

Abstract
The interplay between immunity, inflammation, and metabolic changes is a growing field of research. Toll-like receptors and NOD-like receptors are families of innate immune receptors, and their role in the human immune response is well documented. Exciting new evidence is emerging with regard to their role in the regulation of metabolism and the activation of inflammatory pathways during the progression of metabolic disorders such as type 2 diabetes and atherosclerosis. The proinflammatory cytokine IL-1β appears to play a central role in these disorders. There is also evidence that metabolites such as NAD+ (acting via deacetylases such as SIRT1 and SIRT2) and succinate (which regulates hypoxia-inducible factor 1α) are signals that regulate innate immunity. In addition, the extracellular overproduction of metabolites such as uric acid and cholesterol crystals acts as a signal sensed by NLRP3, leading to the production of IL-1β. These observations cast new light on the role of metabolism during host defense and inflammation.
Keywords: Inflammation, Innate Immunity, Metabolic Diseases, Metabolic Regulation, Metabolism, Signal Transduction
Introduction
The innate immune system acts as the first line of defense against invading pathogens. Several families of receptors called pattern recognition receptors (PRRs)2 recognize highly conserved pathogen-associated molecular patterns as well as host-derived danger signals called damage-associated molecular patterns. These PRRs include the Toll-like receptors (TLRs) and Nod-like receptors. Activation of these PRRs initiates downstream signaling pathways that culminate in phenotypic changes in cell types such as macrophages. Profound changes in gene expression occur, leading to the production of a wide range of host defense and inflammatory factors, most notably cytokines such as IL-1β, TNF, and IL-6. One of the best described TLRs is TLR4, which recognizes the Gram-negative bacterial product LPS and initiates two signaling pathways: the MyD88-dependent pathway, which leads to NF-κB activation and regulates many inflammatory genes, and the MyD88-independent pathway, which leads to IRF3 activation and regulates the expression of genes, including those encoding type 1 interferons (1). A subset of Nod-like receptors, including NLRP3, activate multiprotein complexes called inflammasomes, which lead to the activation of caspase-1 and the cleavage of pro-IL-1β and pro-IL-18 to their active form (2). These immune receptors are associated with host defense against pathogens and in the progression of inflammatory diseases such as rheumatoid arthritis and Crohn syndrome (3, 4). More recent evidence suggests a role for these receptors in the progression of metabolic disorders such as type 2 diabetes (T2D) and atherosclerosis (5). Recent studies provide evidence that TLR4 and NLRP3 induce metabolic changes in cells, with the levels of key metabolites such as NAD+ and succinate being altered (6, 7). Here, we discuss the possibility that these alterations act as signals required for cellular activation, providing an important causal molecular link between metabolism and innate immunity. That metabolism and innate immunity might be linked is perhaps not surprising, as both involve exogenous “stressors” being sensed, leading to maintenance of homeostasis. Recent studies have revealed intriguing molecular associations between the two processes, which could give rise to substantial new insights into the pathogenesis of inflammatory diseases.
The Warburg Effect in Activated Macrophages
The production of proinflammatory cytokines and other host defense proteins requires substantial energy consumption, so it is unsurprising that cells activated via TLRs or NLRP3 undergo a metabolic shift. As long ago as 1986, Newsholme and co-workers (8) demonstrated that activated macrophages have increased hexokinase activity along with increased glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase activities, suggesting an increase in glycolysis and the pentose phosphate pathway in these cells. Glycolysis is also increased during phagocytosis (8). Otto Warburg first described aerobic glycolysis in 1927 (9). Before Warburg's observation, glycolysis was described largely as an anaerobic phenomenon that occurred when oxygen levels were low, limiting the metabolism of pyruvate by the acid (TCA) cycle in the mitochondria during oxidative phosphorylation. Warburg demonstrated, however, that in tumors under normoxia, lactate production from glycolysis dominates, a phenomenon that became known as the “Warburg effect” (9). Warburg went on to conclude, however, in 1958 that this “cancer metabolism” did not occur in leukocytes, his opinion being that the observation of aerobic glycolysis in these cells was an artifact of preparation (10). This proved to be an incorrect conclusion because we now know that a switch in metabolism from oxidative phosphorylation to glycolysis occurs following activation of macrophages and dendritic cells (DCs) by LPS (6, 11), in a similar manner to the Warburg effect observed in tumor cells (9). A metabolomic screen and microarray analysis in macrophages activated by LPS have confirmed this switch from oxidative phosphorylation to glycolysis. Increases in the levels of intermediates in glycolysis and the pentose phosphate pathway occur and clearly correlate with changes in gene expression of the relevant enzymes. Importantly, this switch in the metabolic state directly impacts on the inflammatory state of the cell. Inhibition of glycolysis by 2-deoxyglucose (2-DG) decreases the production of IL-1β in response to LPS stimulation but has no effect on TNFα or IL-6 expression, although precisely how 2-DG inhibition of glycolysis achieves this effect is not wholly known (6). A key question is what does this shift in glycolysis mean for macrophage activation? It is likely that the increase in glycolysis allows for a rapid increase in ATP production since, although less productive in terms of ATP synthesis than the TCA cycle, glycolysis can be strongly induced (12). This increase in ATP is required for metabolic processes such as biosynthesis but is also needed to maintain the mitochondrial membrane potential, which falls in LPS-activated cells (13). This allows the macrophages (or DCs) to remain alive and persist during the host defense response. The increase in the pentose phosphate pathway will allow for the production of intermediates for biosynthesis, such as purines and pyrimidines. However, the shift in metabolism has a more specific consequence, which concerns alterations in TCA cycle intermediates such as succinate and citrate. This also relates to “Warburg metabolism” as it is currently understood.
Interestingly, anti-inflammatory M2 macrophages have decreased glycolysis and utilize oxidative metabolism instead. In adaptive immunity, T lymphocytes also exhibit different metabolic profiles depending on their inflammatory states. Proinflammatory TH17 cells are highly glycolytic, whereas anti-inflammatory T cells such as regulatory T (Treg) cells are characterized by oxidative metabolism. This suggests that a movement toward high glycolysis is indicative of inflammatory cells, whereas a shift toward oxidative phosphorylation is a hallmark of anti-inflammatory cells. Inhibition of glycolysis by 2-DG results in a switch from TH17 cells to Treg cells, suggesting that the plasticity of T cells may be controlled by metabolic cues (14). In addition, one reason for Warburg metabolism in Th1 cells is to sequester GAPDH from certain mRNAs, notably the mRNA encoding IFNγ (15). This releases the IFNγ mRNA from inhibition by GAPDH and provides a compelling rationale for the increase in glycolysis in Th1 cells. Finally, the memory T cell population has oxidative phosphorylation as its major metabolic process but shifts to glycolysis on becoming effector T cells (16). This once again suggests a close link between immune and metabolic signals. The metabolic status of T cells is comprehensively reviewed in Ref. 17.
Succinate as an Inflammatory Signal
Warburg had originally reported that mitochondrial metabolism was severely attenuated in tumors (9). More recently, however, this has been reassessed, and it is now evident that complex changes occur in TCA cycle intermediates in tumors, including alterations in succinate and citrate, with citrate in particular being withdrawn from the TCA cycle for lipid biosynthesis (18). Similarly in LPS-activated macrophages, although there is an overall decrease in TCA cycle activity, there is a marked increase in the TCA cycle intermediate succinate (6). This increase in succinate leads to the inhibition of prolyl hydroxylases (PHDs). Succinate impedes PHD activity by product inhibition. It prevents the decarboxylation of α-ketoglutarate (α-KG) to succinate, an essential co-reaction in the hydroxylation of targets by PHDs. PHDs can no longer hydroxylate hypoxia-inducible factor 1α (HIF1α), resulting in its stabilization. HIF1α has been shown to bind directly to the IL-1β promoter and is required for sustained IL-1β production. 2-DG somehow depresses the increase in succinate, explaining the selective inhibition of IL-1β induction by 2-DG. TNFα, which lacks an HIF1α-binding site in its promoter, is unaffected (6). The increase in succinate is from glutamine metabolism through anaplerosis via α-KG and also via the so-called “GABA shunt,” which converts glutamine to succinate via GABA (6). The importance of these metabolic changes following LPS stimulation was also examined in vivo. The administration of 2-DG prior to LPS treatment decreases the serum levels of IL-1β in mice (6). Importantly, succinate therefore acts as a signal in LPS-activated macrophages, leading to HIF1α activation under normoxia as shown in Fig. 1. LPS also leads to an increase in succinylation, a post-translational modification in which a succinyl group is added to a lysine residue of a protein. Increased succinylation of several proteins was observed, including malate dehydrogenase, GAPDH, glutamate carrier 1, l-lactate dehydrogenase A chain, and transaldolase (6). The consequence of succinylation of these proteins is not yet understood, but it is likely to cause a conformational change that will affect protein function.
FIGURE 1.
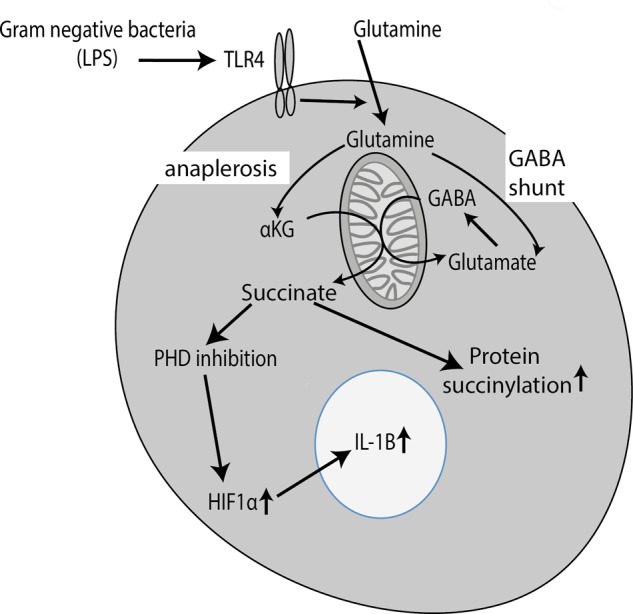
Succinate is a signal generated in response to activation of TLR4 by LPS, leading to HIF1α activation. Activation of TLR4 by LPS leads to a profound change in metabolism, including increased glycolysis and the pentose phosphate pathway (not shown). In addition, LPS alters the TCA cycle such that there is an increase in succinate. This occurs via the alteration in glutamine metabolism via both anaplerosis to α-KG and the GABA shunt. Succinate then inhibits PHDs, increasing HIF1α and promoting the expression of IL-1β and other genes. The increase in succinate may also promote protein succinylation, the consequences of which are not yet known.
Role of NAD+, Sirtuins, and AMP-dependent Protein Kinase in Inflammation and NLRP3 Regulation
Another metabolic factor, NAD+, is proving to be of great interest as a regulator of inflammation via the sirtuin deacetylases, which depend on NAD+ for activity, giving us a link to the NLRP3 inflammasome, a key regulator of the proinflammatory cytokine IL-1β and also the nutrient sensor AMP-dependent protein kinase (AMPK), which appears to be able to limit inflammation. A good example of this is the disease gout, which is caused by elevated levels of uric acid in the blood, resulting normally from a purine-rich diet. The uric acid crystallizes to form monosodium urate crystals, which deposit in the joints. Several studies have proposed a role for NLRP3 in the recognition of monosodium urate, resulting in the activation of IL-1β (19). A recent study has revealed an intriguing link between altered metabolism and NLRP3 activation in gout. Activation of macrophages with agents such as uric acid will cause a decrease in NAD+ via mitochondrial damage, limiting the activity of the NAD+-dependent deacetylase SIRT2 (7). A substrate for SIRT2 is acetylated tubulin, and the authors demonstrated that the decrease in SIRT2 activity, occurring because of the decrease in NAD+, allows acetylated tubulin to persist, promoting its polymerization. The NLRP3 inflammasome associates with the tubulin cytoskeleton and localizes to the mitochondria, where reactive oxygen species (ROS) lead to NLRP3 activation. These data provide one explanation for why colchicine might be effective in gout, as it destabilizes the tubulin cytoskeleton. In addition, however, these findings reveal the drop in NAD+ (and increase in NADH) to be another metabolic signal leading to inflammation, in this case, NLRP3 activation, as shown in Fig. 2. Because uric acid is an NLRP3 activator, the drop in NAD+ could occur downstream of NLRP3, setting up a positive feedback loop. It should be pointed out, however, that Joosten et al. (20) saw no difference in NLRP3-deficient mice in response to uric acid crystals, suggesting a role for an alternative inflammasome in the activation of IL-1β, which could still involve tubulin. What is clear is that IL-1β inhibitors (e.g. the IL-1 receptor antagonist or the anti-IL-1β antibody canakinumab) have proven very effective in treating gout, and a major trial is under way testing IL-1β inhibition as an intervention to decrease the risk of heart attack from atherosclerosis (21–23).
FIGURE 2.
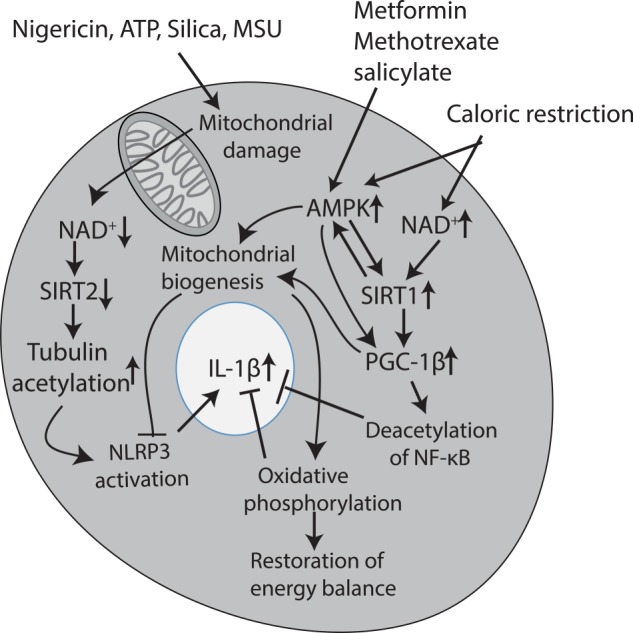
NAD+, AMPK, and SIRT1 as key inflammatory regulators. Agents such as nigericin, ATP, and uric acid crystals lead to mitochondrial damage, which in turn leads to a decrease in NAD+. This limits SIRT2 activity, allowing acetylation of tubulin to persist, which in turn enhances NLRP3 activity by promoting the trafficking of NLRP3 to the mitochondria, enhancing its activation. Because these agents are all NLRP3 activators, this could create a positive feedback loop sustaining NLRP3 activation, leading to IL-1β production. Caloric restriction has the opposite effect and leads to an increase in NAD+. This leads to the activation of SIRT1, a deacetylase that will promote mitochondrial biogenesis and function via PGC-1β and inhibit NF-κB via deacetylation of the p65 subunit. Both of these events will have a net anti-inflammatory effect, with the promotion of mitochondrial function, also inhibiting NLRP3 activation. The increase in oxidative phosphorylation (which appears to be anti-inflammatory probably via an increase in NAD+) will restore the energy balance. Caloric restriction will also activate AMPK, which can also promote mitochondrial biogenesis. There is evidence for cross-talk between SIRT1 and AMPK. Finally, several anti-inflammatory agents, notably methotrexate and salicylate, have been shown to activate AMPK, as has the anti-T2D agent metformin. Metformin may mediate its effects in T2D via this process because limiting inflammation in this way could restore insulin sensitivity. MSU, monosodium urate.
NLRP3 activation by extracellular metabolites has also been implicated in several other diseases such as atherosclerosis. Macrophages infiltrate atherosclerotic plaques and form foam cells, generating multiple inflammatory mediators thought to be pathogenic, notably IL-1β (24). The NLRP3 inflammasome is activated in these macrophages during the development of atherosclerotic plaques by LDLs that accumulate in the artery walls and lead to the production of cholesterol crystals (24, 25). The activation of TLRs (possibly by free fatty acids or oxidized LDL) and NLRP3 leads to the production of active IL-1β (26). IL-1β levels increase in the arterial plaques, and the levels of IL-1β directly correlate with disease severity (27, 28). Patients with a polymorphism in the IL-1 receptor antagonist, which increases IL-1β production, have increased risk of developing atherosclerosis (29). The role of NAD+ and the sirtuins in the development of atherosclerosis has yet to be investigated. Interestingly, NAD(P)H oxidase is known to increase during the development of atherosclerosis. This leads to increased production of superoxide anions but would also result in increased levels of NAD+ (30).
NAD+ levels also regulate another sirtuin, SIRT1, and multiple recent studies have shown that SIRT1 limits inflammation. First, it promotes a switch from glycolysis to fatty acid oxidation (31) by repressing the expression of peroxisome proliferator-activated receptor-γ, a transcription factor that regulates several genes involved in fat storage (32), and activating PGC-1β (peroxisome proliferator-activated receptor-gamma coactivator 1β), a key transcription factor in mitochondrial biogenesis that leads to increased oxidative metabolism (33). This could contribute to a net anti-inflammatory effect via further increases in NAD+ and also via inhibition of NLRP3 activation, which requires mitochondrial damage and ROS production from mitochondria. SIRT1 directly impacts inflammation by deacetylating and inactivating the p65 subunit of NF-κB, limiting the expression of NF-κB-dependent proinflammatory genes (34). A fall in NAD+, resulting in deceased activity of SIRT1, will therefore promote NF-κB activation. Knockdown experiments have indeed demonstrated an anti-inflammatory role for SIRT1 in macrophages. SIRT1 knockdown leads to an increase is several proinflammatory cytokines in response in TNF, with the most dramatic effect being on IL-1β expression (35). Also of note is the observation that SIRT1 is cleaved by caspase-1 and this cleavage is most likely due to NLRP3 inflammasome activation (36). TLR4 activation has been shown to induce nicotinamide phosphoribosyltransferase (NAMPT), which synthesizes NAD+, thus activating SIRT1, which is probably part of a negative feedback loop (37). These studies therefore suggest a close link between the regulation of SIRT1 activity and inflammation.
There is also a link between the activation of SIRT1 and AMPK. AMPK is a key regulator of energy metabolism and glucose homeostasis. AMPK is activated following phosphorylation by LKB1 by a wide variety of stimuli, including caloric restriction, cellular stress, exercise, and several hormones, cytokines, and adipokines. AMPK restores energy balance by promoting catabolic processes while inhibiting energy-consuming processes (38). AMPK therefore promotes oxidative metabolism, which is associated with an anti-inflammatory state, rather than the glycolytic state associated with inflammation or tumor progression. Several anti-inflammatory drugs such as salicylate and methotrexate have been shown to activate AMPK (39, 40). Yuan et al. (41) demonstrated that the treatment of obese mice with salicylate results in a significant decrease in blood glucose levels, whereas clinical trials with salsalate, a non-acetylated prodrug of salicylate, showed a reduction in blood glucose levels in patients with T2D (42). In obesity and T2D, the accumulation of saturated fatty acids such as palmitate results in deactivation of AMPK, and this decreases autophagy and NO production and increases mitochondrial dysfunction and mitochondrial ROS production (43). This leads, in turn, to activation of the NLRP3 inflammasome. Activation of AMPK by salicylate can therefore overcome the mitochondrial dysfunction associated with T2D, decreasing NLRP3 activation. The anti-diabetic agent metformin has also been shown to activate AMPK (44), and this may limit inflammation, again impacting on T2D. LPS has been shown to decrease AMPK activation, whereas activation of AMPK results in a decrease in NF-κB activation and TNFα production in macrophages following stimulation with inflammatory stimuli such as LPS (45). Knockdown of AMPK enhances LPS-induced IL-12p40 expression in DCs (11). AMPK-α1−/− mice are more prone to severe experimental autoimmune encephalomyelitis in comparison with controls, whereas spleen cells isolated from these mice secrete more IFNγ and less IL-17 following antigen stimulation. Interestingly, glycolysis is increased in T cells from AMPK-α1−/− mice, but this increase in glycolysis is not sufficient to promote bias toward CD4+ proinflammatory T cells (46), suggesting that enhanced glycolysis is necessary, but not sufficient, to promote a proinflammatory bias. AMPK also down-regulates HIF1α, as HIF1α expression is significantly up-regulated in AMPK-deficient mouse embryonic fibroblasts (47). All of this evidence points to a role for AMPK in the regulation of inflammation.
In relation to SIRT1, upon activation, SIRT1 results in deacetylation of LKB1, promoting its movement into the cytosol and its activation, leading to phosphorylation and activation of AMPK (48). Knockdown of SIRT1 results in decreased AMPK activation, and the AMPK activator resveratrol requires the presence of SIRT1 and LKB1 for its inhibitory effects (49). AMPK may also activate SIRT1 in a positive feedback loop (50). It is likely that NAD+, which increases upon caloric restriction, most likely via decreased glycolysis and enhanced oxidative phosphorylation from β-oxidation of stored fatty acids (51), will, via SIRT1, lead to AMPK activation, thereby contributing to a net anti-inflammatory effect. This is shown in Fig. 2.
Perspective
Recent studies strongly indicate that the metabolic state of immune cells is critical to their function; proinflammatory cells such as M1 macrophages, activated DCs, and TH17 cells are more glycolytic, whereas anti-inflammatory cells such as Treg cells and M2 macrophages have more oxidative phosphorylation. Why this shift occurs is not fully known because Treg cells and M2 macrophages are no less active in terms of gene expression that TH17 cells or M1 macrophages. Again, this may relate to the metabolic products of glycolysis and the TCA cycle having different signaling roles. There is still much to learn about the exact consequences of these metabolic alterations for cellular activity. However, the metabolites generated are likely not simply consequences of catabolism or anabolism but instead act as intracellular signals. The examples discussed here of succinate, HIF1α, NAD+, and sirtuins are part of a broader picture, including other metabolites such as α-KG (which is used by up to 33 enzymes (52), all of which modify chromatin) and butyrate (which inhibits histone deacetylases), and epigenetic modifications are likely to be a key consequence for these changes, as already indicated in tumors (53). The importance of these metabolic signals in vivo is still being investigated, but several pieces of evidence suggest that these metabolic signals play a central role in inflammation. As mentioned above, the inhibition of glycolysis by 2-DG decreases the levels of the proinflammatory cytokine IL-1β, and the GABA inhibitor vigabatrin protects mice in a sepsis model (6). Sirtuins are known to be involved in prolonging the life span, and their anti-inflammatory effects are likely to contribute to this increased life span. As the well known physicist Richard Feynman once said, “There is a pleasure in recognising old things from a new viewpoint.” These new insights into “old pathways” could give rise to a substantial increase in our understanding of the pathogenesis of inflammatory diseases, which might ultimately give rise to better treatments.
This work was supported by Science Foundation Ireland and the European Research Council (to L. A. J. O.).
- PRR
- pattern recognition receptor
- TLR
- Toll-like receptor
- T2D
- type 2 diabetes
- TCA
- tricarboxylic acid
- DC
- dendritic cell
- 2-DG
- 2-deoxyglucose
- Treg
- regulatory T
- PHD
- prolyl hydroxylase
- α-KG
- α-ketoglutarate
- HIF1α
- hypoxia-inducible factor 1α
- AMPK
- AMP-dependent protein kinase
- ROS
- reactive oxygen species.
REFERENCES
- 1. McGettrick A. F., O'Neill L. A. (2004) The expanding family of MyD88-like adaptors in Toll-like receptor signal transduction. Mol. Immunol. 41, 577–582 [DOI] [PubMed] [Google Scholar]
- 2. Bryant C., Fitzgerald K. A. (2009) Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 19, 455–464 [DOI] [PubMed] [Google Scholar]
- 3. Connolly D. J., O'Neill L. A. (2012) New developments in Toll-like receptor targeted therapeutics. Curr. Opin. Pharmacol. 12, 510–518 [DOI] [PubMed] [Google Scholar]
- 4. Hennessy E. J., Parker A. E., O'Neill L. A. (2010) Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307 [DOI] [PubMed] [Google Scholar]
- 5. Wen H., Ting J. P., O'Neill L. A. (2012) A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat. Immunol. 13, 352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., Haneklaus M., Caffrey B. E., Pierce K., Walmsley S., Beasley F. C., Cummins E., Nizet V., Whyte M., Taylor C. T., Lin H., Masters S. L., Gottlieb E., Kelly V. P., Clish C., Auron P. E., Xavier R. J., O'Neill L. A. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Misawa T., Takahama M., Kozaki T., Lee H., Zou J., Saitoh T., Akira S. (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 [DOI] [PubMed] [Google Scholar]
- 8. Newsholme P., Curi R., Gordon S., Newsholme E. A. (1986) Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 239, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warburg O., Wind F., Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warburg O., Gawehn K., Geissler A. W. (1958) Metabolism of leukocytes. Z. Naturforsch. B 13B, 515–516 [PubMed] [Google Scholar]
- 11. Krawczyk C. M., Holowka T., Sun J., Blagih J., Amiel E., DeBerardinis R. J., Cross J. R., Jung E., Thompson C. B., Jones R. G., Pearce E. J. (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vazquez A., Liu J., Zhou Y., Oltvai Z. N. (2010) Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst. Biol. 4, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garedew A., Henderson S. O., Moncada S. (2010) Activated macrophages utilize glycolytic ATP to maintain mitochondrial membrane potential and prevent apoptotic cell death. Cell Death Differ. 17, 1540–1550 [DOI] [PubMed] [Google Scholar]
- 14. Shi L. Z., Wang R., Huang G., Vogel P., Neale G., Green D. R., Chi H. (2011) HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang C. H., Curtis J. D., Maggi L. B., Jr., Faubert B., Villarino A. V., O'Sullivan D., Huang S. C., van der Windt G. J., Blagih J., Qiu J., Weber J. D., Pearce E. J., Jones R. G., Pearce E. L. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Windt G. J., Everts B., Chang C. H., Curtis J. D., Freitas T. C., Amiel E., Pearce E. J., Pearce E. L. (2012) Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Neill L. A., Hardie D. G. (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 [DOI] [PubMed] [Google Scholar]
- 18. Icard P., Poulain L., Lincet H. (2012) Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 1825, 111–116 [DOI] [PubMed] [Google Scholar]
- 19. Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 [DOI] [PubMed] [Google Scholar]
- 20. Joosten L. A., Netea M. G., Mylona E., Koenders M. I., Malireddi R. K., Oosting M., Stienstra R., van de Veerdonk F. L., Stalenhoef A. F., Giamarellos-Bourboulis E. J., Kanneganti T. D., van der Meer J. W. (2010) Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. 62, 3237–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGonagle D., Tan A. L., Shankaranarayana S., Madden J., Emery P., McDermott M. F. (2007) Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann. Rheum. Dis. 66, 1683–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlesinger N., De Meulemeester M., Pikhlak A., Yücel A. E., Richard D., Murphy V., Arulmani U., Sallstig P., So A. (2011) Canakinumab relieves symptoms of acute flares and improves health-related quality of life in patients with difficult-to-treat gouty arthritis by suppressing inflammation: results of a randomized, dose-ranging study. Arthritis Res. Ther. 13, R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ridker P. M., Thuren T., Zalewski A., Libby P. (2011) Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 162, 597–605 [DOI] [PubMed] [Google Scholar]
- 24. Hansson G. K. (2005) Inflammation, atherosclerosis, and coronary artery disease. New Engl. J. Med. 352, 1685–1695 [DOI] [PubMed] [Google Scholar]
- 25. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tannahill G. M., O'Neill L. A. (2011) The emerging role of metabolic regulation in the functioning of Toll-like receptors and the NOD-like receptor Nlrp3. FEBS Lett. 585, 1568–1572 [DOI] [PubMed] [Google Scholar]
- 27. Galea J., Armstrong J., Gadsdon P., Holden H., Francis S. E., Holt C. M. (1996) Interleukin-1β in coronary arteries of patients with ischemic heart disease. Arterioscler. Thromb. Vasc. Biol. 16, 1000–1006 [DOI] [PubMed] [Google Scholar]
- 28. Moyer C. F., Sajuthi D., Tulli H., Williams J. K. (1991) Synthesis of IL-1α and IL-1 β by arterial cells in atherosclerosis. Am. J. Pathol. 138, 951–960 [PMC free article] [PubMed] [Google Scholar]
- 29. Olofsson P. S., Sheikine Y., Jatta K., Ghaderi M., Samnegård A., Eriksson P., Sirsjö A. (2009) A functional interleukin-1 receptor antagonist polymorphism influences atherosclerosis development. The interleukin-1β:interleukin-1 receptor antagonist balance in atherosclerosis. Circ. J. 73, 1531–1536 [DOI] [PubMed] [Google Scholar]
- 30. Violi F., Basili S., Nigro C., Pignatelli P. (2009) Role of NADPH oxidase in atherosclerosis. Future Cardiol. 5, 83–92 [DOI] [PubMed] [Google Scholar]
- 31. Liu T. F., Vachharajani V. T., Yoza B. K., McCall C. E. (2012) NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 287, 25758–25769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Picard F., Kurtev M., Chung N., Topark-Ngarm A., Senawong T., Machado De Oliveira R., Leid M., McBurney M. W., Guarente L. (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 429, 771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin J., Handschin C., Spiegelman B. M. (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 [DOI] [PubMed] [Google Scholar]
- 34. Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., Mayo M. W. (2004) Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yoshizaki T., Milne J. C., Imamura T., Schenk S., Sonoda N., Babendure J. L., Lu J. C., Smith J. J., Jirousek M. R., Olefsky J. M. (2009) SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol. Cell. Biol. 29, 1363–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vandanmagsar B., Youm Y. H., Ravussin A., Galgani J. E., Stadler K., Mynatt R. L., Ravussin E., Stephens J. M., Dixit V. D. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu T. F., Yoza B. K., El Gazzar M., Vachharajani V. T., McCall C. E. (2011) NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 286, 9856–9864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005) AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 [DOI] [PubMed] [Google Scholar]
- 39. Hawley S. A., Fullerton M. D., Ross F. A., Schertzer J. D., Chevtzoff C., Walker K. J., Peggie M. W., Zibrova D., Green K. A., Mustard K. J., Kemp B. E., Sakamoto K., Steinberg G. R., Hardie D. G. (2012) The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beckers A., Organe S., Timmermans L., Vanderhoydonc F., Deboel L., Derua R., Waelkens E., Brusselmans K., Verhoeven G., Swinnen J. V. (2006) Methotrexate enhances the antianabolic and antiproliferative effects of 5-aminoimidazole-4-carboxamide riboside. Mol. Cancer Ther. 5, 2211–2217 [DOI] [PubMed] [Google Scholar]
- 41. Yuan M., Konstantopoulos N., Lee J., Hansen L., Li Z. W., Karin M., Shoelson S. E. (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of IKKB. Science 293, 1673–1677 [DOI] [PubMed] [Google Scholar]
- 42. Goldfine A. B., Fonseca V., Jablonski K. A., Pyle L., Staten M. A., Shoelson S. E., and TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team (2010) The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med. 152, 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pilon G., Dallaire P., Marette A. (2004) Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase. A new mechanism of action of insulin-sensitizing drugs. J. Biol. Chem. 279, 20767–20774 [DOI] [PubMed] [Google Scholar]
- 44. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang Z., Kahn B. B., Shi H., Xue B. Z. (2010) Macrophage α1 AMP-activated protein kinase (α1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 285, 19051–19059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. MacIver N. J., Blagih J., Saucillo D. C., Tonelli L., Griss T., Rathmell J. C., Jones R. G. (2011) The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J. Immunol. 187, 4187–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shackelford D. B., Vasquez D. S., Corbeil J., Wu S., Leblanc M., Wu C. L., Vera D. R., Shaw R. J. (2009) mTOR and HIF-1α-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 11137–11142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lan F., Cacicedo J. M., Ruderman N., Ido Y. (2008) SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Price N. L., Gomes A. P., Ling A. J., Duarte F. V., Martin-Montalvo A., North B. J., Agarwal B., Ye L., Ramadori G., Teodoro J. S., Hubbard B. P., Varela A. T., Davis J. G., Varamini B., Hafner A., Moaddel R., Rolo A. P., Coppari R., Palmeira C. M., de Cabo R., Baur J. A., Sinclair D. A. (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 15, 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dhahbi J. M., Mote P. L., Wingo J., Tillman J. B., Walford R. L., Spindler S. R. (1999) Calories and aging alter gene expression for gluconeogenic, glycolytic, and nitrogen-metabolizing enzymes. Am. J. Physiol. 277, E352–E360 [DOI] [PubMed] [Google Scholar]
- 52. Loenarz C., Schofield C. J. (2011) Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 36, 7–18 [DOI] [PubMed] [Google Scholar]
- 53. Donohoe D. R., Collins L. B., Wali A., Bigler R., Sun W., Bultman S. J. (2012) The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]