

Abstract
Purpose.
We investigated the evidence of recent positive selection in the human phototransduction system at single nucleotide polymorphism (SNP) and gene level.
Methods.
SNP genotyping data from the International HapMap Project for European, Eastern Asian, and African populations was used to discover differences in haplotype length and allele frequency between these populations. Numeric selection metrics were computed for each SNP and aggregated into gene-level metrics to measure evidence of recent positive selection. The level of recent positive selection in phototransduction genes was evaluated and compared to a set of genes shown previously to be under recent selection, and a set of highly conserved genes as positive and negative controls, respectively.
Results.
Six of 20 phototransduction genes evaluated had gene-level selection metrics above the 90th percentile: RGS9, GNB1, RHO, PDE6G, GNAT1, and SLC24A1. The selection signal across these genes was found to be of similar magnitude to the positive control genes and much greater than the negative control genes.
Conclusions.
There is evidence for selective pressure in the genes involved in retinal phototransduction, and traces of this selective pressure can be demonstrated using SNP-level and gene-level metrics of allelic variation. We hypothesize that the selective pressure on these genes was related to their role in low light vision and retinal adaptation to ambient light changes. Uncovering the underlying genetics of evolutionary adaptations in phototransduction not only allows greater understanding of vision and visual diseases, but also the development of patient-specific diagnostic and intervention strategies.
Keywords: Homo sapiens, evolution, recent positive selection, vision, phototransduction
Evidence of recent positive selection in the human phototransduction system was evaluated in European, Eastern Asian, and African populations. Six phototransduction genes associated with retinal adaption to changing light levels were found to have strong evidence of recent positive selection.
Introduction
Understanding the genetic basis of evolutionary adaptations in vision will help understanding vision and its diseases, as well as our understanding of selective processes.1 One approach to look for evidence of recent selection in humans is observation of the distortion in polymorphism patterns across the genome among different human populations. Even the patterns at neutral sites can reveal evidence of selection caused by selective pressure applied to the alleles at genetically linked sites. Thus, observation of selection is possible within haplotype blocks, regions of the genome where relatively little historic recombination has occurred.2 These regions are the hallmarks of the linkage disequilibrium, which has persisted throughout the lineage of anatomically modern humans, most likely arising less than 150 to 200 thousand years ago (ka).3 In contrast, Homo sapiens only recently (within the last 100 ka, for the most likely out of Africa models) has distributed itself from its origin over the globe.4 During this migration, selective pressure was present constantly and has been shown to persist to the last century.5,6 The new habitats that H. sapiens encountered were substantially different from the African habitat, which probably consisted of savannah-like environments.7,8 Recent evolutionary adaptations that are advantageous in these new habitats have been detected in non–sub-Saharan African populations, including in the lactose tolerance gene LCT,9 salt regulation genes at the CYP3A cluster,10 disease resistance genes, G6PD and CASP12,11,12 and pigmentation genes SLC24A5 and MATP.13,14
Although some investigators have hypothesized that selective pressure has resulted in structural differences in visual organs,15 to our knowledge relatively little work has been done to identify the specific set of vision-related genes that may be under selective pressure. The basic structure of the single-lens camera eye, its ciliary photoreceptor cells, and the process of phototransduction have been highly conserved for at least 400 million years (Ma), with only modest structural differences between gnathostome species.16 Because of the visual system's role in overall fitness, many of the involved genes also have been highly conserved.16,17 However, the Asian and European habitats differed from the African in the lower mean illuminance levels, diurnal and seasonal illuminance cycles, cover provided in forested areas, changes in illuminance associated with the use of covered shelters, and increased variance in ground cover reflectivity.7,8,18–20 These new environments could have exerted selective pressure on the functional systems in the human eye.
Specifically, we focus on evidence of selection within the genes involved in phototransduction. Visual phototransduction is the biologic process by which light is converted into an electric signal in the photoreceptor cells in the retina of the eye. It involves, at a minimum, absorption of a photon by an opsin holoprotein-associated chromophore, activation of a GTP binding protein that in turn activates phosphodiesterase, conversion of cGMP to GMP, and closing of the constitutively open cGMP gated channels.21 The known set of interacting proteins responsible for phototransduction are encoded by the following genes: RHO, OPN1SW, OPN1MW, OPN1LW, SAG, GRK1, GRK7, RCVRN, GNAT1, GNAT2, GNGT1, GNGT2, GNB1, RGS9, RGS16, PDE6A, PDE6B, PDE6C, PDE6D, PDE6G, PDE6H, GUCY2D, GUCY2F, GUCA1A, GUCA1B, SLC24A1, CNGA1, CNGA3, CNGB1, CNGB3, and CACNA1F. See Figure 1 for an illustration of the interactions of phototransduction genes in rod photoreceptors.
Figure 1.

Phototransduction pathway in rod cells. The schematic illustrates the interactions between proteins during phototransduction. The genes encoding each protein in the pathway are overlaid on the schematic.
Because comprehensive functional data at the molecular and organismal scales on these genes currently are unavailable, we used the classic genetic determination of selection, that is, patterns of allelic variation that are not consistent with a neutral model of segregation. Specifically, we leveraged the data from the large-scale genotyping endeavors of the International HapMap Project and the 1000 Genomes project to identify patterns of allelic variation among populations.22,23
Patterns of allelic variation can differ from the expectations of neutrality in multiple ways. Two important characteristics of allelic variation in the presence of selection are long haplotypes and large variance in allele frequencies among populations.24–26 Long haplotypes arise from selected variants quickly reaching high frequency before recombination breaks the associations with nearby polymorphisms.25 The variance in allele frequencies among populations is a result of the differential selection occurring in these populations—a variant and its linked polymorphisms rise in frequency in one population relative to others because of greater selective pressure in that population relative to the others.26 Here, we evaluated polymorphisms using two metrics that attempt to measure each of these aspects of allelic variation from neutrality. Extended haplotype homozygosity per site (EHHS) and its related metrics are used to quantify the length of haplotypes surrounding each polymorphism and compare these lengths across populations to find sites where differential selection is occurring. Fixation index (Fst) is used to quantify the level of differential selection by comparing directly the observed level of variance in allele frequency across the populations to the expected level.
The hypotheses of this pilot study are that Asians and Europeans underwent selective pressure to adapt to changing visual environments, and that the selective pressure on the phototransduction system can be demonstrated using metrics of allelic variation at the gene-level.
Materials and Methods
Single Nucleotide Polymorphism (SNP) Data and Quality Control
The analyses were performed using data generated by the international HapMap project.22 Specifically, SNP genotypes generated during phase 3 (release 28) of the HapMap project for the European in Utah (CEU), Han Chinese in Beijing (CHB), and Africans in Yoruba (YRI) populations were used in this work. The HapMap data used here were generated using assays from 450 individuals (180 in CEU, 90 in CHB, and 180 in YRI). No human or animal testing was performed as part of this study and all analyses were performed on publicly available data.
The HapMap SNPs were filtered based on the following criteria. First, to avoid sex-chromosome related complexities only autosomal SNPs were considered. Second, to maximize discriminatory power, only SNPs genotyped in all of the populations were considered. Finally, SNPs that were missing in a high percentage (>2%) of samples were excluded. The resulting filtered set of SNPs was used in all analyses.
EHHS-Based Statistics
EHHS values were calculated for all SNPs in each of the three populations.25,27 For each SNP, its EHHS signal was calculated over the local region until the value decayed to 0.1.27–29 The integrated EHHS (iES) values for each SNP then were determined by integrating the SNP EHHS signal over the local region. SNP EHHS and iES were calculated using the publicly available REHH package for R.30
Missing genotype values were filled in using a probabilistic algorithm described by Tang et al. before the EHHS was calculated.27 This algorithm assigns the missing genotype for an SNP based upon the genotype at the previous SNP and the conditional probability calculated using the nonmissing data at those two SNPs.27
The ln(Rsb) statistic, which compares EHHS for the same SNP in two different populations, was calculated as the log ratio of the SNP iES value in the population of interest to the iES value in the YRI population for the CEU and CHB populations as this is assumed to be the ancestral population.27 Finally, the ln(Rsb) values for all SNPs within each population were normalized to have a median of zero and variance of one. The median, rather than the mean, was used for normalization to reduce the effects of outlier ln(Rsb) values.
Fst Statistics
Fst was calculated from the genotype type data using a previously described method.31,32 Briefly, this method estimated Fst using the ratio of observed variance in allele frequency across populations to the expected variance as shown in Equation 1.
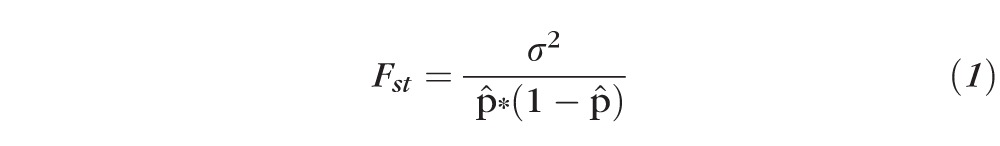 |
The observed allele frequency variance for each SNP, σ2, was calculated directly using HapMap genotype data. The expected variance for each SNP was estimated from the data using the weighted average of allele frequency, p̂, in the populations under comparison.
Fst values were computed for each SNP in the CEU and CHB populations. In each case, the population of interest was compared to the YRI population.27
Extending SNP Level Statistics to the Gene Level
Two procedures were used to extend the above SNP level statistics to summary measurements at the gene level. For ln(Rsb), a gene-level statistic was calculated as the sum of squared ln(Rsb) values for all SNPs within a gene, normalized by the number of SNPs within that gene.
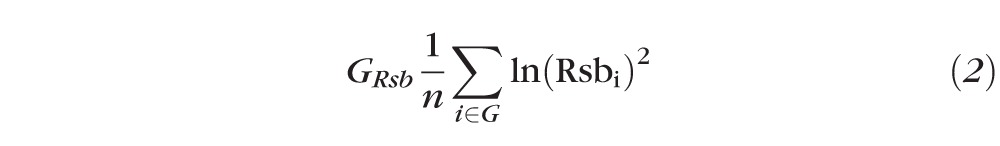 |
Here, the gene-level metric GRsb was computed for gene G based upon the ln(Rsb) value for each SNP i in G and the total number of SNPs in G, n.
For Fst, the gene-level statistic GFst was calculated as the median Fst value of all SNPs within a gene G.
 |
Genomic bounds for known protein-coding genes retrieved from Ensembl (build 65) were used to assign SNP membership to genes.33 GRsb and GFst values were calculated for all known protein-coding genes in the CEU and CHB populations. Using the distributions of GRsb and GFst for all genes, the corresponding percentiles were calculated for each of the phototransduction genes in both populations. These percentiles serve as an estimate of the relative amount of positive selection occurring on each gene.
Definition of Reference Positive and Negative Selection Gene Sets
We chose a set of positively selected, nonphototransduction genes that included EDAR, LCT, OCA2, and ABCB1, as these all have been widely reported to be undergoing recent positive selection.28,29,34–39 We chose a set of negatively selected genes from human housekeeping genes,40 as these genes are constitutively active and any mutations will have systemic effects. Therefore, they are expected to be under negative selective pressure. A previously published meta-analysis of gene expression data across a wide variety of H. sapiens tissues, generated an ordered set of putative housekeeping genes.40 The top genes from this set according to FPEI, a homogeneity metric that expresses the ubiquity of that gene expression, and which contained at least 10 SNPs from the filtered set considered here. The resulting negatively selected genes were LDHA, CFL1, HEATR2, and ATP5L.
Results
After quality control, a resulting set of 1,108,091 SNPs was used for EHHS and Fst analyses. The ln(Rsb) and Fst measurements of SNP-level selection had a low-level but consistent correlation of 0.227 in the CEU population and 0.225 in the CHB population. Heat maps showing the ln(Rsb) and Fst joint distributions for all SNPs are shown in Figure 2. The correlation observed is consistent with previous research that has indicated complementarity between these selection metrics.41,42 That is, while both metrics are useful for quantifying evidence for positive selection, they focus on different genetic signatures associated with selection.
Figure 2.

Joint distribution of ln(Rsb) and Fst for genome-wide SNPs. Heat maps indicating the density of the positive selection measures for 1,108,091 autosomal SNPs in the CEU and CHB populations. Top: distribution across the full range of Fst values. Bottom: distribution zoomed in on Fst values between 0.0 and 0.1.
The Table details the gene-level summary measurements for all autosomal phototransduction genes. The criterion applied to determine the genes under positive selection from the set of all phototransduction genes was the existence of a gene-level metric (GRsb or GFst) greater than the 90th percentile value in either the CEU or CHB population. This criterion was chosen instead of a more conservative one to increase the sensitivity for genes under positive selection. Several phototransduction genes showed evidence of recent positive selection in CHB and/or CEU. The genes PDE6G, GNAT1, SLC24A1, and RGS9 all resulted in gene-level selection measurements greater than 90% of all known protein-coding genes in the CHB population. For the CEU population, this set of genes consisted of RHO, GNB1, GNAT1, and RGS9. The union of these was considered to be the set phototransduction genes under recent positive selection, and consisted of the six genes: RGS9, GNB1, RHO, SLC24A1, PDE6G, and GNAT1.
Table.
GRsb and GFst Statistics for Autosomal Phototransduction Genes for the CHB and CEU Populations
|
Gene |
No. SNPs |
CHB |
CEU |
||||||
|
GRsb |
% |
GFst |
% |
GRsb |
% |
GFst |
% |
||
| RGS9* | 36 | 1.397 | 0.77 | 0.197 | 0.92 | 7.669 | 0.98 | 0.211 | 0.95 |
| GNB1* | 26 | 1.259 | 0.75 | 0.136 | 0.85 | 1.060 | 0.7 | 0.257 | 0.97 |
| RHO* | 2 | 0.312 | 0.28 | 0.108 | 0.79 | 6.381 | 0.97 | 0.370 | 0.99 |
| SLC24A1* | 10 | 4.955 | 0.95 | 0.105 | 0.78 | 1.791 | 0.83 | 0.014 | 0.15 |
| PDE6G* | 6 | 3.631 | 0.92 | 0.082 | 0.7 | 0.226 | 0.18 | 0.063 | 0.65 |
| GNAT1* | 2 | 0.497 | 0.44 | 0.576 | 0.99 | 0.109 | 0.07 | 0.193 | 0.94 |
| PDE6D | 18 | 2.553 | 0.88 | 0.017 | 0.17 | 1.417 | 0.77 | 0.050 | 0.56 |
| CNGB1 | 59 | 0.333 | 0.3 | 0.087 | 0.73 | 0.467 | 0.41 | 0.077 | 0.72 |
| RGS16 | 1 | 0.403 | 0.37 | 0.035 | 0.37 | 0.654 | 0.54 | 0.128 | 0.87 |
| SAG | 25 | 0.798 | 0.62 | 0.120 | 0.82 | 0.364 | 0.32 | 0.016 | 0.18 |
| GNGT1 | 128 | 0.760 | 0.6 | 0.026 | 0.27 | 0.691 | 0.56 | 0.038 | 0.45 |
| GNGT2 | 10 | 0.111 | 0.07 | 0.040 | 0.42 | 0.305 | 0.26 | 0.005 | 0.06 |
| GNAT2 | 5 | 1.045 | 0.7 | 0.028 | 0.29 | 0.592 | 0.5 | 0.019 | 0.21 |
| PDE6A | 36 | 0.479 | 0.43 | 0.032 | 0.34 | 0.505 | 0.43 | 0.070 | 0.69 |
| PDE6B | 11 | 0.269 | 0.23 | 0.009 | 0.09 | 1.099 | 0.71 | 0.058 | 0.62 |
| GUCY2D | 7 | 2.272 | 0.86 | 0.035 | 0.37 | 0.294 | 0.25 | 0.032 | 0.38 |
| GUCA1A | 21 | 1.815 | 0.82 | 0.117 | 0.81 | 1.607 | 0.80 | 0.014 | 0.15 |
| GUCA1B | 10 | 2.009 | 0.84 | 0.147 | 0.86 | 0.681 | 0.55 | 0.093 | 0.77 |
| OPN1SW | 3 | 0.036 | 0.02 | 0.001 | 0.02 | 0.549 | 0.47 | 0.065 | 0.66 |
| RCVRN | 9 | 0.055 | 0.03 | 0.007 | 0.07 | 0.536 | 0.46 | 0.016 | 0.18 |
| CNGA1 | 32 | 0.140 | 0.1 | 0.029 | 0.3 | 0.169 | 0.12 | 0.018 | 0.19 |
| GRK7 | 10 | 0.132 | 0.09 | 0.093 | 0.75 | 0.136 | 0.11 | 0.051 | 0.56 |
| PDE6H | 9 | 0.821 | 0.63 | 0.006 | 0.06 | 0.049 | 0.04 | 0.006 | 0.07 |
| PDE6C | 42 | 0.877 | 0.65 | 0.030 | 0.31 | 0.379 | 0.33 | 0.074 | 0.71 |
| CNGA3 | 26 | 2.055 | 0.84 | 0.025 | 0.26 | 2.130 | 0.86 | 0.032 | 0.38 |
| CNGB3 | 95 | 0.700 | 0.56 | 0.063 | 0.57 | 0.549 | 0.47 | 0.037 | 0.44 |
The raw values for each gene are shown along with the gene percentile within each population.
Achieved GRsb or GFst above 90th percentile in at least one of the populations.
SNP-level selection measurements of known positively-selected, negatively-selected, and phototransduction genes are shown in Figure 3. Here, ln(Rsb) and Fst values of each SNP in these genes are plotted against their position within the genes. The chromosomal location of each gene is shown along with SNP-level values for the CEU and CHB populations are shown. Plots of SNP-level values for the set of known positively-selected (Fig. 3A) and negatively-selected genes (Fig. 3B) are included for comparison. As expected, the known positively-selected genes exhibited high magnitude SNP-level selection metrics along their lengths relative to the negatively-selected genes and most of the phototransduction genes.
Figure 3. .

Signal of positive selection across genes. SNP ln(Rsb) and Fst values across the length of some positively-selected (A), negatively-selected (B), and phototransduction genes (C).
Within the set of phototransduction genes under recent positive selection (Fig. 3C), SNP-level ln(Rsb) and Fst values revealed distinct levels and modes of selection acting on the genes. The SNP-level measurements for gene RGS9 showed elevated values across the length of the gene as well as distinct peaks along the length of the gene, covering several exons near the 3′ end of the gene. The SNP-level measurements of gene GNB1, on the other hand, revealed a lower magnitude, uniform selection across the entire length of the gene. Interpretation of SNP-level measurements for some of these genes (e.g., RHO, GNAT1) is limited by the lack of dense SNP coverage.
Discussion
Our results indicated that there is evidence for selective pressure in the genes involved in retinal phototransduction, and that traces of this selective pressure can be demonstrated using SNP- and gene-level ln(Rsb) and Fst metrics of allelic variation.
The joint distributions of ln(Rsb) and Fst measurements (Fig. 1) are consistent with the fact that these measure different aspects of selection. Specifically, ln(Rsb) measures the differences in haplotype length surrounding a SNP, while Fst measures the variance in allele variances at the SNP. Here, both were used to increase the overall sensitivity of finding phototransduction genes undergoing recent positive selection. Additionally, these SNP-level measurements were extended to simple gene-level metrics so that evidence for positive selection could be evaluated at the gene level.
Genes shown previously to be under positive selection were used as positive controls. Three of these four genes (EDAR, OCA2, and LCT) corresponded to a GRsb or GFst value that was at the 90th percentile or higher in at least one the populations. Figure 3 also shows that the selection signals at SNPs in the genomic regions surrounding these genes were consistently high in most cases. The fourth gene, ABCB1, did not achieve GRsb or GFst above the 90th percentile, but has consistent, low-level evidence of selection across its length. In contrast, the SNPs in genes serving as negative controls (LDHA, CFL1, HEATR, and ATP5L) had greatly diminished signals.
The set of retinal phototransduction genes with relatively strong evidence for recent positive selection at the gene level consists of RGS9, RHO, GNB1, PDE6G, GNAT1, and SLC24A1, of which the RGS9 has the most striking magnitude of Fst and ln(Rsb) metrics at the SNP level. GNB1 exhibited high magnitude SNP-level Fst signals, but a consistently negative ln(Rsb) signal, possibly indicating selected variants arose and quickly increased in frequency in the African population due to selective pressures not encountered by populations leaving Africa.
Determining specific genes under positive selection by extending the SNP-level selection measurements to the gene-level summary metrics allows existing knowledge of gene functions and disease associations to be leveraged in developing hypotheses of the functional significance of the positive selection. Deleterious mutations in the selected retinal phototransduction genes have been associated with reduced visual acuity in low light environments and abnormalities in retinal adaptation to changes in ambient light. Specifically, mutations in RGS9 have been found to be associated with bradyopsia, a disease characterized by extreme delay in retinal adaptation to bright and dark light.43 Mutations in PDE6G in mouse models have been shown associated with delayed retinal adaptation to rapid changes in light level in mice.44 The genes GNAT1, RHO, and SLC24A1 have been associated with either dominant or recessive forms of congenital stationary night blindness.45–47 Among these selected genes, GNB1 is notable for its lack of specificity to phototransduction. It is expressed in a wide variety of tissue and is involved many biological processes.48 Selection acting on this gene may be caused by pressure on those processes, or could have functional consequences beyond phototransduction. It also should be noted that this selected set consists of genes active in phototransduction in rods only (RHO, GNAT1, GNB1, and PDE6G) or in rods and cones (RGS9 and SLC24A1). No cone-specific genes were found among the most highly selected set of phototransduction genes.
Based on functional annotation and known disease associations of the selected genes, the proteins encoded by these genes are likely to have an important role in low light vision and retinal adaptation to ambient light changes. The rod-focused nature of the observed selection may suggest that improved acuity in low and intermediate light (scotopic and mesopic) rather than higher light (photopic) conditions was an important driver of the observed selection. Previous work on two of the selected genes (RGS9 and PDE6G) also shows that mutations in these genes have dramatic effects on retinal adaptation to changing light levels and more recent work also has found a signature of selection acting on RGS9.49 This suggests that an increased ability or immediacy in adapting to changing light levels also could have driven the selection. Based only on the observed levels of selection on these genes, we cannot distinguish between these hypotheses or preclude the possibility that some other functional consideration is driving the selection.
Upon migrating to higher latitudes, H. sapiens would have encountered very different lighting environments than what was typical of their previous habitats. Lower mean illuminance levels, more pronounced diurnal and seasonal cycles, forest or foliage cover, and the use of covered shelters would have increased the importance of maintaining visual acuity in low light conditions. Additionally, an increased exposure to rapidly changing light levels would have resulted from the traversing through forested areas and snow-covered tundras as well as entering or exiting covered shelters. Changes in the environmental albedo (proportion of light reflected) also could have increased the exposure to rapidly changing light levels and visual scenes with a high dynamic range of illumination. The albedo values typical of higher latitude ground cover, from deep forests to light vegetation and tundra to complete snow cover (0.08–0.9), exhibit much higher variance compared to those of the savannah-like environments encountered by ancient humans in Africa (0.15–0.30).18–20,50 H. sapiens capable of maintaining high visual acuity in low light conditions and/or rapid retinal adaptation to changing light levels during hunting and predator avoidance would have a selective advantage in these habitats.
The Fst and EHHS-based quantitative metrics used here are able to distinguish the observed allelic patterns that are likely due to selective pressure from those expected under neutral selection. While these results suggested that a subset of phototransduction genes have been under recent positive selection, they cannot confirm the functional significance of this selection. Therefore, this study is limited by the lack of ability to identify uniquely the predominant driving force behind the observed positive selection in the phototransduction system. Without direct functional evidence, it is impossible to determine the relative importance of increased visual acuity in low light conditions, more rapid adaptation to changing light levels, or even some other functional change in conferring a selective advantage.
The gene-level selection metrics presented are subject to a few limitations that should be noted. The evaluation of the numeric metrics of selections could be performed only at known SNPs, and several genes (RHO, GNAT1, and PDE6G) contained only a very small number of SNPs. Any noise in the SNP-level measurements could result in serious biases in the gene-level metrics. In addition, the gene-level selection metrics presented may not be ideal for characterizing the most recently occurring selection. In general, more recent selection is associated with large regions (hundreds of thousands to millions of bases) exhibiting elevated selection metrics because insufficient time has passed for these regions to be fragmented thoroughly by recombination events. Because many of the genes considered span much smaller regions (tens of thousands of bases), it is not always clear based solely on the gene-level metrics that the genes with high gene-level metrics fall within a larger region of recent selection. These large regions also may overlap multiple genes, making it difficult to determine which are responsible for driving the selection. A qualitative assessment of the selected genes can help reveal their relationship with the larger regions of selection. Examination of the surrounding genomic region reveals that the genes RHO, RGS9, PDE6G, and SLC24A1 occur within large regions of selection spanning at least several hundred thousand base pairs (see Supplementary Figs. S1–S6). The region corresponding to RGS9, in particular, is centered on this gene and overlaps few others.
The methods we use also lack the resolution to localize the selection to specific variants within the selected genes. The polymorphic variants (SNPs) used in this analysis are unlikely to be directly functional, but instead are in linkage disequilibrium with the functional variants. Identification and confirmation of the functional variants in these genes is the next logical step. A set of variants within the genes undergoing selection could be generated using sequence data from members of the three populations considered here. Requiring that sequence variants have substantially different frequencies across populations and application of commonly used variant filters would result in a set of putative selected variants. Functional study of these variants then could be performed to determine the impact of these variants on the observed positive selection. Functional evidence could be gathered either by defining the phenotype in human subjects using functional tests, such as low light acuity or photostress testing,43 or animal models engineered to harbor the same variants found in different human populations. This evidence would help to determine if the selected variants were likely to impart an advantage in low light vision, retinal adaptation to changing light levels, or have some other role.
Preliminary analysis of the data from the Exome Sequencing Project (available in the public domain at http://evs.gs.washington.edu/EVS/)51 has identified several plausibly functional variations in different prevalence between the European American and African American populations. The comprehensive identification of selected functional variants in phototransduction genes is the focus of ongoing investigation, the details of which are beyond the scope of this study.
In summary, we have found evidence for selective pressure in the genes involved in retinal phototransduction in H. sapiens, at the SNP and gene levels, and outlined plausible hypotheses for the functional impact of this selective pressure. Further, we have proposed experiments for determining the selected genetic variants and functional impact of the selective pressure acting on the human phototransduction system.
Supplementary Material
Acknowledgments
Supported by Grants NEI-EY017066, R01 EY018853, R01 EY019112 (MDA), NEI-EY017451 (RFM, TES), and NEI-EY018825 (TES) from the National Eye Institute and National Institutes of Health; BR-GE-0608-0457-UIA (TES) from the Foundation Fighting Blindness; and VA-5I01CX000119 (MDA, MC, TES) from the Veterans' Administration.
Disclosure: M. Christopher, None; T.E. Scheetz, None; R.F. Mullins, None; M.D. Abràmoff, None
References
- 1. Teshima KM, Coop G, Przeworski M. How reliable are empirical genomic scans for selective sweeps? Genome Res. 2006; 16: 702–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang ET, Kodama G, Baldi P, Moyzis RK. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc Natl Acad Sci U S A. 2006; 103: 135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Przeworski M. The signature of positive selection at randomly chosen loci. Genetics. 2002; 160: 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aquadro CF. Bauer DuMont V, Reed FA. Genome-wide variation in the human and fruitfly: a comparison. Curr Opin Genet Dev. 2001; 11: 627–634 [DOI] [PubMed] [Google Scholar]
- 5. Milot E, Mayer FM, Nussey DH, Boisvert M, Pelletier F, Reale D. Evidence for evolution in response to natural selection in a contemporary human population. Proc Natl Acad Sci U S A. 2011; 108: 17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Byars SG, Ewbank D, Govindaraju DR, Stearns SC. Natural selection in a contemporary human population. Proc Natl Acad Sci U S A. 2010; 107: 1787–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cerling TE, Wynn JG, Andanje SA, et al. Woody cover and hominin environments in the past 6 million years. Nature. 2011; 476: 51–56 [DOI] [PubMed] [Google Scholar]
- 8. De Blij HJ, Muller PO. Physical Geography of the Global Environment. 2nd ed. New York, NY: J. Wiley; 1996: xviii, 599 [Google Scholar]
- 9. Bersaglieri T, Sabeti PC, Patterson N, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004; 74: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thompson EE, Kuttab-Boulos H, Witonsky D, Yang L, Roe BA, Di Rienzo A. CYP3A variation and the evolution of salt-sensitivity variants. Am J Hum Genet. 2004; 75: 1059–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saunders MA, Hammer MF, Nachman MW. Nucleotide variability at G6pd and the signature of malarial selection in humans. Genetics. 2002; 162: 1849–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xue YL, Daly A, Yngvadottir B, et al. Spread of an inactive form of caspase-12 in humans is due to recent positive selection. Am J Hum Genet. 2006; 78: 659–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamason RL, Mohideen MAPK, Mest JR, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science. 2005; 310: 1782–1786 [DOI] [PubMed] [Google Scholar]
- 14. Beleza S, Dos Santos AM, McEvoy B, et al. The timing of pigmentation lightening in Europeans. Mol Biol Evol. 2013; 30: 24–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pearce E, Dunbar R. Latitudinal variation in light levels drives human visual system size. Biol Lett. 2012; 8: 90–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamb TD, Collin SP, Pugh EN. Evolution of the vertebrate eye: opsins, photoreceptors, retina and eye cup. Nat Rev Neurosci. 2007; 8: 960–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ogura A, Ikeo K, Gojobori T. Comparative analysis of gene expression for convergent evolution of camera eye between octopus and human. Genome Res. 2004; 14: 1555–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoffmann WA, Jackson RB. Vegetation – climate feedbacks in the conversion of tropical savanna to grassland. J Climate. 2000; 13: 1593–1602 [Google Scholar]
- 19. Loranty MM, Goetz SJ, Beck PSA. Tundra vegetation effects on pan-Arctic albedo. Environ Res Lett. 2011; 6: 029601–029601 [Google Scholar]
- 20. Bonfils C, de Noblet-Ducoure N, Braconnot P, Joussaume S. Hot desert albedo and climate change: Mid-Holocene monsoon in North Africa. J Climate. 2001; 14: 3724–3737 [Google Scholar]
- 21. Arshavsky VY, Burns ME. Photoreceptor signaling: supporting vision across a wide range of light intensities. J Biol Chem. 2012; 287: 1620–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altshuler DM, Gibbs RA, Peltonen L, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010; 467: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Consortium TGP. A map of human genome variation from population-scale sequencing. Nature. 2010; 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grossman SR, Shlyakhter I, Karlsson EK, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010; 327: 883–886 [DOI] [PubMed] [Google Scholar]
- 25. Sabeti PC, Reich DE, Higgins JM, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002; 419: 832–837 [DOI] [PubMed] [Google Scholar]
- 26. Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002; 12: 1805–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tang K, Thornton KR, Stoneking M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007; 5: e171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lao O, de Gruijter JM, van Duijn K, Navarro A, Kayser M. Signatures of positive selection in genes associated with human skin pigmentation as revealed from analyses of single nucleotide polymorphisms. Ann Hum Genet. 2007; 71: 354–369 [DOI] [PubMed] [Google Scholar]
- 29. Lopez Herraez D, Bauchet M, Tang K, et al. Genetic variation and recent positive selection in worldwide human populations: evidence from nearly 1 million SNPs. PLoS One. 2009; 4: e7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gautier M. Vitalis R. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012; 28: 1176–1177 [DOI] [PubMed] [Google Scholar]
- 31. Wright S. Genetical structure of populations. Nature. 1950; 166: 247–249 [DOI] [PubMed] [Google Scholar]
- 32. Neigel JE. Is Fst obsolete? Conserv Genet. 2002; 3: 167–173 [Google Scholar]
- 33. Flicek P, Amode MR, Barrell D, et al. Ensembl 2012. Nucl Acids Res. 2012; 40: D84–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H, Ding K, Zhang Y, Jin L, Kullo IJ, He F. Comparative and evolutionary pharmacogenetics of ABCB1: complex signatures of positive selection on coding and regulatory regions. Pharmacogenet Genomics. 2007; 17: 667–678 [DOI] [PubMed] [Google Scholar]
- 35. de Gruijter JM, Lao O, Vermeulen M, et al. Contrasting signals of positive selection in genes involved in human skin-color variation from tests based on SNP scans and resequencing. Invest Genet. 2011; 2: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bryk J, Hardouin E, Pugach I, et al. Positive selection in East Asians for an EDAR allele that enhances NF-kappaB activation. PLoS One. 2008; 3: e2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sabeti PC, Varilly P, Fry B, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007; 449: 913–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bersaglieri T, Sabeti PC, Patterson N, et al. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004; 74: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sabeti PC, Schaffner SF, Fry B, et al. Positive natural selection in the human lineage. Science. 2006; 312: 1614–1620 [DOI] [PubMed] [Google Scholar]
- 40. Chang CW, Cheng WC, Chen CR, et al. Identification of human housekeeping genes and tissue-selective genes by microarray meta-analysis. PLoS One. 2011; 6: e22859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Biswas S, Akey JM. Genomic insights into positive selection. Trends Genet. 2006; 22: 437–446 [DOI] [PubMed] [Google Scholar]
- 42. Qanbari S, Gianola D, Hayes B, et al. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011; 12: 318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nishiguchi KM, Sandberg MA, Kooijman AC, et al. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature. 2004; 427: 75–78 [DOI] [PubMed] [Google Scholar]
- 44. Tsang SH, Burns ME, Calvert PD, et al. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998; 282: 117–121 [DOI] [PubMed] [Google Scholar]
- 45. Dryja TP, Hahn LB, Reboul T, Arnaud B. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nature Genet. 1996; 13: 358–360 [DOI] [PubMed] [Google Scholar]
- 46. Dryja TP, Berson EL, Rao VR, Oprian DD. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nature Genet. 1993; 4: 280–283 [DOI] [PubMed] [Google Scholar]
- 47. Riazuddin SA, Shahzadi A, Zeitz C, et al. A mutation in SLC24A1 implicated in autosomal-recessive congenital stationary night blindness. Am J Hum Genet. 2010; 87: 523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. National Center for Biotechnology Information GNB1 guanine nucleotide binding protein (G protein), beta polypeptide 1 [Homo sapiens (human)]. Available at: http://www.ncbi.nlm.nih.gov/gene/2782. Accessed March 25, 2013. [Google Scholar]
- 49. Liu X, Ong RT, Pillai EN, et al. Detecting and characterizing genomic signatures of positive selection in global populations. Am J Hum Genet. 2013; 92: 866–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Markvart T, Castañer L. Practical Handbook of Photovoltaics: Fundamentals and Applications. New York, NY: Elsevier Advanced Technology; 2003: xiv, 984. [Google Scholar]
- 51. NHLBI Exome Sequencing Project Exome Variant Server. 2012. Available at: http://evs.gs.washington.edu/EVS/. Accessed November 15, 2012 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.