

Background: UDP-glucose dehydrogenase (Ugd) from E. coli K-12 is reported to be activated by tyrosine phosphorylation.
Results: Ugd displayed NAD substrate inhibition that could be relieved by the addition of a nucleotide triphosphate in the absence of kinase.
Conclusion: Nucleotide triphosphates are allosteric activators of Ugd.
Significance: Tyrosine phosphorylation is not required for Ugd activation.
Keywords: Bacterial Metabolism, Carbohydrate Biosynthesis, Carbohydrate Glycoconjugate, Enzymes, Escherichia coli, Glycobiology
Abstract
UDP-glucose dehydrogenase (Ugd) generates UDP-glucuronic acid, an important precursor for the production of many hexuronic acid-containing bacterial surface glycostructures. In Escherichia coli K-12, Ugd is important for biosynthesis of the environmentally regulated exopolysaccharide known as colanic acid, whereas in other E. coli isolates, the same enzyme is required for production of the constitutive group 1 capsular polysaccharides, which act as virulence determinants. Recent studies have implicated tyrosine phosphorylation in the activation of Ugd from E. coli K-12, although it is not known if this is a feature shared by bacterial Ugd proteins. The activities of Ugd from E. coli K-12 and from the group 1 capsule prototype (serotype K30) were compared. Surprisingly, for both enzymes, site-directed Tyr → Phe mutants affecting the previously proposed phosphorylation site retained similar kinetic properties to the wild-type protein. Purified Ugd from E. coli K-12 had significant levels of NAD substrate inhibition, which could be alleviated by the addition of ATP and several other nucleotide triphosphates. Mutations in a previously identified UDP-glucuronic acid allosteric binding site decreased the binding affinity of the nucleotide triphosphate. Ugd from E. coli serotype K30 was not inhibited by NAD, but its activity still increased in the presence of ATP.
Introduction
UDP-glucuronic acid (UDPGA)2 is an essential substrate for biosynthesis of several important glycoconjugates. In multicellular eukaryotes, UDPGA is a precursor for the synthesis of proteoglycans and glycosaminoglycans (1). In both Gram-positive and Gram-negative bacteria, UDPGA is a substrate in the production of many bacterial surface glycostructures, including capsular polysaccharides (CPSs), secreted exopolysaccharides, lipopolysaccharides (LPSs), and teichuronic acids. Bacterial surface glycoconjugates are often critical virulence factors, so UDPGA synthesis can have a direct effect on bacterial pathogenicity. For instance, UDPGA is essential for the formation of hexuronic acid-containing CPSs that often protect bacteria from phagocytosis (2, 3). It is also required to synthesize 4-aminoarabinose, which is used to modify the lipid A core of some bacterial LPSs, resulting in resistance to polycationic compounds (4). UDP-glucose dehydrogenase (Ugd) is the enzyme responsible for generating the activated nucleotide sugar from UDP-glucose (UDPG), coincident with the reduction of NAD (5). The enzyme is conserved in prokaryotes and eukaryotes (6). Human Ugd forms a hexameric ternary complex (7) that can be regulated by feedback inhibition of a downstream product, UDP-xylose (8). Bacterial Ugd enzymes exist as dimers (9–11). In recent years, the structures of a few bacterial homologs of Ugd have been determined, and the kinetics and modes of regulation have been investigated. In the context of this study, the structure of Klebsiella pneumoniae Ugd (Fig. 1B), in complex with substrate and cofactor (11), is particularly important. One form of regulation that has come under scrutiny is activation of bacterial Ugd by tyrosine phosphorylation by bacterial tyrosine kinases (BY-kinase) (12).
FIGURE 1.

Sequence alignment and complementation of CWG875 by UgdK-12 and UgdK30. A, amino acid sequences were aligned by Clustal (59). Identical and similar residues are highlighted in black and gray, respectively (by Boxshade). The proposed site of tyrosine phosphorylation (Tyr-71) is indicated by a green asterisk. Lys-323 and Arg-324 are indicated by yellow and blue asterisks, respectively. B, model of UgdK-12 displayed as a dimer (protomers colored light blue and pink). NAD (red sticks), UDPG (purple sticks), and tyrosine 71 (green sticks) are displayed. Lys-323 and Arg-324 are colored yellow and blue, respectively. C, surface representation of the model of UgdK-12 colored as in B. D, immunoblotting of proteinase K-treated cell lysates of CWG875 expressing Ugd homologs (and Y71F mutants of each) probed with antibodies specific for the K30 capsular polysaccharide. See Table 1 for plasmid assignments. Plasmid pBAD24 was used for sample labeled vector.
Bacterial BY-kinases are frequently found as part of the machinery catalyzing the assembly of a large and diverse subset of extracellular polysaccharides (12–14). They are sometimes found as a domain of a subfamily of polysaccharide co-polymerase proteins associated with capsular and exopolysaccharide production systems. These proteins possess a transmembrane modulator domain and a cytoplasmic catalytic domain with tyrosine kinase activity. In Escherichia coli, these two domains are consolidated into a single polypeptide (designated Wzc) (15, 16), but the domains are encoded as separate polypeptides in Gram-positive bacteria (12). In E. coli capsule systems, the polysaccharide co-polymerase protein transmembrane domain interacts with an octameric outer membrane channel (Wza) to form part of the export system for completed polysaccharides (17, 18). The purified BY-kinase domains from Bacillus subtilis (YwqC, the transmembrane modulator, and PtkA, the kinase) have been reported to phosphorylate the B. subtilis Ugd homolog (UgdBs) in vitro, resulting in an ∼2-fold increase in UgdBs activity (19). This result led to the proposal that phosphorylated UgdBs is the active form of the enzyme. Similar experiments implicated phosphorylation in the regulation of Ugd activity in E. coli K-12 (20).
E. coli isolates may contain two BY-kinases. The Wzc protein is part of the assembly pathway for serotype-specific group 1 capsular polysaccharides, or colanic acid, which is produced by isolates lacking group 1 capsules, including E. coli K-12 (21). Colanic acid is an environmentally regulated exopolysaccharide, which is activated by the Rcs regulon (22). Group 1 CPSs are constitutively expressed virulence determinants (23) and are encoded by gene clusters that occupy the same chromosomal location as the colanic acid genes (24). Consequently, possession of colanic acid and group 1 capsule assembly machinery appears to be mutually exclusive. The second BY-kinase, Etk, is encoded at an unlinked locus that is not expressed in all isolates (25) but can contribute to the formation of group 1 CPSs (26). Etk and Wzc are structural homologs (27, 28).
The ugd gene is located near the colanic acid/group 1 CPS locus. It has been proposed that Ugd from E. coli K-12 (UgdK-12) can be phosphorylated by either Etk or Wzc (29); phosphorylation by Wzc is correlated with production of colanic acid, whereas phosphorylation by Etk promotes the synthesis of 4-aminoarabinose for lipid A modification. This study was initially undertaken to investigate the possible role of Ugd phosphorylation in the production of a constitutively expressed CPS. In vitro characterization indicated a significant difference in the activities of the two Ugd homologs; UgdK-12 displays significant levels of NAD substrate inhibition, although Ugd from E. coli serotype K30 (UgdK30) does not. We could not establish an obvious role for phosphorylation in modulating activity of Ugd, but kinetic analyses revealed a novel mechanism of activation that is NTP-dependent.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
The bacterial strains and plasmids used in this study are listed in Table 1. All bacterial cultures were grown at 37 °C in lysogeny broth media (30) containing arabinose (0.02–0.1%), galactose (0.04%), and antibiotics (kanamycin at 50 μg/ml and/or ampicillin at 100 μg/ml and/or chloramphenicol at 34 μg/ml), as required. For plasmid nomenclature, ApR, KmR, CmR, and GmR represent ampicillin resistance, kanamycin resistance, chloramphenicol resistance, and gentamicin resistance, respectively.
TABLE 1.
Bacterial strains and plasmids
| Description | Ref. | |
|---|---|---|
| Strains | ||
| E.coli DH5α | φ80d deoR lacZΔM15 endA1 recA1 hsdR17 (rK−mK−) supE44 thi-1 gyrA96 relA1 Δ (lacZYA-argF)U169 F− | |
| E. coli E69 | Prototroph, serotype O9a:K30:H12 | I. Orskov |
| E.coli W3110 | F− λ− IN (rrnD-rrnE)1 rph-1 | |
| CWG285 | E69 wzcK30::aacC1; wza22min::aadA (polar on etp and etk); GmR, SpR | 26 |
| CWG875 | E69 ΔgalE ugd::aaC1; GmR | This study |
| CWG876 | W3110 Δwzc Δetk ugd::kan; KmR | This study |
| Plasmids | ||
| pBAD18, pBAD24, pBAD33 | Arabinose-inducible expression vectors; KmR or ApR or CmR | 56 |
| pWQ173 | Allelic exchange suicide vector with pheS gene; CmR | 23 |
| pWQ500 | pHC79 derivative containing E. coli E69 chromosomal region surrounding rcsAK30; ApR | 57 |
| pWQ629 | pUC19 derivative containing E. coli E69 chromosomal region surrounding ugdK30 | 37, 58 |
| pWQ728 | pWQ173 derivative containing ugdK30 interrupted with gentamicin resistance cassette at SacI site; GmR, CmR | This study |
| pWQ729 | pBAD24 derivative expressing His6-UgdK-12; ApR | This study |
| pWQ730 | pBAD24 derivative expressing His6-UgdK30; ApR | This study |
| pWQ731 | pWQ729 derivative containing Y71F mutation; ApR | This study |
| pWQ732 | pWQ730 derivative containing Y71F mutation; ApR | This study |
| pWQ733 | pWQ729 derivative containing K323A mutation; ApR | This study |
| pWQ734 | pWQ729 derivative containing R324A mutation; ApR | This study |
| pWQ735 | pWQ729 derivative containing KR → AA mutations at residues 323–324; ApR | This study |
| pWQ736 | pBAD18 derivative expressing His6-WzcK-12 residues 447–704; ApR | This study |
| pWQ737 | pBAD18 derivative expressing His6-WzcK30 residues 447–704; ApR | This study |
| pWQ738 | pBAD33 derivative expressing His6-UgdK-12; CmR | This study |
| pWQ739 | pBAD33 derivative expressing His6-UgdK30; CmR | This study |
Construction of Chromosomal Deletion Mutants in E. coli W3110 and E. coli E69
E. coli CWG876, a derivative of E. coli K-12 strain W3110 containing Δwzc Δetk and ugd::kan mutations, was constructed using the λRed recombinase system (31) in a multistep process. Each gene was first completely replaced with a kanamycin resistance cassette, and then the cassette was excised using the pCP20 helper plasmid (32), before subsequent genes were deleted. The kanamycin resistance cassette was not removed from the chromosome in the final mutant strain as the marker had no bearing on subsequent experiments. Gene deletions were confirmed by PCR.
Mutation of ugd in E. coli serotype K30 affected growth of the organism, presumably due to the accumulation of incomplete capsular polysaccharide repeat units. This led to the accumulation of second-site suppressor mutants, as seen in comparable biosynthetic systems (33, 34). To make CWG875, an E. coli serotype K30 (strain E69) derivative with a ugd deletion, the galE gene was first deleted to generate a strain where K30 capsular polysaccharide production was conditional. GalE (UDP-galactose-4-epimerase) is required for synthesis of the precursor for the galactose residues for the K30 repeat units, one of which is the first sugar of the repeat unit (35). The deletion of galE stops the synthesis of the CPS at the initial step of building the repeat unit on a carrier lipid thereby preventing the production of any synthesis intermediates whose accumulation could affect cell fitness and/or survival. To provide permissive conditions for K30 antigen production, the absence of GalE is overcome by addition of galactose to the growth medium (36). The galE gene was replaced with the kanamycin resistance cassette using the λRed recombinase system, and the cassette was excised, as above. The ugdK30 gene was disrupted with a gentamicin resistance cassette, using a suicide delivery vector (pWQ173), using a method reported elsewhere (23). Under nonpermissive conditions for CPS synthesis, the ugd mutation was tolerated. This provided a background where function of plasmid-encoded Ugd proteins could be tested in vivo based on their ability to restore CPS production under permissive conditions.
Amplification and Mutagenesis of ugd Gene
The ugdK-12 gene was PCR-amplified from E. coli DH5α genomic DNA with primers JB2 and JB6 that incorporate the coding sequence for an N-terminal hexahistidine (His6) tag. The resulting PCR product was cloned into pBAD24 to generate pWQ729. The ugdK30 gene was amplified from pWQ629 (37), incorporating the His6 tag and cloned into pBAD24, generating pWQ730. The QuikChange mutagenesis protocol (Stratagene/Agilent Technologies) was used to make site-directed mutants of UgdK-12 and UgdK30. Mutagenic primer pairs (primer sequences available upon request) were synthesized by Sigma, and DNA was amplified using Pfu Ultra DNA polymerase (Stratagene/Agilent Technologies). Mutations were confirmed by DNA sequencing at the Advanced Analysis Centre (University of Guelph).
DNA fragments encoding His6-tagged UgdK-12 and UgdK30 were excised from pWQ729 and pWQ730, respectively, by digestion with BamHI and HindIII and then cloned into pBAD33 producing pWQ738 and pWQ739, respectively. These pBAD33-based plasmids were compatible with the rcsA-containing pWQ500 for co-transformations of W3110.
Ugd Purification
His6-tagged UgdK-12 and UgdK30 proteins were expressed and purified primarily from CWG876 (exceptions to this are noted where relevant). Overnight cultures were diluted 1:50 into 1 liter of lysogeny broth media and grown at 37 °C to an A600 = 0.4–0.6, at which time arabinose was added to a final concentration of 0.02%. After 3 more hours of growth at 37 °C, cells were collected by centrifugation at 5,000 × g for 10 min at 4 °C. Cell pellets were resuspended in 20 mm sodium phosphate, pH 7.0, containing Complete Mini, EDTA-free protease inhibitor tablets (Roche Diagnostics), RNase (0.375 mg/ml, Roche Diagnostics), and DNase (0.375 mg/ml, Roche Diagnostics). The cells were then lysed by sonication. After centrifugation (1 h at 100,000 × g) to remove cell debris and unbroken cells, the lysate was incubated with nickel-nitrilotriacetic acid resin (Qiagen) for 1 h with mixing at 4 °C. The resin was poured into a column, and the flow-through was collected. Loosely bound proteins were removed with sequential washes of Buffer A (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 10 mm β-mercaptoethanol, and 10% glycerol) containing increasing concentrations of imidazole (10 and 40 mm). Ugd was then eluted with Buffer A containing 250 mm imidazole. The elution buffer in the protein samples was replaced with Buffer B (50 mm Tris-HCl, pH 7.5, containing 100 mm NaCl, 1 mm EDTA, 1 mm DTT, and 10% glycerol) by passage over a PD-10 desalting column (GE Healthcare). Protein concentration was determined using protein assay (Bio-Rad).
Ugd Activity and Kinetics Analysis
The UDP-glucose dehydrogenase activity of Ugd was measured using a version of a spectrophotometric assay (38), modified to facilitate the use of 96-well plates. The dehydrogenase reaction contained Buffer C (100 mm glycine, pH 9.5, 100 mm NaCl, 0.1 mg/ml acetylated BSA, and 10% glycerol). NAD and UDPG were varied as required (maximal concentrations were 5 mm for both). Reactions were monitored by absorbance at 340 nm in a temperature-controlled BMG FLUOStar OPTIMA plate reader. Km, Ksi, and Vmax values were calculated using the GraphPad Prism4 software program (GraphPad Software, Inc.), by fitting the data to the equation Y = Vmax·X (Km + X·(1 + X/Ksi)), where Y is the enzyme velocity; X is the substrate concentration; Vmax is the maximum enzyme velocity; Km is the Michaelis-Menten constant, and Ksi is the substrate inhibition constant. Km and Ksi values are expressed in the same units as the substrate concentration (X). Reactions were performed in triplicate (unless otherwise noted).
For pretreatment with kinase, nucleotides, and/or cation additives, purified Ugd (1 μg of Ugd per 125 μl reaction) was incubated for 10 min at 37 °C with the appropriate additive in the presence of Buffer B. To measure the dehydrogenase activity, 30 μl of the pretreatment reactions (or an equivalent concentration of Ugd) was combined with reaction mixture in a final volume of 200 μl.
SDS-PAGE and Western Blotting
Proteins were examined by SDS-PAGE using 10% resolving gels. K30 CPS production was monitored by immunoblotting of proteinase K-treated whole-cell lysates (39). SDS-PAGE and immunoblotting with anti-K30 antibodies (40) were performed as described previously (41).
In Vivo Phosphorylation Status of Ugd
Production of colanic acid was induced in wild-type E. coli K-12 strain, W3110, with the introduction of plasmid pWQ500, encoding the transcriptional regulator RcsA (57). Overexpression of His6-tagged Ugd proteins (encoded on plasmids pWQ738 and pWQ739) in cells carrying pWQ500 was also done to improve the detection of a potential tyrosyl-phosphorylated Ugd. Cell lysates were separated by SDS-PAGE, and proteins were visualized by immunoblotting with antibodies specific to phosphotyrosine (PY20, Sigma).
Structural Model of UgdK-12
The model of UgdK-12 was generated by SWISS-MODEL using the “automated mode.” The template was provided by the structure of the protein from K. pneumoniae (UgdKp) in complex with NAD and uridine 5′-monophosphate (PDB 3PLR (11)). To obtain a better representation of a molecule of UDPG in the active site, the structure of UgdKp in complex with UDPGA (PDB 3PJG) was aligned to the UgdK-12 model by PyMOL (PyMOL Molecular Graphics System Version 0.99rc6, Schrödinger, LLC), and an oxygen atom on C6 of the glucose moiety of UDPGA molecule was removed.
RESULTS
UgdK-12 Can Functionally Replace UgdK30 in Biosynthesis of K30 CPS
The Ugd proteins from E. coli K-12 and the K30 serotype strain, E69 (hereafter referred to as UgdK-12 and UgdK30, respectively), are recently diverged homologs sharing 82% sequence identity and 93% similarity (Fig. 1A). Both proteins possess the Tyr-71 residue, which is the proposed site of tyrosine phosphorylation in UgdK-12 (29). However, there is some sequence diversity in the region of this residue (Fig. 1A), giving rise to the possibility that the two proteins may differ in either the efficiency or the effects of phosphorylation at this position. Activity of UgdK30 is required for the production of K30 CPS in E. coli E69, so CPS production from a ugd deletion strain reflects the in vivo functioning of plasmid-encoded Ugd proteins. An additional ΔgalE mutation in this background made CPS production conditional on addition of galactose to the growth medium (see “Experimental Procedures”).
Expression of His6-tagged UgdK-12 restored K30 CPS production in CWG875 (ΔgalE Δugd), as shown by immunoblotting cellular extracts with K30-specific antibodies (Fig. 1D). Therefore, any regulatory elements required for UgdK-12 are met in the group 1 capsule-producing background. Surprisingly, a Tyr → Phe mutation at the proposed site of phosphorylation, Tyr-71, did not have any detectable effect on the ability of UgdK-12 to function in K30 CPS synthesis. In previous reports, a UgdK-12 Y71F mutant showed very low activity in vitro (29). In addition, when examined in an E. coli K-12 background where colanic acid expression was activated, the Y71F mutant protein resulted in drastically reduced levels of colanic acid and a corresponding loss of the mucoid colony phenotype. In CWG875, the Tyr-71 residue of UgdK30 was also not essential for K30 CPS production. In the context of a constitutively expressed capsular polysaccharide, the proposed phosphorylation site of Ugd was therefore not required in either of the Ugd homologs.
Ugd Is Active in the Absence of Phosphorylation
These results suggested that either redundant phosphorylation sites exist or that Ugd phosphorylation is not required for K30 capsule production. To rule out the possibility of phosphorylation occurring on a different residue in Ugd, the genes encoding the only two known BY-kinases in E. coli K-12 were deleted to generate strain CWG876 (W3110 Δwzc Δetk ugd::kan). The dehydrogenase activities of purified Ugd proteins isolated from CWG876 were analyzed at fixed substrate concentrations (Fig. 2A). Consistent with the in vivo activities of the Y71F mutants, both Ugd homologs expressed in the kinase-deficient background were enzymatically active, although the activity of UgdK-12 was considerably lower than UgdK30. The activity of UgdK-12 isolated from CWG875 (i.e. a BY-kinase-positive strain) was indistinguishable from the same protein prepared from the kinase-deficient background. Furthermore, we could not detect any phosphorylation of UgdK-12 in vivo in E. coli under conditions where the wzc kinase gene was derepressed and colanic acid production was activated by the overexpression of the transcriptional regulator RcsA (data not shown).
FIGURE 2.
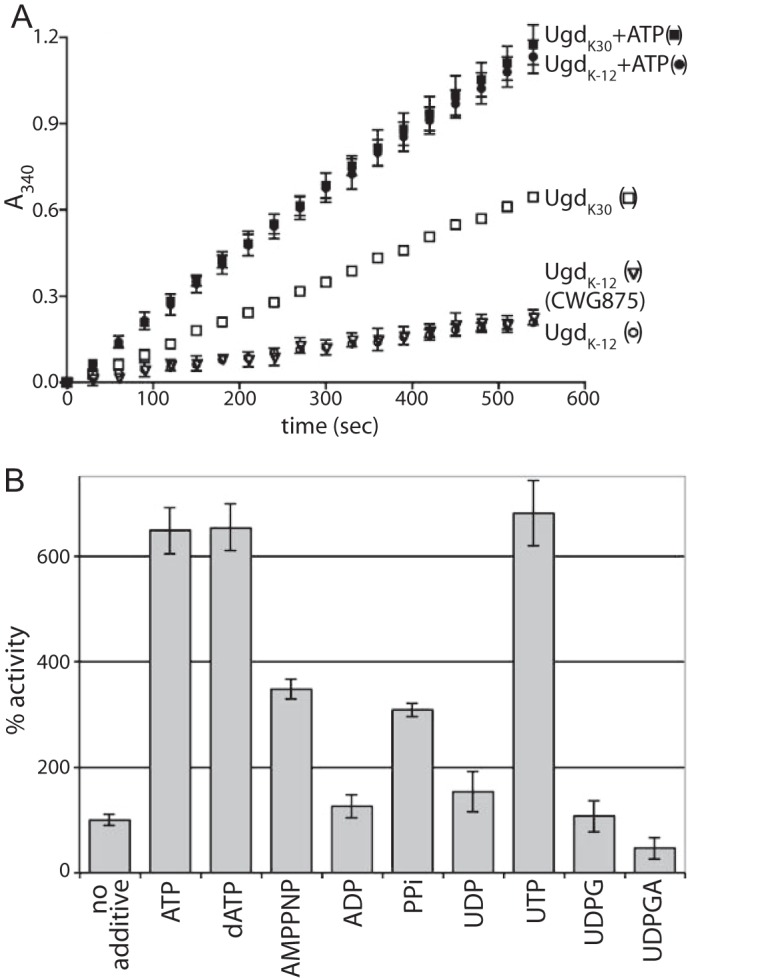
Activation of Ugd dehydrogenase activity. A, activities of UgdK-12 and UgdK30 purified from CWG876 were monitored over 10 min in the presence or absence of 1 mm ATP, at fixed concentrations of 5 mm NAD and 5 mm UDPG. The activity of UgdK-12 purified from CWG875 was included as a control for a BY-kinase-positive strain. B, other nucleotides and pyrophosphate were tested for the ability to affect UgdK-12 activity. All were added to a concentration of 1 mm. The slope of a line produced by plotting the A340 versus time (as in A) was calculated and normalized to the “no additive” reaction, which was arbitrarily set to 100%. All reactions were tested in triplicate (except for the UgdK30 reaction, which was done in duplicate), and standard deviations are shown as error bars. The NAD and UDPG substrates were fixed at 5 mm for all reactions (except for the reaction with added UDPG).
Ugd Activity Is Stimulated by Triphosphate Esters
It was previously reported that incubation of UgdK-12 with the kinase domain of Wzc and ATP resulted in a modest increase in Ugd activity (20). We observed similar results (data not shown), but this was inconsistent with the results described above. To investigate this phenomenon in more detail, the contents of the phosphorylation reactions were examined systematically. Surprisingly, an ∼6-fold increase in UgdK-12 activity was observed when only ATP was added to the reaction (Fig. 2A). A substantial increase in UdgK30 activity was also observed when ATP was added, but in this case, the increase was not as large, possibly because UgdK30 has a higher basal activity than UgdK-12. To rule out the possibility that enzyme activation resulted from trace amounts of phosphorylation by the activity of a contaminating kinase, the effect of AMPPNP was examined. This nonhydrolyzable analog of ATP also caused an increase in the activity of UgdK-12, although to a lower extent than ATP (Fig. 2B). Other nucleotides were tested for the ability to activate UgdK-12; all of the tested nucleotide triphosphates (including dATP) were able to increase the activity of UgdK-12. In contrast, nucleotide diphosphates and the nucleotide sugar substrate, UDPG, did not affect the activity. Consistent with product inhibition observed with other Ugd homologs (11, 42), UDPGA caused a decrease in UgdK-12 activity. As any tested NTP (or dNTP) was able to cause the activity increase, and NDP was not able to activate, the triphosphate ester moiety appears to be the shared “activating” feature. Highlighting the importance of the phosphate groups, pyrophosphate was able to increase the activity to an intermediate level (on par with AMPPNP).
Kinetic Parameters for Ugd Dehydrogenase Activity
The demonstrable lack of requirement for a kinase in Ugd activation indicated that there is some other underlying mechanism for the NTP-dependent activation of UgdK-12. To investigate this further, the kinetics of the enzyme were analyzed. At a fixed UDPG concentration, the dehydrogenase activity of UgdK-12 decreased at NAD concentrations above 1 mm (Fig. 3), indicating substrate inhibition. A kinetic model that includes substrate inhibition gave a satisfactory fit for the plot of activity versus NAD concentration and resulted in a calculated Ksi for NAD of 1.4 ± 0.2 mm (Table 2). Conversely, the activity of UgdK30 reached a plateau at high concentrations of NAD, and the resulting plot conformed more closely to classical Michaelis-Menten kinetics. When analyzed with a substrate inhibition model, the UgdK30 Ksi for NAD was five times higher than that for UgdK-12 (Table 2). In summary, there may be some NAD substrate inhibition for UgdK30, but it plays a relatively minor role at the concentrations used in these experiments. The other kinetic parameters were relatively similar for the two homologs (see Table 2). Additionally, UgdK-12 purified from wild-type serotype K30 and wild-type W3110 (both of which contain endogenous etk and wzc genes) showed kinetic parameters virtually identical to UgdK-12 purified from related BY-kinase deletion strains, CWG285 and CWG876 (Table 3).
FIGURE 3.
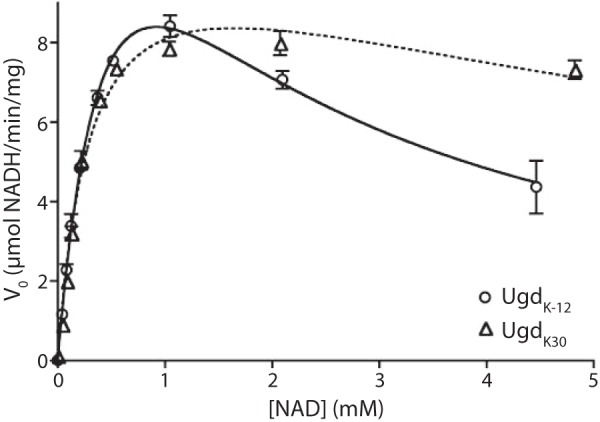
NAD kinetics for UgdK-12 and UgdK30. The initial reaction velocities of UgdK-12 and UgdK30 were calculated over a range of NAD concentrations at a fixed UDPG concentration (5 mm). Lines representing the best fit of substrate inhibition kinetics were plotted for both homologs. Error bars represent the standard deviation of reactions done in triplicate.
TABLE 2.
Kinetic parameters for NAD of UgdK-12 and UgdK30 isolated from CWG876
| kcat | Km | Ksi | |
|---|---|---|---|
| s−1 | mm | mm | |
| UgdK-12 | 7.1 ± 0.5 | 0.62 ± 0.07 | 1.4 ± 0.2 |
| UgdK30 | 4.2 ± 0.2 | 0.31 ± 0.03 | 8.7 ± 1.6 |
| UgdK-12 Y71Fa | 7.3 ± 0.9 | 0.50 ± 0.10 | 1.4 ± 0.3 |
| UgdK-12 K323A | 2.7 ± 0.2 | 0.43 ± 0.05 | 2.8 ± 0.4 |
| UgdK-12 + [ATP] (μm) | |||
| 25 | 5.9 ± 0.4 | 0.45 ± 0.05 | 2.6 ± 0.3 |
| 50 | 6.2 ± 0.4 | 0.62 ± 0.06 | 2.8 ± 0.4 |
| 100 | 5.3 ± 0.2 | 0.45 ± 0.03 | 5.4 ± 0.7 |
| 250 | 5.1 ± 0.3 | 0.45 ± 0.04 | 13.4 ± 3.7 |
| 500 | 5.0 ± 0.2 | 0.35 ± 0.03 | 24.9 ± 7.9 |
| 750 | 5.6 ± 0.3 | 0.44 ± 0.04 | 30.9 ± 15.2 |
| 1000 | 5.5 ± 0.2 | 0.38 ± 0.03 | 47.4 ± 27.0 |
a UgdK-12 Y71F was tested in duplicate; all others were tested in triplicate.
TABLE 3.
Kinetic parameters for NAD of UgdK-12 isolated from wild-type and BY-kinase deletion strains
| kcat | Km | Ksi | |
|---|---|---|---|
| s−1 | mm | mm | |
| Wild-type E69 | 5.7 ± 0.4 | 0.45 ± 0.05 | 1.3 ± 0.2 |
| CWG285 | 5.4 ± 0.4 | 0.35 ± 0.04 | 1.5 ± 0.2 |
| Wild-type W3110 | 6.1 ± 0.4 | 0.45 ± 0.04 | 1.9 ± 0.2 |
| CWG876 | 7.1 ± 0.5 | 0.62 ± 0.07 | 1.4 ± 0.2 |
Consistent with the ability of the Y71F mutant of UgdK-12 to substitute in vivo for wild-type protein, the kinetic parameters of this mutant were similar to the wild-type protein (Table 2). However, the purified UgdK-12 Y71F mutant protein was highly prone to aggregation and loss of activity in the absence of glycerol in the reaction buffer (Fig. 4). To overcome any potential problems with aggregation, glycerol was included in the buffers in all of the experiments presented here (unless otherwise noted).
FIGURE 4.
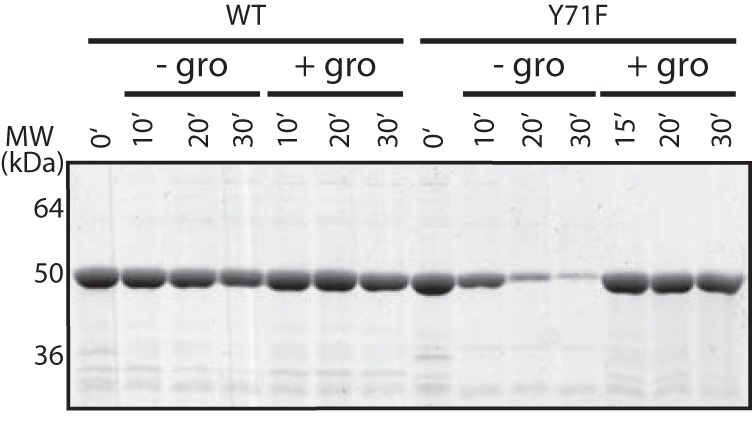
Stability of UgdK-12. Wild-type and Y71F mutant UgdK-12 proteins were incubated at 37 °C in the presence/absence of glycerol (gro, 10% v/v final concentration) for the times indicated. Samples were subjected to centrifugation at maximum speed (16,100 × g) for 5 min in a microcentrifuge. The resulting supernatants were separated by SDS-PAGE and stained with Simply Blue (Invitrogen).
To determine whether the NTP-dependent activation of UgdK-12 influenced the kinetics of the dehydrogenase, the NAD reduction kinetics were analyzed in the presence of increasing ATP (Fig. 5). As the concentration of ATP increased, the substrate inhibition due to NAD diminished, resulting in a corresponding ATP-dependent increase in Ksi for NAD (Table 2). At higher ATP concentrations, the activity plot for UgdK-12 appeared to follow Michaelis-Menten kinetics. Replacing ATP with AMPPNP or pyrophosphate had a similar effect on the NAD substrate inhibition (data not shown), consistent with the observation that this effect is not specific to ATP.
FIGURE 5.
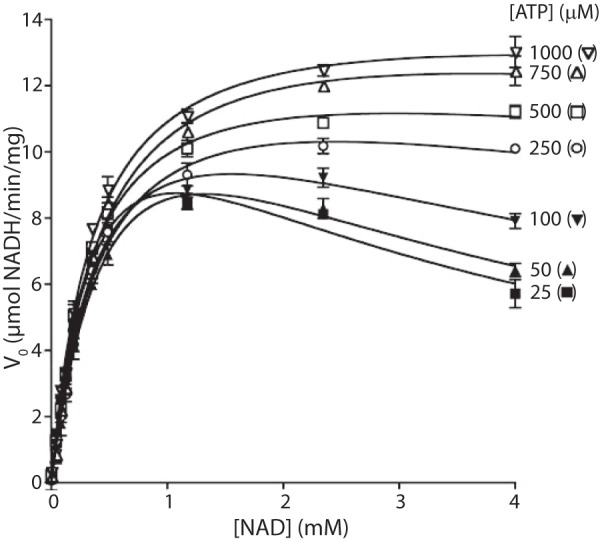
ATP addition removes the NAD substrate inhibition of UgdK-12 in a concentration-dependent manner. The substrate inhibition kinetics of UgdK-12 were monitored and plotted, as in Fig. 3, with increasing ATP concentrations. There was an outlier at 100 μm NAD for the 750 μm ATP run that was removed.
During assay optimization, it was found that the inclusion of MgCl2 in the reaction buffer could reverse the NTP-dependent activation of UgdK-12, in a concentration-dependent manner (Fig. 6A). This was not specific to Mg2+ as a similar effect was found for other divalent cations, including Ca2+, Mn2+, Zn2+, and Ni2+ (data not shown). Monovalent cations did not affect UgdK-12 activity (data not shown). Chelating the divalent cation with EDTA (Fig. 6) (or EGTA, data not shown) restored the NTP-dependent activation of UgdK-12.
FIGURE 6.
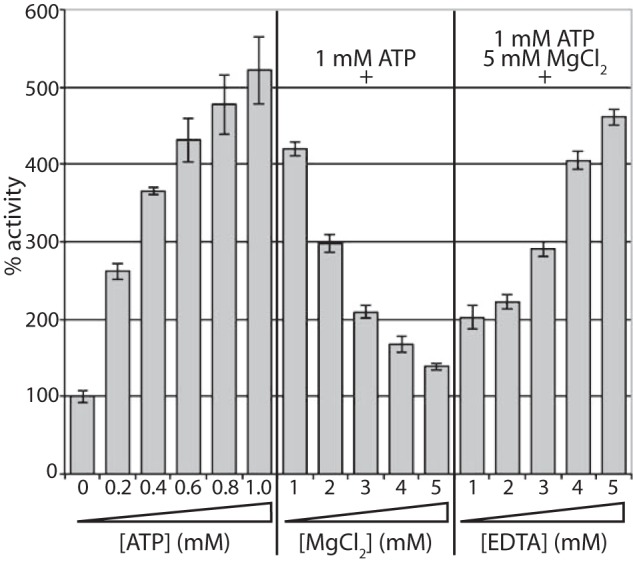
Effect of ATP, Mg2+, and EDTA on UgdK-12 activity. The activity of UgdK-12 was calculated relative to the no additive control, as in Fig. 2B, with increasing ATP, Mg2+, and EDTA. All reactions were performed in triplicate.
Mutagenesis of a Putative Allosteric Binding Site on UgdK-12
The structure of Ugd from K. pneumoniae NTUH-K2044 (UgdKp) has been determined (11). UgdKp shares 83% sequence identity and 93% sequence similarity with UgdK-12 and only differs from UgdK30 by three amino acids. The UgdKp-UDPGA complex structure (PDB 3PJG) contained an additional molecule of UDPGA bound at a second site, distinct from the catalytic site. This second UDPGA-binding site lies between the C-terminal Rossmann fold domain (UDP-glucose binding) and the helical subdomain linking this domain to the N-terminal NAD-binding Rossmann fold domain (11). In the UgdKp-UDPGA structure, the conformations of Cys-253, Lys-256, and Asp-257, all critical for catalysis (60, 61), were shifted out of the catalytic site, and this state of Ugd was therefore proposed to represent the structural state of the enzyme under conditions of known product inhibition (11). Because UDPGA, NAD, and the various nucleotide triphosphates share a common nucleotide structure, it is reasonable to hypothesize that all of these molecules share a single allosteric binding site. This site is defined by the presence of a cluster of basic residues, including Lys-323 and Arg-324 of UgdKp (Fig. 1C), which hydrogen bonds to the pyrophosphate linkage of UDPGA. Each of these critical basic residues was replaced (individually and together) with alanine. The R324A single and KR → AA double mutants of UgdK-12 purified in much lower amounts relative to wild type; they were prone to degradation and had negligible activity (data not shown). However, the K323A mutant purified similar to wild-type UgdK-12, with respect to both yield and stability (data not shown). NAD kinetics for the K323A mutant were similar to wild-type UgdK-12 (Table 2), albeit with a slightly lower kcat. When NAD and UDPG were kept constant and ATP was varied, a velocity versus ATP concentration plot generated curves that fit Michaelis-Menten kinetics for both the wild-type and K323A mutant proteins (data not shown). The maximal velocities were similar (kcat = 4.6 ± 0.1 and 3.3 ± 0.2 μmol of NADH/min/mg for wild-type and K323A mutant proteins, respectively) but the ATP concentration at one-half the maximum velocity was about 6-fold higher for the K323A mutant (K½ = 0.14 ± 0.01 and 0.83 ± 0.16 mm ATP, wild-type and mutant proteins, respectively). The higher K½ value caused by the K323A mutation could therefore be explained by a lower binding affinity for ATP compared with UgdK-12.
DISCUSSION
Ugd synthesizes a critical precursor for the biosynthesis of many bacterial glycans. Although the Ugd homologs from two strains of E. coli share a high degree of sequence conservation, they show different kinetic properties; substrate inhibition due to NAD was only a significant factor for UgdK-12. The kinetics of UgdK30 could be modeled to include NAD substrate inhibition (Fig. 3 and Table 2), but the inhibitory effect was very limited for this homolog and is unlikely to have a profound effect on its in vivo activity. The measured cellular concentration of NAD in E. coli is in the range of 0.8–1.5 mm depending on the growth phase (43), which is at or slightly below the Ksi NAD for UgdK-12 (1.4 ± 0.2 mm). The inhibitory effect of NAD at these cellular concentrations will likely impinge on UgdK-12 activity.
In previous studies, purified, nonphosphorylated UgdK-12 was found to have relatively low basal activity (20, 29), but the reaction buffers for those experiments included 5 mm NAD. Based on the kinetic data presented here, the low basal activity could be explained by the substrate inhibition due to the high levels of NAD in the reaction mixtures. Lacour et al. (29) also found that UgdK-12 Y71F was folded properly but was almost completely inactive. In this study, inclusion of glycerol in the reaction buffers was essential to stabilize the Y71F mutant and maintain activity. Under these conditions, the mutant and wild-type enzymes possess comparable kinetic properties (Table 2), including NTP-dependent activation
The mechanism of NAD substrate inhibition is not known for UgdK-12. There are several models available to explain substrate inhibition (44), but one attractive model for UgdK-12 invokes the possibility that multiple molecules of NAD enter the active site. The structure of UgdKp (which shares 93% sequence similarity with UgdK-12) has been solved from a co-crystal of the enzyme with NAD, revealing that the binding pocket for NAD is relatively open (11). At high concentrations of NAD, multiple molecules of the cofactor may fit into the site in nonproductive geometries, giving rise to the observed decrease in dehydrogenase activity. UgdK30 shares a higher degree of conservation with UgdKp (differing by only three amino acids), consistent with the proposal that the capsule biosynthesis/export genes lie in a region that may have been horizontally transferred between E. coli and K. pneumoniae (24). UgdK30 is much less susceptible to NAD substrate inhibition, so the open binding site as observed in UgdKp is not sufficient by itself to cause substrate inhibition. Additional structural differences in the UgdK-12 NAD binding pocket may account for the observed differences in substrate inhibition. However, binding of NAD to an allosteric site that negatively impacts the catalytic site is another possible mechanism to explain the substrate inhibition of UgdK-12.
Allosteric regulation of Ugd is a well established phenomenon. In particular, UgdKp shows product inhibition by UDPGA, where UDGPA binds a second pocket adjacent to the catalytic site, and reorganizes several key catalytic residues (11). The compromised binding of ATP (and, to a lesser extent, NAD) in the UDPGA allosteric site of the K323A mutant argues that all three effectors share a common, or overlapping, binding site(s). In particular, it is interesting to note that the uracil-binding portion of this pocket interacts with the nucleotide via Phe-345 and the aliphatic portion of Lys-323, but the pocket does not form hydrogen bonds directly to the nucleotide and is sufficiently open that pyrimidines or purines should be accommodated. This suggests a model where diverse nucleotides can bind at this site, with one of two potential effects. Binding UDPGA or NAD results in the preferential stabilization of the catalytic loop in a conformation incompatible with efficient turnover (Fig. 7). Alternatively, binding of ATP or related trinucleotides may fail to stabilize the unproductive conformation of this loop, while still competing with NAD and UDPGA for the same binding site. Interactions formed by Asp-257 and Lys-259 with the sugar hydroxyls of UDPGA (and possibly also the ribose of NAD) may be critical determinants in whether the unproductive conformation of the catalytic loop is stabilized. Differences in susceptibility to inhibition may reflect the relative stability of the catalytic loop in its two possible conformations.
FIGURE 7.

Conceptual model for Ugd regulation. A, in the presence of high concentrations of UDPGA or NAD (shown), these molecules can bind in the regulatory site. Through interactions with residues such as Lys-256 and Asp-257, the mobile loop, including α11, is stabilized in a conformation where Cys-253 is misplaced from the catalytic site, resulting in a conformation that is not competent for turnover (this conformation is seen in UDPGA-bound UgdKp, PDB 3PJG (11)). B, if a nucleotide triphosphate is bound in this site (ATP depicted), then interactions mediated by the γ-phosphate with residues on α11 (most likely Lys-256 and Lys-259) stabilize this region as a long α-helix in an alternative conformation. This conformation is seen in NAD plus UDP-bound UgdKp (PDB 3PLR (11)), except that ATP was modeled in analogy to the second UDPGA in 3PJG. In this conformation, Cys-253 is correctly positioned for attacking the aldehyde moiety of UDPG, and catalysis can proceed. Note that this conformation is likely also stable in the absence of NTP binding, as catalysis is not dependent upon the presence of NTPs under low NAD concentrations.
Kinetic analysis of UgdK-12 demonstrated that the NTP-dependent activation and NAD substrate inhibition were linked, i.e. the addition of NTP offset the effect of substrate inhibition in a concentration-dependent manner (Fig. 5 and Table 2). The NTP-dependent activation was not nucleotide base-specific as both UTP and ATP could activate UgdK-12. The lack of activation with NDP addition indicated that the triphosphate moiety was important. However, because pyrophosphate was shown to act as a moderate activator, it is conceivable that only the terminal diphosphate (β- and γ-phosphates of the NTP) is required. An alternative model is that the third phosphate is required to compensate for unfavorable interactions mediated by the nucleotide. Divalent cations greatly reduce the effectiveness of ATP in relieving inhibition, probably because the NTP-cation complex lacks sufficient electronegativity to bind favorably in the highly electropositive binding pocket.
The concentration of ATP required to remove substrate inhibition is lower than the reported intracellular concentration of 3 mm for E. coli (45), so it is likely that any substrate inhibition of UgdK-12 would be diminished in vivo, especially considering that the source of stimulation is not confined to ATP (Fig. 2B). However, confidently assessing the contributions is difficult because the competing effects are further complicated by the concentration of free Mg2+ in E. coli (reported to be between 1 and 2 mm (46)), which would at least partly attenuate the NTP-dependent stimulatory effect. Any local variation in the concentrations of ATP and Mg2+ (and NAD) in the microenvironments of UgdK-12 could also exert an effect. In essence, small alterations in the local concentrations for any of these molecules could enable the modulation of Ugd activity affecting the production of UDPGA and subsequent downstream products (e.g. extracellular polysaccharide).
The data reported here indicate that, in contrast to previous publications (20, 29), phosphorylation of Tyr-71 is not required for Ugd activity. The structure of UgdKp (which shares high sequence identity with UgdK-12) positions Tyr-71 relatively deep within the NAD-binding site (Fig. 1C) (11). There is some evidence that BY-kinases bind their substrates as extended peptides. In the crystal of the tyrosine kinase domain of the Staphylococcus aureus Wzc homolog, CapB, the tyrosine-rich C terminus of one monomer adopted an extended conformation to bind in the active site of an adjacent monomer (47). Therefore, phosphorylation of Tyr-71 in Ugd would require local unfolding to place the tyrosine in the catalytic site of the kinase. In contrast, phosphorylation of UDP-N-acetylmannosamine dehydrogenase (Cap5O) by the Cap5B BY-kinase from S. aureus occurs at a tyrosine residue that is surface-exposed in the solved structure (48). The activity of the modified enzyme is increased 2–3-fold above the basal (unmodified) level. The Ugd homolog from B. subtilis (UgdBs) could be phosphorylated in vitro leading to an increase (2-fold) in dehydrogenase activity (19). Although no structure is available for UgdBs, a structural model generated by structure-based sequence alignment located the proposed tyrosine residue on the surface of the protein (49), potentially making it accessible to kinase. Consistent with this, phosphorylated UgdBs was identified in a phosphoproteomics study (50), although it was not clear if it was the tyrosine or an adjacent serine that was phosphorylated. UgdK-12 was not identified in a similar phosphoproteomics study in E. coli, but it is unclear whether the BY-kinases were active under the conditions used (51); notably, neither of the autophosphorylating BY-kinases (Etk and Wzc) (25, 52) were detected in the phosphoproteome under the same conditions. The absence of any effect of Tyr-71 mutations could be explained by phosphorylation at a different site. For example, TuaD (another Ugd homolog in B. subtilis involved in teichuronic acid production) was activated upon phosphorylation but does not have a tyrosine residue in the position equivalent to the Tyr-71 of UgdBs (19). However, we were unable to identify conditions that gave convincing phosphorylation of UgdK30 or UgdK-12 in the E. coli K30 background, despite clear activity for WzcK30 (data not shown). Also, induction of colanic acid production in wild-type E. coli K-12 strain, W3110, caused increased wzc expression, but no concomitant phosphorylation of Ugd was detected (data not shown). Low level, nonspecific phosphorylation of Cap5O was reported and interpreted as a reflection of the flexibility of the Cap5B kinase (48). Whether this extends to Wzc/Etk is unclear. The regulation of extracellular polysaccharide production is a complex process. The data reported here indicate that different homologs of Ugd exhibit altered properties in two isolates of E. coli despite a highly conserved sequence. The regulation of ugd in E. coli K-12 is a multilayered process. In some bacteria, the ugd gene itself is regulated by as many as three different phosphorelay systems. In E. coli, ugdK-12 was found to be within the top 10 genes that were up-regulated by the RcsC/YojN/RcsB/RcsA phosphorelay system as part of the Rcs regulon (53); the regulon includes a wide range of genes associated with cell envelope assembly and homeostasis, including colanic acid production (22). UgdK-12 regulation is also impacted by both PmrA/PmrB and PhoP/PhoQ, and this may influence the need for RcsA (54). The data presented here demonstrate that unique features of the enzyme kinetics of UgdK-12 also provide an additional level of post-translational control, potentially allowing instantaneous response to particular environmental cues. In contrast, group 1 CPSs are constitutively expressed in E. coli and K. pneumoniae, where they are important virulence factors. The cps locus is not responsive to RcsC/YojN/RcsB/(RcsA) (23) in E. coli with group 1 CPSs. Enhanced CPS production in these strains results from Rcs-mediated up-regulation of the galF gene, whose product is implicated in the biosynthesis of a UDP-glucose precursor (55). The sequences upstream of ugdK-12 and ugdK30 differ, with the latter lacking an evident PmrA box (data not shown) suggesting different regulation. In these bacteria, UgdK30 is active under conditions where Rcs-mediated up-regulation is not anticipated, and it lacks the subtle modulation of activity seen in UgdK-12. Why two Ugd homologs from different E. coli strains would differ in post-translational regulatory properties remains unclear.
Acknowledgments
We thank Jason Carere and Scott Mazurkewich for technical advice regarding enzyme kinetics.
This work was supported by Canadian Institutes of Health Research Operating Grant FRN-9623 (to C. W.) and National Science and Engineering Research Council of Canada Grant 327280 (to M. S. K.).
- UDPGA
- UDP-glucuronic acid
- AMPPNP
- 5′-(β,γ-imido)triphosphate
- CPS
- capsular polysaccharide
- PDB
- Protein Data Bank
- UDPG
- UDP-glucose
- Ugd
- UDP-glucose dehydrogenase
- BY-kinase
- bacterial tyrosine kinase.
REFERENCES
- 1. Clarkin C. E., Allen S., Kuiper N. J., Wheeler B. T., Wheeler-Jones C. P., Pitsillides A. A. (2011) Regulation of UDP-glucose dehydrogenase is sufficient to modulate hyaluronan production and release, control sulfated GAG synthesis, and promote chondrogenesis. J. Cell Physiol. 226, 749–761 [DOI] [PubMed] [Google Scholar]
- 2. Alvarez D., Merino S., Tomás J. M., Benedí V. J., Albertí S. (2000) Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O side chain-deficient Klebsiella pneumoniae clinical isolates. Infect. Immun. 68, 953–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wessels M. R., Goldberg J. B., Moses A. E., DiCesare T. J. (1994) Effects on virulence of mutations in a locus essential for hyaluronic acid capsule expression in group A streptococci. Infect. Immun. 62, 433–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raetz C. R., Reynolds C. M., Trent M. S., Bishop R. E. (2007) Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strominger J. L., Kalckar H. M., Axelrod J., Maxwell E. S. (1954) Enzymatic oxidation of uridine diphosphate glucose to uridine diphosphate glucuronic acid. J. Am. Chem. Soc. 76, 6411–6412 [Google Scholar]
- 6. Egger S., Chaikuad A., Kavanagh K. L., Oppermann U., Nidetzky B. (2010) UDP-glucose dehydrogenase: structure and function of a potential drug target. Biochem. Soc. Trans. 38, 1378–1385 [DOI] [PubMed] [Google Scholar]
- 7. Egger S., Chaikuad A., Kavanagh K. L., Oppermann U., Nidetzky B. (2011) Structure and mechanism of human UDP-glucose 6-dehydrogenase. J. Biol. Chem. 286, 23877–23887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kadirvelraj R., Sennett N. C., Polizzi S. J., Weitzel S., Wood Z. A. (2011) Role of packing defects in the evolution of allostery and induced fit in human UDP-glucose dehydrogenase. Biochemistry 50, 5780–5789 [DOI] [PubMed] [Google Scholar]
- 9. Hung R. J., Chien H. S., Lin R. Z., Lin C. T., Vatsyayan J., Peng H. L., Chang H. Y. (2007) Comparative analysis of two UDP-glucose dehydrogenases in Pseudomonas aeruginosa PAO1. J. Biol. Chem. 282, 17738–17748 [DOI] [PubMed] [Google Scholar]
- 10. Schiller J. G., Bowser A. M., Feingold D. S. (1973) Partial purification and properties of UDPG dehydrogenase from Escherichia coli. Biochim. Biophys. Acta 293, 1–10 [DOI] [PubMed] [Google Scholar]
- 11. Chen Y. Y., Ko T. P., Lin C. H., Chen W. H., Wang A. H. (2011) Conformational change upon product binding to Klebsiella pneumoniae UDP-glucose dehydrogenase: a possible inhibition mechanism for the key enzyme in polymyxin resistance. J. Struct. Biol. 175, 300–310 [DOI] [PubMed] [Google Scholar]
- 12. Grangeasse C., Cozzone A. J., Deutscher J., Mijakovic I. (2007) Tyrosine phosphorylation: an emerging regulatory device of bacterial physiology. Trends Biochem. Sci. 32, 86–94 [DOI] [PubMed] [Google Scholar]
- 13. Cuthbertson L., Mainprize I. L., Naismith J. H., Whitfield C. (2009) Pivotal roles of the outer membrane polysaccharide export and polysaccharide copolymerase protein families in export of extracellular polysaccharides in Gram-negative bacteria. Microbiol. Mol. Biol. Rev. 73, 155–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shi L., Kobir A., Jers C., Mijakovic I. (2010) Bacterial protein-tyrosine kinases. Curr. Proteomics 7, 188–194 [Google Scholar]
- 15. Drummelsmith J., Whitfield C. (1999) Gene products required for surface expression of the capsular form of the group 1 K antigen in Escherichia coli (O9a: K30). Mol. Microbiol. 31, 1321–1332 [DOI] [PubMed] [Google Scholar]
- 16. Stevenson G., Andrianopoulos K., Hobbs M., Reeves P. R. (1996) Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J. Bacteriol. 178, 4885–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Collins R. F., Beis K., Dong C., Botting C. H., McDonnell C., Ford R. C., Clarke B. R., Whitfield C., Naismith J. H. (2007) The 3D structure of a periplasm-spanning platform required for assembly of group 1 capsular polysaccharides in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 104, 2390–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong C., Beis K., Nesper J., Brunkan-Lamontagne A. L., Clarke B. R., Whitfield C., Naismith J. H. (2006) Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature 444, 226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mijakovic I., Poncet S., Boël G., Mazé A., Gillet S., Jamet E., Decottignies P., Grangeasse C., Doublet P., Le Maréchal P., Deutscher J. (2003) Transmembrane modulator-dependent bacterial tyrosine kinase activates UDP-glucose dehydrogenases. EMBO J. 22, 4709–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grangeasse C., Obadia B., Mijakovic I., Deutscher J., Cozzone A. J., Doublet P. (2003) Autophosphorylation of the Escherichia coli protein kinase Wzc regulates tyrosine phosphorylation of Ugd, a UDP-glucose dehydrogenase. J. Biol. Chem. 278, 39323–39329 [DOI] [PubMed] [Google Scholar]
- 21. Whitfield C. (2006) Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem. 75, 39–68 [DOI] [PubMed] [Google Scholar]
- 22. Majdalani N., Gottesman S. (2005) The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59, 379–405 [DOI] [PubMed] [Google Scholar]
- 23. Rahn A., Whitfield C. (2003) Transcriptional organization and regulation of the Escherichia coli K30 group 1 capsule biosynthesis (cps) gene cluster. Mol. Microbiol. 47, 1045–1060 [DOI] [PubMed] [Google Scholar]
- 24. Rahn A., Drummelsmith J., Whitfield C. (1999) Conserved organization in the cps gene clusters for expression of Escherichia coli group 1 K antigens: relationship to the colanic acid biosynthesis locus and the cps genes from Klebsiella pneumoniae. J. Bacteriol. 181, 2307–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ilan O., Bloch Y., Frankel G., Ullrich H., Geider K., Rosenshine I. (1999) Protein tyrosine kinases in bacterial pathogens are associated with virulence and production of exopolysaccharide. EMBO J. 18, 3241–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wugeditsch T., Paiment A., Hocking J., Drummelsmith J., Forrester C., Whitfield C. (2001) Phosphorylation of Wzc, a tyrosine autokinase, is essential for assembly of group 1 capsular polysaccharides in Escherichia coli. J. Biol. Chem. 276, 2361–2371 [DOI] [PubMed] [Google Scholar]
- 27. Bechet E., Gruszczyk J., Terreux R., Gueguen-Chaignon V., Vigouroux A., Obadia B., Cozzone A. J., Nessler S., Grangeasse C. (2010) Identification of structural and molecular determinants of the tyrosine kinase Wzc and implications in capsular polysaccharide export. Mol. Microbiol. 77, 1315–1325 [DOI] [PubMed] [Google Scholar]
- 28. Lee D. C., Zheng J., She Y. M., Jia Z. (2008) Structure of Escherichia coli tyrosine kinase Etk reveals a novel activation mechanism. EMBO J. 27, 1758–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lacour S., Bechet E., Cozzone A. J., Mijakovic I., Grangeasse C. (2008) Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One 3, e3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bertani G. (1951) Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62, 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cherepanov P. P., Wackernagel W. (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic resistance determinant. Gene 158, 9–14 [DOI] [PubMed] [Google Scholar]
- 33. Xayarath B., Yother J. (2007) Mutations blocking side chain assembly, polymerization, or transport of a Wzy-dependent Streptococcus pneumoniae capsule are lethal in the absence of suppressor mutations and can affect polymer transfer to the cell wall. J. Bacteriol. 189, 3369–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yuasa R., Levinthal M., Nikaido H. (1969) Biosynthesis of cell wall lipopolysaccharide in mutants of Salmonella. V. A mutant of Salmonella typhimurium defective in the synthesis of cytidine diphosphoabequose. J. Bacteriol. 100, 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Whitfield C., Paiment A. (2003) Biosynthesis and assembly of group 1 capsular polysaccharides in Escherichia coli and related extracellular polysaccharides in other bacteria. Carbohydr. Res. 338, 2491–2502 [DOI] [PubMed] [Google Scholar]
- 36. Boels I. C., Ramos A., Kleerebezem M., de Vos W. M. (2001) Functional analysis of the Lactococcus lactis galU and galE genes and their impact on sugar nucleotide and exopolysaccharide biosynthesis. Appl. Environ. Microbiol. 67, 3033–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jayaratne P., Bronner D., MacLachlan P. R., Dodgson C., Kido N., Whitfield C. (1994) Cloning and analysis of duplicated rfbM and rfbK genes involved in the formation of GDP-mannose in Escherichia coli O9:K30 and participation of rfb genes in the synthesis of the group I K30 capsular polysaccharide. J. Bacteriol. 176, 3126–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pagni M., Lazarevic V., Soldo B., Karamata D. (1999) Assay for UDPglucose-6-dehydrogenase in phosphate-starved cells: gene tuaD of Bacillus subtilis 168 encodes the UDP-glucose-6-dehydrogenase involved in teichuronic acid synthesis. Microbiology 145, 1049–1053 [DOI] [PubMed] [Google Scholar]
- 39. Hitchcock P. J., Brown T. M. (1983) Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dodgson C., Amor P., Whitfield C. (1996) Distribution of the rol gene encoding the regulator of lipopolysaccharide O-chain length in Escherichia coli and its influence on the expression of group I capsular K antigens. J. Bacteriol. 178, 1895–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reid A. N., Whitfield C. (2005) Functional analysis of conserved gene products involved in assembly of Escherichia coli capsules and exopolysaccharides: evidence for molecular recognition between Wza and Wzc for colanic acid biosynthesis. J. Bacteriol. 187, 5470–5481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Campbell R. E., Sala R. F., van de Rijn I., Tanner M. E. (1997) Properties and kinetic analysis of UDP-glucose dehydrogenase from group A streptococci. Irreversible inhibition by UDP-chloroacetol. J. Biol. Chem. 272, 3416–3422 [DOI] [PubMed] [Google Scholar]
- 43. Dhamdhere G., Zgurskaya H. I. (2010) Metabolic shutdown in Escherichia coli cells lacking the outer membrane channel TolC. Mol. Microbiol. 77, 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reed M. C., Lieb A., Nijhout H. F. (2010) The biological significance of substrate inhibition: a mechanism with diverse functions. BioEssays 32, 422–429 [DOI] [PubMed] [Google Scholar]
- 45. Buckstein M. H., He J., Rubin H. (2008) Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 190, 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alatossava T., Jütte H., Kuhn A., Kellenberger E. (1985) Manipulation of intracellular magnesium content in polymyxin B nonapeptide-sensitized Escherichia coli by ionophore A23187. J. Bacteriol. 162, 413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Olivares-Illana V., Meyer P., Bechet E., Gueguen-Chaignon V., Soulat D., Lazereg-Riquier S., Mijakovic I., Deutscher J., Cozzone A. J., Laprévote O., Morera S., Grangeasse C., Nessler S. (2008) Structural basis for the regulation mechanism of the tyrosine kinase CapB from Staphylococcus aureus. PLoS Biol. 6, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gruszczyk J., Fleurie A., Olivares-Illana V., Béchet E., Zanella-Cleon I., Moréra S., Meyer P., Pompidor G., Kahn R., Grangeasse C., Nessler S. (2011) Structure analysis of the Staphylococcus aureus UDP-N-acetylmannosamine dehydrogenase Cap5O involved in capsular polysaccharide biosynthesis. J. Biol. Chem. 286, 17112–17121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Petranovic D., Grangeasse C., Macek B., Abdillatef M., Gueguen-Chaignon V., Nessler S., Deutscher J., Mijakovic I. (2009) Activation of Bacillus subtilis Ugd by the BY-kinase PtkA proceeds via phosphorylation of its residue tyrosine 70. J. Mol. Microbiol. Biotechnol. 17, 83–89 [DOI] [PubMed] [Google Scholar]
- 50. Macek B., Mijakovic I., Olsen J. V., Gnad F., Kumar C., Jensen P. R., Mann M. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 51. Macek B., Gnad F., Soufi B., Kumar C., Olsen J. V., Mijakovic I., Mann M. (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 52. Vincent C., Doublet P., Grangeasse C., Vaganay E., Cozzone A. J., Duclos B. (1999) Cells of Escherichia coli contain a protein-tyrosine kinase, Wzc, and a phosphotyrosine-protein phosphatase, Wzb. J. Bacteriol. 181, 3472–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ferrières L., Clarke D. J. (2003) The RcsC sensor kinase is required for normal biofilm formation in Escherichia coli K-12 and controls the expression of a regulon in response to growth on a solid surface. Mol. Microbiol. 50, 1665–1682 [DOI] [PubMed] [Google Scholar]
- 54. Mouslim C., Groisman E. A. (2003) Control of the Salmonella ugd gene by three two-component regulatory systems. Mol. Microbiol. 47, 335–344 [DOI] [PubMed] [Google Scholar]
- 55. Ho J. Y., Lin T. L., Li C. Y., Lee A., Cheng A. N., Chen M. C., Wu S. H., Wang J. T., Li T. L., Tsai M. D. (2011) Functions of some capsular polysaccharide biosynthetic genes in Klebsiella pneumoniae NTUH K-2044. PLoS One 6, e21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guzman L. M., Belin D., Carson M. J., Beckwith J. (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177, 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Keenleyside W. J., Jayaratne P., MacLachlan P. R., Whitfield C. (1992) The rcsA gene of Escherichia coli O9:K30:H12 is involved in the expression of the serotype-specific group I K (capsular) antigen. J. Bacteriol. 174, 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Drummelsmith J., Amor P. A., Whitfield C. (1997) Polymorphism, duplication, and IS1-mediated rearrangement in the chromosomal his-rfb-gnd region of Escherichia coli strains with group IA and capsular K antigens. J. Bacteriol. 179, 3232–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sommer B. J, Barycki J. J., Simpson M. A. (2004) Characterization of human UDP-glucose dehydrogenase. CYS-276 is required for the second of two successive oxidations. J. Biol. Chem. 279, 23590–23596 [DOI] [PubMed] [Google Scholar]
- 61. Easley K. E., Sommer B. J., Boanca G., Barycki J. J., Simpson M. A. (2007) Characterization of human UDP-glucose dehydrogenase reveals critical catalytic roles for lysine 220 and aspartate 280. Biochemistry 46, 369–378 [DOI] [PubMed] [Google Scholar]