrogates B cell tolerance in anti-double-stranded DNA B cells. Journal of immunology. 2002;168:4142–4153. doi: 10.4049/jimmunol.168.8.4142. [DOI] [PubMed] [Google Scholar]
- 23.Brard F, Shannon M, Prak EL, Litwin S, Weigert M. Somatic mutation and light chain rearrangement generate autoimmunity in anti-single-stranded DNA transgenic MRL/lpr mice. The Journal of experimental medicine. 1999;190:691–704. doi: 10.1084/jem.190.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. The Journal of experimental medicine. 1999;189:1799–1814. doi: 10.1084/jem.189.11.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Li L, Kumar KR, Xie C, Lightfoot S, Zhou XJ, Kearney JF, Weigert M, Mohan C. Lupus susceptibility genes may breach tolerance to DNA by impairing receptor editing of nuclear antigen-reactive B cells. Journal of immunology. 2007;179:1340–1352. doi: 10.4049/jimmunol.179.2.1340. [DOI] [PubMed] [Google Scholar]
- 26.Mannoor K, Matejuk A, Xu Y, Beardall M, Chen C. Expression of Natural Autoantibodies in MRL-lpr Mice Protects from Lupus Nephritis and Improves Survival. Journal of immunology. 2012;188:3628–3638. doi: 10.4049/jimmunol.1102859. [DOI] [PubMed] [Google Scholar]
- 27.Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, Diaz M. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. Journal of immunology. 2007;178:7422–7431. doi: 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang C, Zhao ML, Scearce RM, Diaz M. Activation-induced deaminase-deficient MRL/lpr mice secrete high levels of protective antibodies against lupus nephritis. Arthritis and rheumatism. 2011;63:1086–1096. doi: 10.1002/art.30230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Werwitzke S, Trick D, Kamino K, Matthias T, Kniesch K, Schlegelberger B, Schmidt RE, Witte T. Inhibition of lupus disease by anti-double-stranded DNA antibodies of the IgM isotype in the (NZB x NZW)F1 mouse. Arthritis and rheumatism. 2005;52:3629–3638. doi: 10.1002/art.21379. [DOI] [PubMed] [Google Scholar]
- 31.Liang Z, Chen C, Mohan C. Molecular signatures of anti-nuclear antibodies: contributions of specific light chain residues and a novel New Zealand Black V kappa 1 germline gene. Journal of immunology. 2003;171:3886–3894. doi: 10.4049/jimmunol.171.7.3886. [DOI] [PubMed] [Google Scholar]
- 32.Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, Gonzalez JB, Pascual V, Stichweh D, Wardemann H, Nussenzweig MC. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9727–9732. doi: 10.1073/pnas.0803644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Jacobi AM, Wang T, Diamond B. Pathogenic autoantibodies in systemic lupus erythematosus are derived from both self-reactive and non-self-reactive B cells. Mol Med. 2008;14:675–681. doi: 10.2119/2008-00066.Zhang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNA autoantibodies. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9258–9263. doi: 10.1073/pnas.0500132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilfillan S, Dierich A, Lemeur M, Benoist C, Mathis D. Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science. 1993;261:1175–1178. doi: 10.1126/science.8356452. [DOI] [PubMed] [Google Scholar]
- 36.Guo W, Smith D, Aviszus K, Detanico T, Heiser RA, Wysocki LJ. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J Exp Med. 2010;207:2225–2237. doi: 10.1084/jem.20092712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vyse TJ, Rozzo SJ, Drake CG, Izui S, Kotzin BL. Control of multiple autoantibodies linked with a lupus nephritis susceptibility locus in New Zealand black mice. Journal of immunology. 1997;158:5566–5574. [PubMed] [Google Scholar]
- 38.Tsao BP, Cantor RM, Kalunian KC, Chen CJ, Badsha H, Singh R, Wallace DJ, Kitridou RC, Chen SL, Shen N, Song YW, Isenberg DA, Yu CL, Hahn BH, Rotter JI. Evidence for linkage of a candidate chromosome 1 region to human systemic lupus erythematosus. The Journal of clinical investigation. 1997;99:725–731. doi: 10.1172/JCI119217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molano ID, Redmond S, Sekine H, Zhang XK, Reilly C, Hutchison F, Ruiz P, Gilkeson GS. Effect of genetic deficiency of terminal deoxynucleotidyl transferase on autoantibody production and renal disease in MRL/lpr mice. Clinical Immunology. 2003;107:186–197. doi: 10.1016/s1521-6616(03)00035-4. [DOI] [PubMed] [Google Scholar]
- 40.Feeney AJ, Lawson BR, Kono DH, Theofilopoulos AN. Terminal deoxynucleotidyl transferase deficiency decreases autoimmune disease in MRL-Fas(lpr) mice. Journal of immunology. 2001;167:3486–3493. doi: 10.4049/jimmunol.167.6.3486. [DOI] [PubMed] [Google Scholar]
- 41.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. The Journal of experimental medicine. 1994;180:1295–1306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conde C, Weller S, Gilfillan S, Marcellin L, Martin T, Pasquali JL. Terminal deoxynucleotidyl transferase deficiency reduces the incidence of autoimmune nephritis in (New Zealand Black x New Zealand White)F1 mice. Journal of immunology. 1998;161:7023–7030. [PubMed] [Google Scholar]
- 43.Guth AM, Zhang X, Smith D, Detanico T, Wysocki LJ. Chromatin specificity of anti-double-stranded DNA antibodies and a role for Arg residues in the third complementarity-determining region of the heavy chain. J Immunol. 2003;171:6260–6266. doi: 10.4049/jimmunol.171.11.6260. [DOI] [PubMed] [Google Scholar]
- 44.Krishnan MR, Jou NT, Marion TN. Correlation between the amino acid position of arginine in VH-CDR3 and specificity for native DNA among autoimmune antibodies. Journal of immunology. 1996;157:2430–2439. [PubMed] [Google Scholar]
- 45.Li Z, Schettino EW, Padlan EA, Ikematsu H, Casali P. Structure-function analysis of a lupus anti-DNA autoantibody: central role of the heavy chain complementarity-determining region 3 Arg in binding of double- and single-stranded DNA. European Journal of Immunology. 2000;30:2015–2026. doi: 10.1002/1521-4141(200007)30:7<2015::AID-IMMU2015>3.0.CO;2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith DS, Creadon G, Jena PK, Portanova JP, Kotzin BL, Wysocki LJ. Di- and trinucleotide target preferences of somatic mutagenesis in normal and autoreactive B cells. Journal of immunology. 1996;156:2642–2652. [PubMed] [Google Scholar]
- 47.Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annual Review of Immunology. 1994;12:487–520. doi: 10.1146/annurev.iy.12.040194.002415. [DOI] [PubMed] [Google Scholar]
- 48.Shlomchik MJ, Aucoin AH, Pisetsky DS, Weigert MG. Structure and function of anti-DNA autoantibodies derived from a single autoimmune mouse. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:9150–9154. doi: 10.1073/pnas.84.24.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. The Journal of experimental medicine. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Connolly K, Roubinian JR, Wofsy D. Development of murine lupus in CD4-depleted NZB/NZW mice. Sustained inhibition of residual CD4+ T cells is required to suppress autoimmunity. Journal of immunology. 1992;149:3083–3088. [PubMed] [Google Scholar]
- 51.Peng SL, Cappadona J, McNiff JM, Madaio MP, Owen MJ, Hayday AC, Craft J. Pathogenesis of autoimmunity in alphabeta T cell-deficient lupus-prone mice. Clinical and Experimental Immunology. 1998;111:107–116. doi: 10.1046/j.1365-2249.1998.00424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peng SL, Madaio MP, Hughes DP, Crispe IN, Owen MJ, Wen L, Hayday AC, Craft J. Murine lupus in the absence of alpha beta T cells. Journal of immunology. 1996;156:4041–4049. [PubMed] [Google Scholar]
- 53.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sweet RA, Ols ML, Cullen JL, Milam AV, Yagita H, Shlomchik MJ. Facultative role for T cells in extrafollicular Toll-like receptor-dependent autoreactive B-cell responses in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7932–7937. doi: 10.1073/pnas.1018571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: a major immunogen for pathogenic autoantibody-inducing T cells of lupus. The Journal of experimental medicine. 1993;177:1367–1381. doi: 10.1084/jem.177.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaliyaperumal A, Mohan C, Wu W, Datta SK. Nucleosomal peptide epitopes for nephritis-inducing T helper cells of murine lupus. The Journal of experimental medicine. 1996;183:2459–2469. doi: 10.1084/jem.183.6.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi Y, Kaliyaperumal A, Lu L, Southwood S, Sette A, Michaels MA, Datta SK. Promiscuous presentation and recognition of nucleosomal autoepitopes in lupus: role of autoimmune T cell receptor alpha chain. The Journal of experimental medicine. 1998;187:367–378. doi: 10.1084/jem.187.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michaels MA, Kang HK, Kaliyaperumal A, Satyaraj E, Shi Y, Datta SK. A defect in deletion of nucleosome-specific autoimmune T cells in lupus-prone thymus: role of thymic dendritic cells. Journal of immunology. 2005;175:5857–5865. doi: 10.4049/jimmunol.175.9.5857. [DOI] [PubMed] [Google Scholar]
- 59.Singh RR, Kumar V, Ebling FM, Southwood S, Sette A, Sercarz EE, Hahn BH. T cell determinants from autoantibodies to DNA can upregulate autoimmunity in murine systemic lupus erythematosus. The Journal of experimental medicine. 1995;181:2017–2027. doi: 10.1084/jem.181.6.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hahn BH, Singh RR, Tsao BP, Ebling FM. Peptides from Vh regions of antibodies to DNA activate T cell help to upregulate autoantibody synthesis. Lupus. 1997;6:330–332. doi: 10.1177/096120339700600329. [DOI] [PubMed] [Google Scholar]
- 61.Weiss S, Bogen B. B-lymphoma cells process and present their endogenous immunoglobulin to major histocompatibility complex-restricted T cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:282–286. doi: 10.1073/pnas.86.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bartnes K, Hannestad K. Engagement of the B lymphocyte antigen receptor induces presentation of intrinsic immunoglobulin peptides on major histocompatibility complex class II molecules. European Journal of Immunology. 1997;27:1124–1130. doi: 10.1002/eji.1830270512. [DOI] [PubMed] [Google Scholar]
- 63.Snyder CM, Zhang X, Wysocki LJ. Negligible class II MHC presentation of B cell receptor-derived peptides by high density resting B cells. Journal of immunology. 2002;168:3865–3873. doi: 10.4049/jimmunol.168.8.3865. [DOI] [PubMed] [Google Scholar]
- 64.Rudensky AY, V, Yurin L. Immunoglobulin-specific T-B cell interaction. I. Presentation of self immunoglobulin determinants by B lymphocytes. European Journal of Immunology. 1989;19:1677–1683. doi: 10.1002/eji.1830190923. [DOI] [PubMed] [Google Scholar]
- 65.Holmoy T, Fredriksen AB, Thompson KM, Hestvik AL, Bogen B, Vartdal F. Cerebrospinal fluid T cell clones from patients with multiple sclerosis: recognition of idiotopes on monoclonal IgG secreted by autologous cerebrospinal fluid B cells. European Journal of Immunology. 2005;35:1786–1794. doi: 10.1002/eji.200425417. [DOI] [PubMed] [Google Scholar]
- 66.Hestvik AL, Vartdal F, Fredriksen AB, Thompson KM, Kvale EO, Skorstad G, Bogen B, Holmoy T. T cells from multiple sclerosis patients recognize multiple epitopes on Self-IgG. Scandinavian journal of immunology. 2007;66:393–401. doi: 10.1111/j.1365-3083.2007.01955.x. [DOI] [PubMed] [Google Scholar]
- 67.Detanico T, Heiser RA, Aviszus K, Bonorino C, Wysocki LJ. Self-tolerance checkpoints in CD4 T cells specific for a peptide derived from the B cell antigen receptor. Journal of immunology. 2011;187:82–91. doi: 10.4049/jimmunol.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eyerman MC, Zhang X, Wysocki LJ. T cell recognition and tolerance of antibody diversity. J Immunol. 1996;157:1037–1046. [PubMed] [Google Scholar]
- 69.Guo W, Smith D, Guth A, Aviszus K, Wysocki LJ. T cell tolerance to germline-encoded antibody sequences in a lupus-prone mouse. J Immunol. 2005;175:2184–2190. doi: 10.4049/jimmunol.175.4.2184. [DOI] [PubMed] [Google Scholar]
- 70.Bogen B, Jorgensen T, Hannestad K. T helper cell recognition of idiotopes on lambda 2 light chains of M315 and T952: evidence for dependence on somatic mutations in the third hypervariable region. European Journal of Immunology. 1985;15:278–281. doi: 10.1002/eji.1830150313. [DOI] [PubMed] [Google Scholar]
- 71.Eyerman MC, Wysocki L. T cell recognition of somatically-generated Ab diversity. Journal of immunology. 1994;152:1569–1577. [PubMed] [Google Scholar]
- 72.Zhang X, Smith DS, Guth A, Wysocki LJ. A receptor presentation hypothesis for T cell help that recruits autoreactive B cells. J Immunol. 2001;166:1562–1571. doi: 10.4049/jimmunol.166.3.1562. [DOI] [PubMed] [Google Scholar]
- 73.Munthe LA, Os A, Zangani M, Bogen B. MHC-restricted Ig V region-driven T-B lymphocyte collaboration: B cell receptor ligation facilitates switch to IgG production. J Immunol. 2004;172:7476–7484. doi: 10.4049/jimmunol.172.12.7476. [DOI] [PubMed] [Google Scholar]
- 74.Snyder CM, Aviszus K, Heiser RA, Tonkin DR, Guth AM, Wysocki LJ. Activation and tolerance in CD4(+) T cells reactive to an immunoglobulin variable region. The Journal of experimental medicine. 2004;200:1–11. doi: 10.1084/jem.20031234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vyse TJ, Kotzin BL. Genetic susceptibility to systemic lupus erythematosus. Annual Review of Immunology. 1998;16:261–292. doi: 10.1146/annurev.immunol.16.1.261. [DOI] [PubMed] [Google Scholar]
- 76.Wakeland EK, Liu K, Graham RR, Behrens TW. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397–408. doi: 10.1016/s1074-7613(01)00201-1. [DOI] [PubMed] [Google Scholar]
- 77.Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Advances in immunology. 2006;92:1–69. doi: 10.1016/S0065-2776(06)92001-X. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 79.Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, Nakano T. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med. 2003;197:657–667. doi: 10.1084/jem.20021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O’Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med. 1999;189:1307–1313. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burnet FM. Autoimmunity and Autoimmune Disease. Medical and Technical Publishing Co Ttd; Lancaster, UK: 1972. [Google Scholar]
- 82.Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, Blanche S, Bartunkova J, Vilmer E, Fischer A, Le Deist F, Rieux-Laucat F. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–1418. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- 83.Oliveira JB, Bidere N, Niemela JE, Zheng L, Sakai K, Nix CP, Danner RL, Barb J, Munson PJ, Puck JM, Dale J, Straus SE, Fleisher TA, Lenardo MJ. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8953–8958. doi: 10.1073/pnas.0702975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takagi M, Shinoda K, Piao J, Mitsuiki N, Matsuda K, Muramatsu H, Doisaki S, Nagasawa M, Morio T, Kasahara Y, Koike K, Kojima S, Takao A, Mizutani S. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–2890. doi: 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- 85.Niemela JE, Lu L, Fleisher TA, Davis J, Caminha I, Natter M, Beer LA, Dowdell KC, Pittaluga S, Raffeld M, Rao VK, Oliveira JB. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117:2883–2886. doi: 10.1182/blood-2010-07-295501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hao Z, Hampel B, Yagita H, Rajewsky K. T cell-specific ablation of Fas leads to Fas ligand-mediated lymphocyte depletion and inflammatory pulmonary fibrosis. J Exp Med. 2004;199:1355–1365. doi: 10.1084/jem.20032196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hao Z, Duncan GS, Seagal J, Su YW, Hong C, Haight J, Chen NJ, Elia A, Wakeham A, Li WY, Liepa J, Wood GA, Casola S, Rajewsky K, Mak TW. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity. 2008;29:615–627. doi: 10.1016/j.immuni.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, Fink PJ, Chervonsky AV. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fukuyama H, Adachi M, Suematsu S, Miwa K, Suda T, Yoshida N, Nagata S. Requirement of Fas expression in B cells for tolerance induction. Eur J Immunol. 2002;32:223–230. doi: 10.1002/1521-4141(200201)32:1<223::AID-IMMU223>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 90.Fukuyama H, Adachi M, Suematsu S, Miwa K, Suda T, Yoshida N, Nagata S. Transgenic expression of Fas in T cells blocks lymphoproliferation but not autoimmune disease in MRL-lpr mice. J Immunol. 1998;160:3805–3811. [PubMed] [Google Scholar]
- 91.Komano H, Ikegami Y, Yokoyama M, Suzuki R, Yonehara S, Yamasaki Y, Shinohara N. Severe impairment of B cell function in lpr/lpr mice expressing transgenic Fas selectively on B cells. Int Immunol. 1999;11:1035–1042. doi: 10.1093/intimm/11.7.1035. [DOI] [PubMed] [Google Scholar]
- 92.Cuda CM, Agrawal H, Misharin AV, Haines GK, 3rd, Hutcheson J, Weber E, Schoenfeldt JA, Mohan C, Pope RM, Perlman H. Requirement of myeloid cell-specific Fas expression for prevention of systemic autoimmunity in mice. Arthritis and rheumatism. 2012;64:808–820. doi: 10.1002/art.34317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maher ER, Yates JR, Ferguson-Smith MA. Statistical analysis of the two stage mutation model in von Hippel-Lindau disease, and in sporadic cerebellar haemangioblastoma and renal cell carcinoma. J Med Genet. 1990;27:311–314. doi: 10.1136/jmg.27.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chompret A, Brugieres L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, Hua D, Ligot L, Dondon MG, Bressac-de Paillerets B, Frebourg T, Lemerle J, Bonaiti-Pellie C, Feunteun J. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–1937. doi: 10.1054/bjoc.2000.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–198. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 97.Jachez B, Montecino-Rodriguez E, Fonteneau P, Loor F. Partial expression of the lpr locus in the heterozygous state: presence of autoantibodies. Immunology. 1988;64:31–36. [PMC free article] [PubMed] [Google Scholar]
- 98.Mastache EF, Lindroth K, Fernandez C, Gonzalez-Fernandez A. Somatic hypermutation of Ig genes is affected differently by failures in apoptosis caused by disruption of Fas (lpr mutation) or by overexpression of Bcl-2. Scand J Immunol. 2006;63:420–429. doi: 10.1111/j.1365-3083.2006.001758.x. [DOI] [PubMed] [Google Scholar]
- 99.Takahashi Y, Ohta H, Takemori T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity. 2001;14:181–192. doi: 10.1016/s1074-7613(01)00100-5. [DOI] [PubMed] [Google Scholar]
- 100.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 101.Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805–811. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- 102.Blier PR, Bothwell A. A limited number of B cell lineages generates the heterogeneity of a secondary immune response. Journal of immunology. 1987;139:3996–4006. [PubMed] [Google Scholar]
- 103.Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman C, Varela I, Nik-Zainal S, Davies HR, Ordonez GR, Mudie LJ, Latimer C, Edkins S, Stebbings L, Chen L, Jia M, Leroy C, Marshall J, Menzies A, Butler A, Teague JW, Mangion J, Sun YA, McLaughlin SF, Peckham HE, Tsung EF, Costa GL, Lee CC, Minna JD, Gazdar A, Birney E, Rhodes MD, McKernan KJ, Stratton MR, Futreal PA, Campbell PJ. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, Ha C, Johnson S, Kennemer MI, Mohan S, Nazarenko I, Watanabe C, Sparks AB, Shames DS, Gentleman R, de Sauvage FJ, Stern H, Pandita A, Ballinger DG, Drmanac R, Modrusan Z, Seshagiri S, Zhang Z. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 105.Pleasance ED, Cheetham RK, Stephens PJ. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 110.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975:15. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]