

Background: CCN1, a matricellular protein composed of four modular domains, is essential for proper vascular development.
Results: CCN1 variants containing fewer domains stimulate or inhibit retinal neovascularization under ischemic conditions.
Conclusion: Proteolytic degradation of CCN1 amplifies the angiogenic or angiostatic potential of its exposed domains.
Significance: CCN1 degradation products are potential targets for therapeutic inhibition or monitoring of retinal neovascular diseases.
Keywords: Angiogenesis, Endothelial Cell, Extracellular Matrix Proteins, Ischemia, Matrix Metalloproteinase (MMP), Protein Degradation, Protein Domains, Retina, Cysteine-rich Protein 61 (CCN1), Matricellular Protein
Abstract
CCN1 is a matricellular protein involved in normal vascular development and tissue repair. CCN1 exhibits cell- and context-dependent activities that are reflective of its tetramodular structure phylogenetically linked to four domains found in various matrix proteins. Here, we show that vitreal fluids from patients with proliferative diabetic retinopathy (PDR) were enriched with a two-module form of CCN1 comprising completely or partially the insulin-like growth factor-binding protein (IGFBP) and von Willebrand factor type C (vWC) domains. The two- and three-module forms comprising, in addition to IGFBP and vWC, the thrombospondin type 1 (TSP1) repeats are CCN1 degradome products by matrix metalloproteinase-2 and -14. The functional significance of CCN1 and its truncated variants was determined in the mouse model of oxygen-induced retinopathy, which simulates neovascular growth associated with PDR and assesses treatment outcomes. In this model, lentivirus-mediated expression of either CCN1 or the IGFBP-vWC-TSP1 form reduced ischemia-induced neovascularization, whereas ectopic expression of the IGFBP-vWC variant exacerbated pathological angiogenesis. The IGFBP-vWC form has potent proangiogenic properties promoting retinal endothelial cell growth, migration, and three-dimensional tubular structure formation, whereas the IGFBP-vWC-TSP1 variant suppressed cell growth and angiogenic gene expression. Both IGFBP-vWC and IGFBP-vWC-TSP1 forms exhibited predictable variations of their domain folding that enhanced their functional potential. These data provide new insights into the formation and activities of CCN1-truncated variants and raise the predictive value of the form containing completely or partially the IGFBP and vWC domains as a surrogate marker of CCN1 activity in PDR distinguishing pathological from physiological angiogenesis.
Introduction
Ischemic retinal diseases are triggered by vaso-obliteration and/or vessel remodeling caused by various factors, including hyperglycemia associated with diabetes, thrombosis in vein occlusion, and hyperoxic injury linked to premature birth (1, 2). As compensatory revascularization occurs to replenish regions of poorly perfused retina, an aberrant intravitreal preretinal neovascularization ensues. This is a major sight-threatening complication due to the abnormally formed fibrovascular tissue that can hemorrhage and apply tractional forces to the retina resulting in retinal detachment and possibly blindness, a hallmark of late stage diabetic retinopathy, age-macular degeneration, and retinopathy of prematurity.
Of the various factors implicated in the pathogenesis of neovascularization, extracellular matrix (ECM)2 proteins play a critical role serving as a storage site for many pathological factors that are readily released during matrix degradation as active growth factors (e.g. vascular endothelial growth factor (VEGF), tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β) (3, 4). ECM proteins are also a direct source of angiostatic and proangiogenic regulatory proteins, many of which are cryptic domains of larger molecules. Conformational changes of ECM molecules or their proteolytic processing expose their active domains that often act by binding to integrins to induce vascular cell growth, migration, differentiation, and/or gene reprogramming. Such cryptic peptides as endostatin, tumstatin, canstatin, and endoreppelin are known to have a regulatory role in blood vessel remodeling and hold promise as possible therapeutic agents (5).
The matricellular protein CCN1, also known as cysteine-rich protein 61, is an ECM-associated immediate early gene-encoded protein, which exhibits a highly restricted and dynamic pattern of expression at sites of vascularization and skeletogenesis during development and in pathological states (6). Our previous studies have shown that, in the retina, CCN1 expression was confined to the superficial, secondary, and deep capillary plexuses as they form during the first 2 weeks of postnatal development in mice (7). CCN1 expression was undetectable when the adult vasculature was completely established. Similarly, CCN1 expression has been found to be down-regulated during tissue involution, in avascular tissues, and under conditions associated with vaso-obliteration, which is consistent with a potential role of this protein in vessel formation, stabilization, and integrity (8–10).
Analyses of fluids and tissue biopsies from human clinical specimens have shown increased levels of CCN1 in several ocular vascular complications, including proliferative diabetic retinopathy (PDR) and active ophthalmopathy (11, 12). Similarly, the levels of CCN1 increased in retinal blood vessels at the early stages of diabetes and in late stages of proliferative disorders in mouse models of diabetic and ischemic angiopathies (13, 14). Whether CCN1 expression under pathological conditions recapitulates biological events characteristic of earlier developmental stages, as most fetal genes do, is unknown.
The CCN1 protein is composed of conserved modular domains with sequence similarities to insulin-like growth factor-binding protein (IGFBP), von Willebrand factor type C repeat (vWC), thrombospondin type I repeat (TSP1), and C-terminal (CT) motif containing a cystine-knot region (15). These modules contain binding sites for integrins, including αvβ3, αvβ5, α vβ1, and α6β1, low density lipoprotein receptors, heparan sulfate proteoglycan, growth factor receptors, and/or ECM proteins (16–22).
CCN1 interaction with its binding partners activates signal transduction cascades that culminate in the regulation of functionally related (e.g. Wnt pathway genes) and unrelated genes (e.g. cell cycle and matrix degradation genes) (6, 23). CCN1 can also modulate the activities of several ECM, growth factors, and cytokines, including TGF-β, TNF-α, VEGF, and bone morphogenic proteins through direct physical interaction with these ligands or their receptors (24–26). Through these activities, CCN1 modulates cell adhesion, migration, proliferation, differentiation, and even cell lineage commitment (27, 28).
In this study, we show that CCN1 degradome products containing fewer modules were generated in a context environment characterized by elevated levels of matrix-degradative enzymes both in vivo and in a cell culture model in vitro. We found that truncation of modular domains of CCN1 boosted the functional capabilities of the truncated variants, which then act as either angiostimulators or angioinhibitors in the context of ischemic retinopathy. These studies provided evidence that proteolytic processing altering the activity of the CCN1 protein may occur under pathological conditions, which reinforces the notion that the multimodular structure of CCN1 may be a means of generating variants that exhibit a diverse range of agonistic and antagonistic activities reflective of their context environment.
EXPERIMENTAL PROCEDURES
Sample Collection and Enzyme-linked Immunosorbent Assay (ELISA)
Vitreoretinal fluid washes were collected from patients whose primary diagnosis included active PDR with or without vitreous hemorrhage and nondiabetic patients with ocular trauma, macular hole, or macular pucker. The collection procedure was performed in a standard fashion, similar for all stated conditions as described previously (29). Briefly, vitreous samples were collected using a vitrector probe to evacuate fluid and vitreous from the eye. After running through an inert plastic tubing, the fluid was collected in a plastic cartridge. At the end of the procedure, the collection cartridge was disconnected. The fluid was transferred into sterile plastic tubes containing a mixture of protease inhibitors (Roche Diagnostics) and centrifuged at 1000 × g for 15 min at 4 °C. The collected supernatants were concentrated 10–15-fold by ultrafiltration using Centricon columns (GE Healthcare). Protein concentration was determined using the bicinchoninic acid protein assay kit (Pierce). Levels of CCN1, VEGF, angiopoietin2, and TNF-α were determined by the Quantikine ELISA according to the manufacturer's protocol (R&D Systems, Minneapolis, MN). Recombinant CCN1 was produced using a Rapid Translation System RTS100 kit (Roche Diagnostics) and was used to generate a standard curve for CCN1 quantification. Sensitivity of the assay was determined to be in the range of 0.5 ng/ml.
Generation of CCN1 Deletion Mutants and Recombinant Adenoviral and Lentiviral Vectors
Full-length mouse CCN1 cDNA was obtained from ATCC (Manassas, VA) and cloned into a shuttle vector. Three serially truncated variants of the CCN1 gene, CCN11–3, CCN11–2, CCN11 in which the CT, CT and TSP1, and CT, TSP1, and vWC domains were deleted, respectively, were generated by PCR amplification using the full-length cDNA construct as a template. The recombinant adenoviruses, Ad-CCN1, Ad-CCN11–3, Ad-CCN11–2, and Ad-CCN11, were produced by cotransfecting adenoviral shuttle vector with Ad.OBHRE first generation recombinant adenoviral vector (E1/E3 deleted) viral backbone in which recombinant DNA is driven by the cytomegalovirus (CMV) promoter. The adenovirus encoding the green fluorescent protein (GFP) gene, Ad-GFP, was used as a control for infection. Other adenoviral vectors used in the study include the adenoviral vector expressing the MMP-2 gene (Ad-MMP-2) obtained from Vector Biolabs (Philadelphia, PA) and adenovirus expressing the MMP-14 gene provided by Dr. Jane Sottile (University of Rochester, Rochester, NY) (30). All adenoviruses were replication-deficient and used at 20 multiplicities of infection to transduce retinal endothelial cells (REC).
For in vivo studies, lentiviral vectors were produced and used because of their transgene capacity, rapid onset of expression, and robust and sustained gene expression profile. Lentiviral vectors expressing either CCN1 (lnv-CCN1), CCN11–3 (lnv-CCN11–3), CCN11–2 (lnv-CCN11–2), CCN11 (lnv-CCN11), or luciferase (lnv-luc) were constructed according to a previously established protocol (31). Rhabdovirus vesicular stomatitis virus (VSV-G)-pseudotyped lentiviral vectors were produced by transfection of 20 × 150-mm plates of 293T cells with the following three plasmids: the helper packaging construct pCMVDR8.2, the transfer vector, and the plasmid encoding for envelope protein VSV-G using the calcium phosphate precipitation method. The proportion of retinal cells transduced with lentiviral particles in a series of random sections (6–12) that spanned at least 50% of the retinal surface varied between 45 and 79% (32). The Gene Therapy Resource Program of the NHLBI, National Institutes of Health, produced the gene vectors used in this study. Production of viral vectors was performed in the Preclinical Vector Core Laboratory at the University of Pennsylvania.
Oxygen-induced Retinopathy (OIR)
Ischemic retinopathy was induced in C57BL/6J mice as described by Smith et al. (33). Neonatal mice and their nursing dams were exposed to 75% oxygen in a PRO-OX 110 chamber oxygen controller from Biospherix Ltd. (Redfield, NY) between postnatal day 7 (P7) and P12 producing vaso-obliteration and cessation of vascular development in the capillary beds of the central retina. The mice were exposed to a 12-h cyclical broad spectrum light. The room temperature was maintained at 28 °C. On P12, the mice were placed at room air until P17 when the retinas were assessed for maximum neovascular response. For gene therapy studies, animals were anesthetized by intraperitoneal injection of ketamine (65 mg/kg) and xylazine (35 mg/kg). A lentiviral vector (∼1 μl) was injected into the vitreous in one eye of each animal using a 33-gauge needled-syringe. In the contralateral eye, an equal volume of control vector (lnv-luc) was injected. Cells infected with lnv-GFP at 5–10 multiplicities of infection achieved transduction efficiency of 70–90% as determined by counting the number of GFP-positive cells (32).
Visualization of the Retinal Vasculature and Image Analyses
Quantification of vascular obliteration, retinal vascularization, and preretinal neovascular tufts was performed at P12 and P17 as described previously (7). Eyes were enucleated and fixed in 4% paraformaldehyde for 1 h. Retinas were then dissected, flat mounted through four incisions dividing them into four quadrants, and incubated overnight in 10 μg/ml fluorescently labeled Ulex Europaeus Agglutinin (UEA)-1, a specific marker of endothelial cells. Images were acquired using an Olympus fluorescence microscope. The areas of vascular obliteration were measured by delineating the avascular zone in the central retina and calculating the total area using Photoshop CS5 (Adobe). Similarly, the areas of preretinal neovascularization were calculated by selecting tufts, which appear more brightly stained than normal vasculature based on pixel intensities. Selected regions were then summed to generate total area of neovascularization.
Cells
In vitro studies were performed with rat RECs obtained from Cellpro (San Pedro, CA) and maintained in culture according to the manufacturer's instructions. Cells were characterized by immunostaining using endothelial cell markers, e.g. von Willebrand factor and CD31 obtained from Millipore (Billerica, MA) and Miltenyi Biotec (Bergisch Gladbach, Germany), respectively. The cells were propagated in 35-mm dishes in predefined endothelial growth medium containing 10% fetal bovine serum (FBS) obtained from Atlanta Biological Inc. (San Diego, CA). Cells at 80% confluence were treated as described in the text and further processed for various analyses. Conditioned medium (CM) was obtained from cells transduced with Ad-GFP, Ad-CCN1, Ad-CCN11–3, Ad-CCN11–2, or Ad-CCN11. 12 h after transduction, cells were incubated with low glucose Dulbecco's modified Eagle's medium without FCS for 24 h. Medium was then collected as described in the text.
Proliferation, Adhesion, and Chemotaxis Assays
Proliferation rate of RECs was determined using the CyQUANT® direct cell proliferation assay obtained from Invitrogen according to the manufacturer's protocol. Briefly, 5 × 103 RECs were seeded in 96-well plates. After 16 h of incubation at 37 °C, cells were placed in CM enriched with CCN1, CCN11–3, CCN11–2, or CCN11 and incubated for an additional 16 h at 37 °C. Medium was then discarded, and the plates were placed at −80 °C for 2 h. Dye binding solution was added to the wells, and the plates were incubated at 37 °C for 30 min in the dark. Fluorescence was measured with a microplate reader with excitation at 485 nm and emission at 530 nm using a fluorescein isothiocyanate (FITC) filter set.
In the adhesion assay, cells were seeded in 96-well plates either uncoated or coated overnight (4 °C) with CM-free medium as a control or CM enriched with CCN1, CCN11–3, CCN11–2, or CCN11. The assay was performed in the same adhesion plate for all adhesion substrates tested. Cells were plated at a concentration of 5 × 104 cells/well in 100 μl of serum-free medium containing 0.1% bovine serum albumin (BSA). After 4 h, nonadherent and loosely attached cells were washed off by pouring serum-free medium containing 0.1% BSA in the wells and placing the plate on a shaker for 3 min at medium speed to generate shear forces as the meniscus passes over the attached cells, thus testing cell resistance to detachment. Adhesion was quantified in duplicate by counting adherent cells using an inverted cell culture microscope.
For cell migration, proteins tested for their chemotactic activity were diluted, each in serum-free medium containing 0.5% BSA, and loaded in the lower well of 12-well Transwell plates. The lower well was then covered with a polycarbonate filter (5-μm pore diameter, Nucleopore), which was treated with 2.9% (v/v) glacial acetic acid overnight, and rinsed three times in PBS for 1 h immediately before use. Cells were resuspended in 1.0 ml of pre-equilibrated serum-free medium containing 0.5% BSA and added into each Transwell insert. After incubation for 6 h at 37 °C, the membrane was removed and stained with Diff-Quik. The chemotactic response was determined by counting the total number of cells migrating in 10 randomly selected microscope fields.
Endothelial Tubule Formation/Angiogenesis Assay
The formation of capillary-like tubules by endothelial cells was evaluated using a three-dimensional Matrigel. Briefly, pre-cooled BD MatrigelTM matrix growth factor-reduced (BD Biosciences) was filled into the lower compartment of μ-Slide angiogenesis wells (ibidi GmbH, Martinsried, Germany) on ice. The slides were incubated at 37 °C for 30 min to allow polymerization of the MatrigelTM matrix. Cells (12,000/well) were suspended in serum-free CM or CM enriched with CCN1, CCN11–3, CCN11–2, or CCN11 and plated in wells pre-coated with MatrigelTM for 16 h. Images of the tubular network formed in each well were captured using a high resolution wide field Nikon Eclipse TS100 microscope. Microscope images were then analyzed using AngioTool, a software analysis tool that computes several morphological and spatial parameters, including the number of tubes, their density, length, and lacunarity (34). These experiments were repeated at least three times, and each substrate was tested in triplicate.
MMP-2 Activity Assays
MMP-2 BiotrackTM ELISA activity (GE Healthcare) was used to measure both endogenously active MMP-2 and total MMP-2 following activation of latent MMP-2 by aminophenylmercuric acetate. Briefly, MMP-2 standard samples and vitreal fluid samples (appropriately diluted) were added to ELISA plates precoated with MMP-2 antibodies and incubated for 20 h at 4 °C. Chromogenic substrate was added, and color development was recorded in a microplate spectrophotometer at 405 nm at different time intervals (up to 3 h). The amount of total MMP-2 activity (already active plus activatable pro-MMP-2) was measured by incubation of the captured MMP-2 with 0.5 mm aminophenylmercuric acetate for 3 h at 37 °C before addition of chromogenic substrate. Calculation of the relative amounts of MMP-2 in the protein homogenates was made by comparison with the respective standard curves. For in-gel zymography, conditioned media were mixed with loading buffer without denaturation agents and assayed for MMP activity using gelatin-containing pre-cast gels (Invitrogen) according to the manufacturer's instructions. After electrophoresis, gels were incubated in zymogram renaturation buffer for 1 h, followed by incubation in development buffer at 37 °C for 48 h. Subsequently, gels were stained with 0.1% Coomassie Blue R-250 containing 30% methanol and 10% acetic acid. Destaining was performed in a solution containing 50% methanol and 10% acetic acid.
Antibodies and Western Immunoblotting
For Western immunoblotting, cell culture media were collected, and cell lysates were homogenized in lysis buffer. Protein samples (20 μg) were fractioned in a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane, and Western blot analysis was performed with each of the indicated primary antibodies. Immunodetection was performed using enhanced chemiluminescence (ECL) from Pierce. Protein bands were quantified by densitometric scanning. Polyclonal antibody to CCN1 was described previously (35). Peptides to the most immunogenic part of each of the IGFBP, vWC, and TSP1 domains were synthesized and used to raise anti-IGFBP, anti-vWC, and anti-TSP1 polyclonal antibodies and affinity-purified against the peptide as described previously (21).
RNA Isolation and Quantitative Analysis of mRNA
Total RNA was extracted from cells using RNeasy column purification protocol (Qiagen). Quantitative real time PCR (qPCR) assay was performed to determine the levels of a specific mRNA using TaqMan technology on ABI 7000 sequence detection system from Applied Biosystems (Carlsbad, CA). Highly specific primers were designed using Primer3, a Web-based primer design program. Primers used include the following: 5′TTACTGCTGTACCTCCACC3′ and 5′ACAGGACGGCTTGAAGATG3′ for VEGF; 5′TCCGGGAGAGATTGGTTTCC3′ and 5′CTGGCCTATAAGCCCTGGT3′ for α1 chain of collagen type IV (Col4A1); 5′CAAGTTCCCCGGCGATGTC3′ and 5′TTCTGGTCAAGGTCACCTGTC3′ for MMP-2; 5′TGTCTCCTCTAAACTCCCTCAG3′ and 5′TGCGTAAATCCCAAGCAAAGT3′ for αv integrin subunit; 5′GCGACGGTATTCTGTAAAGTGG3′ and 5′GGACAGGGCTTTGGCAGTT3′ for fibronectin (FN), and 5′GGAACACGTCGTGGGATAATG3′ and 5′GGCAGACTTTGGATGCTTCTT3′ for TNF-α. The cycling parameters for qPCR amplification reactions were as follows: AmpliTaq activation at 95 °C for 10 min, denaturation at 95 °C for 15 s, and annealing/extension at 60 °C for 1 min (40 cycles). Triplicate CT values were analyzed with Microsoft Excel using the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems). The transcript amount (2−ΔΔCT) was obtained by normalizing to the acidic ribosomal phosphoprotein, an endogenous reference that has been shown to be the least sensitive housekeeper gene to oxygen tension in animal models of OIR (36).
Three-dimensional Structure Prediction
The three-dimensional structure of the identified domains of the CCN1 protein sequences were predicted using the Iterative Threading Assembly Refinement or I-TASSER. I-TASSER server uses a sequence-to-structure-to-function paradigm algorithm. It predicts secondary structure, tertiary structure, and functional annotations on ligand-binding sites, enzyme commission numbers, and gene ontology terms. The accuracy of prediction is based on the confidence score of the modeling (37).
Study Approval
Animal studies were carried out in accordance to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the State University of New York-Downstate Medical Center (approval number 12-10279). The use of vitreal fluid washes from nondiabetic and diabetic patients has received institutional review board approval (study number 07-169).
Statistical Analyses
Statistical analyses were performed using the Prism software for Windows Version 4 from GraphPad Inc. (San Diego). To test differences among several means of significance, a one-way analysis of variance with the Newman-Keuls multiple comparison test was used. Where appropriate, post hoc unpaired t test was used to compare two means/groups, and p values <0.05 or <0.01 were considered significant. Student's t test with Bonferroni correction were used for pairwise comparisons when an analysis of variance p value was statistically significant.
RESULTS
CCN1 Degradome Products in Vitreal Fluids
CCN1 protein levels in vitreal fluids of diabetic patients with PDR and nondiabetic controls were determined by ELISA. As shown in Fig. 1A, the mean vitreous levels of CCN1 were significantly higher in patients with PDR than in nondiabetic patients (p < 0.01). The levels of CCN1 were still higher in patients with or without vitreous hemorrhage than nondiabetic patients with hemorrhage (Fig. 1B). However, there was no significant change of CCN1 levels between diabetic patients with and without hemorrhage. Thus, locally synthesized CCN1 likely contributed to CCN1 shedding in vitreous fluids more than blood-borne CCN1, although systemic levels of CCN1 and their fluctuations in diabetic patients are unknown.
FIGURE 1.

Quantitative and qualitative analyses of CCN1 and its truncated variants in vitreous fluids from nondiabetic and diabetic patients with PDR. A, quantitative analysis of CCN1 protein levels (ng/ml) in vitreous fluid samples of nondiabetic (ND) and diabetic patients with PDR as determined by ELISA. B, levels of CCN1 protein in vitreous fluid samples of nondiabetic controls with hemorrhage (ND+H) and PDR samples with (PDR+H) and without (PDR−H) hemorrhage. C, qualitative analysis of the CCN1 protein in vitreous fluid samples of PDR patients (D1, D2, and D3) by Western immunoblotting. Protein extracts were fractioned by electrophoresis and transferred to a nitrocellulose membrane, and immunodetection was performed with the indicated primary antibodies. The 1st panel shows the migration profile of the full-length CCN1 protein and a truncated form composed of the IGFBP and vWC domains from protein lysates infected with adenoviral vectors expressing the corresponding proteins (e.g. Ad-CCN1 and Ad-CCN11–2). Degradome product bands of CCN1 are labeled 1–3. D, loading control for the samples analyzed in C is shown by the use of Coomassie Blue staining of the blots. E, qualitative analysis of the CCN1 protein in vitreous fluid samples of nondiabetic control patients (C1, C2, and C3) by Western immunoblotting. F, loading control for the samples analyzed in E is shown by the use of Coomassie Blue staining of the blots. G, schematic representation of the multimodular structure of the CCN1 protein and molecular weights of its respective domains. SP, signal peptide; IGFBP, insulin-like growth factor binding protein; vWC, von Willebrand type C repeat; TSP1, thrombospondin type I repeat; CT, C-terminal cysteine knot.
Qualitative analysis by Western immunoblotting of CCN1 in the vitreous fluid secretome of PDR patients identified CCN1 as several discrete fragments with apparent molecular masses ranging from 11- to 23-kDa instead of a single 42-kDa band of the full-length protein (Fig. 1C). A polyclonal CCN1 antibody identified in the large majority of samples two bands (labeled 1 and 2 in Fig. 1C) with an apparent molecular mass of 29 and 18 kDa. Very few PDR samples showed an additional 12-kDa band (labeled 3 in Fig. 1C). Specific antibodies raised against either the IGFBP or vWC domain allowed the simultaneous detection of both the 23- and 18-kDa protein bands. A specific antibody against the TSP1 domain failed to detect any of the protein bands suggesting that the major 18- and 23-kDa protein bands contained either partially or completely both IGFBP and vWC modules. The third 12-kDa protein band, which was seldom detected in vitreal fluids, may be derived from a combination of the N-terminal domain and hinge segment (as depicted in Fig. 1G), which are recognizable by neither the IGFBP nor vWC antibody. CCN1 and/or its degradome products were not detected by Western immunoblotting in the large majority of vitreal fluid samples from nondiabetic patients, although one samples showed a faint CCN1 band signal (Fig. 1E). As shown by Coomassie Blue staining of total proteins fractioned by gel electrophoresis, differences between the controls and PDR samples were due to qualitative and quantitative changes of CCN1 fragments rather than sampling differences (Fig. 1, D and F).
Moreover, vitreous fluids from patients with PDR were likewise highly enriched with MMP-2 proteolytic activity as the levels of the active form of MMP-2 were four times higher in PDR fluids than those in control nondiabetic samples (Fig. 2, left panel). In addition, PDR samples contained significantly higher levels of VEGF, a potent permeability factor and upstream activator of MMP-2 gene expression (Fig. 2, right panel). Thus, proteolytic processing of CCN1 by MMPs potentially contributed to the enrichment of vitreal fluids with CCN1 truncated forms. The levels of antiangiogenic growth factors such as angiopoietin-2 were dramatically diminished (data not shown), which is consistent with previous reports (38).
FIGURE 2.
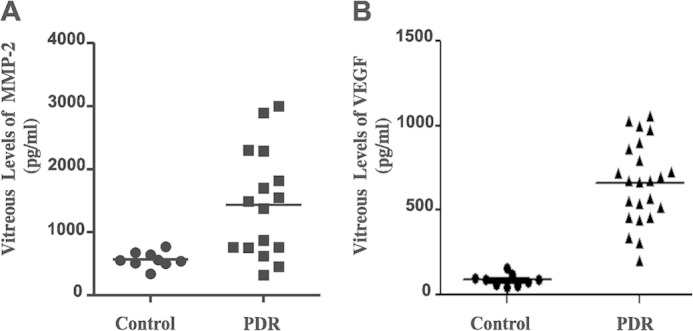
Quantitative analysis of MMP-2 and VEGF levels in vitreal fluids. Left panel, combined levels of latent pro-MMP-2 and active MMP-2 in vitreal fluids from nondiabetic controls and diabetic patients with PDR were measured by ELISA as described under “Experimental Procedures.” The amount of MMP-2 was determined by interpolation from a standard curve. Right panel, levels of VEGF in vitreous fluids samples of controls and diabetic patients with PDR were determined by ELISA.
CCN1 Is a Substrate for MMP-2 and MMP-14
The CCN proteins have previously been identified as candidate substrates for MMPs (39). CCN1, in particular, has been biochemically validated as a substrate of MMP-14 also known as MT1-MMP (40). Utilization of the protease specificity prediction server (PROSPER), a protein sequence mining tool that predicts potential cleavage sites by proteases in a given protein sequence (41), identified within the primary sequence of CCN1 numerous potential cleavage sites by metalloproteases and by aspartic, cysteine, and serine proteases (Fig. 3A). Many of the putative MMP cleavage sites localized either within the hinge regions linking two adjacent modules or within the CT domain. Thus, processing of the CCN1 protein by MMPs potentially releases either the individual IGFBP, vWC, and TSP1 domains or combinations of two or three of those domains either fully or partially cleaved. Potential products of MMP-mediated proteolysis of CCN1 are shown in Fig. 3B.
FIGURE 3.

MMP-2- and MMP-14-induced CCN1 protein proteolytic processing in cultured RECs. A, schematic diagram of the primary sequence of CCN1 with potential cleavage sites by proteases, including MMPs as predicted by protease specificity prediction server (PROSPER). B, schematic representation of the domain composition of potential CCN1 degradome products following processing by MMPs. C, in-gel zymography of MMP-2 activity in conditioned media from cells transduced with Ad-GFP, Ad-MMP-2, Ad-MMP-14, or both Ad-MMP-2 and Ad-MMP-14. Cell were either left untreated or pretreated with the MMP inhibitor SB-3CT (+S) or NNGH (+N). D, Western blot analysis of conditioned medium proteins (20 μg) from cultured cells transduced with Ad-CCN1 and either Ad-MMP-2, Ad-MMP-14 or both. Following viral infection, cells were either left untreated or treated with SB-3CT (+S), NNGH (+N) or both (+SN). Medium was collected after 16 h and analyzed by electrophoresis and immunoblotting with the indicated antibodies. E, loading control for conditioned media analyzed in D is shown by the use of Coomassie Blue staining of the blots. F and G, Western blot analysis of conditioned medium proteins (20 μg) from cultured cells transduced with Ad-GFP or Ad-CCN1 and Ad-MMP-2. Immunodetection was performed with either anti-vWC (F) or anti-TSP1 (G) antibodies.
Given the increased MMP-2 activity in vitreal fluids of PDR patients, we looked for evidence of MMP-2-mediated CCN1 processing in cultured RECs. Adenovirus-mediated expression of MMP-2 in RECs increased active MMP-2 levels in the medium as shown by in-gel zymography (Fig. 3C). Co-expression of both MMP-2 and MMP-14 further increased the active MMP-2 form that is consistent with the role of MMP-14 as an efficient pro-MMP2 activator (42). The active form of MMP-2 nearly disappeared when a selective or a nonspecific inhibitor of MMP-2 (e.g. SB-3CT and NNGH, respectively) was included in the medium. Coexpression of Ad-CCN1 and Ad-MMP-2 induced proteolytic processing of CCN1 and release in the medium of two soluble CCN1 peptide fragments with apparent molecular weights of 29 and 18 kDa (Fig. 3D). The inclusion in the medium of selective or nonselective MMP inhibitors suppressed proteolytic release of these degradome products. Thus, CCN1 fragments did not result from alternative splicing patterns of the CCN1 gene as this is another effective way to generate CCN variants (43). Similarly, co-expression of CCN1 and MMP-14 induced the release of the 29- and 18-kDa truncated variants of CCN1. Interestingly, dual expression of both MMP-2 and MMP-14 resulted in a near total digestion of the full-length CCN1 molecule as no proteolytic products could be detected. Thus, the level of MMP activity may determine the extent of CCN1 protein degradation.
Furthermore, CCN1 degradome product analyses showed that limited proteolysis of CCN1 by either MMP-2 or MMP-14 produced a 29-kDa peptide that was simultaneously recognized by anti-IGFBP, anti-vWC, and anti-TSP1 antibodies and an 18-kDa peptide that was recognized by anti-IGFBP and anti-vWC antibodies only (Fig. 3, D–G). An 8-kDa peptide was also detected with anti-vWC antibodies. Clearly, CCN1 proteolytic processing by MMPs generates individual or combined domain-truncated variants suggesting a potential involvement of MMPs in CCN1 processing in PDR.
Effects of CCN1 and Its Truncated Variants on Retinal Vaso-obliteration and Neovascular Tuft Formation in OIR Mice
We determined the functional significance of CCN1 and CCN1 variant forms lacking specific domains on retinal neovascularization in a mouse model of OIR that simulates the final neovascular stage commonly associated with human ischemic retinopathies, including PDR (44). In this model, physiological angiogenesis is disrupted during hyperoxia-induced vaso-obliteration and recapitulated, although aberrantly, in the subsequent ischemia-induced neovessel formation phase. As shown in Fig. 4, A and B, exposure to hyperoxia reduced CCN1 and VEGF steady state mRNA levels by 75 and 30%, respectively. The subsequent hypoxic/ischemic phase was characterized by an up-regulation of VEGF gene expression (Fig. 4B), yet, under these same vaso-occlusive conditions, CCN1 expression was still reduced by 50% as compared with its levels under normoxic conditions (Fig. 4A). Fluctuations of TNF-α levels were not statistically significant mainly because of the variability of the response among animals (Fig. 4C).
FIGURE 4.

Expression and effects of CCN1 on hyperoxia-induced vaso-obliteration and neovessel formation in the mouse model of OIR. A–C, steady state mRNA levels of CCN1, VEGF, and TNF-α as determined by qPCR in retinas from mice either under normoxic conditions at P12 and P17 or subjected to OIR at the same postnatal days. The data shown are the means ± S.E. of three determinations each performed in triplicate. RNA levels at P12 and P17 in normoxic retinas were set to 100% to facilitate comparison among animals and groups. *, p < 0.01 versus control (12 or P17). **, p < 0.05 versus OIR (P12). D and F, representative flat mount preparations of rhodamine-UEA-1-stained retinas from OIR eyes at P12 (D) and P17 (F) injected with either lnv-luc (panel a) or lnv-CCN1 (panel c). Areas of vaso-obliteration and preretinal neovascular tufts as determined by computer-assisted image analyses are shown in white and yellow, respectively (panel b for panels a and d for panel c). E and G, percentage of avascular and neovascular tuft areas in lnv-luc- and lnv-CCN1-injected eyes. *, p < 0.05 versus lnv-luc. (n = 9). **, p < 0.01 (n = 7).
To determine whether re-expression of CCN1 rescues/prevents the vascular damages associated with OIR, intravitreal injection of a lentiviral vector expressing CCN1 (lnv-CCN1) was performed in mouse pups subjected to OIR. Interestingly, vaso-obliterated areas in retinas from lnv-CCN1-injected eyes were reduced by nearly 30% as compared with those from eyes injected with lnv-luc (Fig. 4, D and E). Normally formed blood vessels were seen over the central region at P12 after lnv-CCN1 injection as compared with the extensive vaso-obliteration clearly evident in retinas injected with the control lnv-luc (Fig. 4D). Similarly, preretinal neovascular tufts, which can be abundantly seen in the central and mid-peripheral retina in lnv-luc-injected eyes, were reduced by 75% in eyes injected with lnv-CCN1 (Fig. 4, F and G). Thus, ectopic expression of the CCN1 gene enhanced normalization of the retinal vasculature following ischemia, while providing a protective effect against ischemia-induced retinal neovascularization.
Next, we examined the rescuing effects of CCN1 variants serially truncated from the C-terminal end on hyperoxia-induced vaso-obliteration and recovery. Modular forms lacking either CT domain alone, CT and TSP1 domains, or CT, TSP1, and vWC domains were cloned into lentiviral vectors to express CCN1-truncated variants referred to as CCN11–3, CCN11–2, and CCN11, respectively (Fig. 5A). Examination of flat-mounted retinas demonstrated several similarities and some fundamental differences following ectopic expression of CCN1 and its derived variants. Injection of either lnv-CCN11–3 or lnv-CCN11 had no significant vaso-protective effect against hyperoxia at P12 as the extent of vaso-obliterated areas was similar to those following lnv-luc injection. Conversely, injection of lnv-CCN11–2 significantly reduced the percentage of avascular areas reflecting a vasoprotective effect similar to that mediated by lnv-CCN1(Fig. 5, B and C). Retinas treated with lnv-CCN11–2 showed limited vaso-obliteration and persistence of retinal vessels and branched capillaries from the main arteries and veins emerging from the optic nerve. Clearly, the protective properties of CCN1 are, at least in part, contained within the IGFBP and vWC domains. At P17, the formation of neovascular tufts was similarly reduced upon injection of either lnv-CCN1 or lnv-CCN11–3. However, injection of lnv-CCN11–2 further exacerbated neovascularization as neovascular tufts nearly extended throughout the retinal surface (Fig. 5, D and E). Thus, although the vasoprotective properties of the CCN1 protein against hyperoxic injury were largely conveyed by the first IGFBP and vWC domains, the TSP1 module seemed to rather provide an angioinhibitory activity. Surprisingly, the CCN11 variant containing the IGFBP domain alone exhibited a significant protective effect against neovascular tuft formation, although to a much lesser extent than CCN1 and CCN11–3.
FIGURE 5.

Effects of CCN1-truncated variants on vaso-obliteration and preretinal neovascularization following OIR. A, schematic representation of CCN1 truncated variants in which the modular domains at the C-terminal end were serially deleted. Each variant form was cloned into a lentiviral vector and further intravitreally injected to mouse pups prior to OIR. B and D, representative flat mount preparations of rhodamine-UEA-1-stained retinas from OIR eyes at P12 (B) and P17 (D). Retinas were from eyes injected with either lnv-CCN11–3 (panel a), lnv-CCN11–2 (panel c), or lnv-CCN11 (panel e). Areas of vaso-obliteration and preretinal neovascular tufts as determined by computer-assisted image analyses are shown in white and yellow, respectively ( panel b for panel a, and panel d for panels c and f for panel e). C and E, compiled data showing percentage of avascularized and neovascular areas in eyes injected with either lnv-luc, lnv-CCN1, lnv-CCN11–3, lnv-CCN11–2, or lnv-CCN11. *, p < 0.05 versus +lnv-luc (n = 9). **, p < 0.05 versus +lnv-luc, (n = 8).
Effects of CCN1 and Its Truncated Variants on the Function and Behavior of Cultured RECs
We determined the cellular bases for the in vivo effects of CCN1-truncated forms using assays routinely used to investigate angiogenesis in vitro by assessing retinal endothelial cell proliferation, migration, tube formation, and gene reprogramming. As shown in Fig, 6A, full-length CCN1 protein enhanced the proliferation rate of RECs in culture. CCN1 induced a 60% increase in cell growth as compared with a 100% increase induced by 5% FBS. Interestingly, the CCN1 variant in which both CT and TSP1 domains were truncated (i.e. CCN11–2) spurred more growth of RECs than the full-length protein inducing up to 89% of the proliferation induced by FBS. Conversely, neither CCN11 nor CCN11–3 enhanced cell proliferation. Thus, the mitogenic activity of CCN1 is largely conveyed by the vWC domain and such activity is altered when vWC is physically linked to the TSP1 domain. Whether individual vWC, TSP1, and CT domains harbor inherent mitogenic activities when taken separately is not clear at the present time. Nevertheless, the potent mitogenic activity of the CCN11–2 variant appears to be physiologically relevant because this variant exacerbated neovessel growth during the ischemic phase of OIR in mice.
FIGURE 6.

Effects of CCN1-truncated variants on cultured RECs. A, RECs were exposed to CM from cells transduced with Ad-GFP, Ad-CCN1, Ad-CCN11–3, Ad-CCN11–2, or Ad-CCN11. The proliferation rate of the cells cultured under these conditions was measured as described under “Experimental Procedures.” The proliferation rate of the cells in 5% FBS was set to 100% to facilitate comparisons among different treatments. *, p < 0.001 versus control. **, p < 0.05 versus 5% FBS. B, effects of CCN1 and its truncated variants on REC adhesion. Cells were seeded in plate wells precoated with increasing concentrations of conditioned serum-free medium or CM enriched with either CCN1, CCN11–3, CCN11–2, or CCN11. Cells were allowed to adhere to the wells, and adherent cells were counted using an inverted cell culture microscope. Data shown are the means ± S.E. of three determinations, each performed in triplicate. C, REC migration was analyzed by chemotaxis assay as described under “Experimental Procedures.” The data shown are the means ± S.E. of three determinations. *, p < 0.05 versus control. **, p < 0.01 versus control. D, effects of CCN1-truncated variants on capillary-like tube formation by RECs. Cells were suspended in either 5% FBS containing medium or CCN1-, CCN11–3-, CCN11–2-, or CCN11-enriched CM and plated in wells pre-coated with Matrigel gel. Tubule formation was assessed after 16 h. Experiments were repeated three times with similar results. E, steady state mRNA levels of VEGF, Col4A1, MMP-2, integrin αv chain, fibronectin (FN), and TNF-α in RECs transduced with either Ad-CCN1, Ad-CCN11–3, Ad-CCN11–2, or Ad-CCN11. Gene expression was quantified by qPCR. The data shown are the means ± S.E. of three determinations each performed in triplicate. RNA levels in CCN1-treated cells were set to 100% to facilitate comparison among experiments. *, p < 0.05 versus CCN1. **, p < 0.01 versus CCN1.
Meanwhile, both CCN1 and CCN11–2 strongly increased REC adhesion in a dose-dependent manner, although the full-length protein exhibited higher adhesive properties (Fig. 6B). Conversely, CCN11–3 was associated with a remarkable reduction of cell adhesion by more than 60% relative to the full-length CCN1. In chemotaxis assay, CCN1 and its variants CCN11–3 and CCN11–2 were nearly equally efficient in inducing REC migration, whereas the chemotactic activity of CCN11 was reduced by 36% as compared with that of CCN1 (Fig. 6C). Matrigel angiogenesis assay showed that CCN1 induced formation of tube-like structures similar to those induced by 5% FBS (Fig. 6D). Both CCN1 and CCN11–2 rapidly induced migration of RECs within and at the surface of Matrigel and formed complex widely lacunated three-dimensional tubular structures. Conversely, cells exposed to either CCN11 or CCN11–3 formed aggregates with short protruding sprouts and defective tubulogenesis, which is consistent with the observed impairment of migration, adhesion, and/or proliferation of RECs exposed to these variants.
We examined the molecular bases for angioinhibition and angiostimulation activities by CCN1-truncated variants through analysis of the expression profile of selected angiogenic gene markers. As shown in Fig. 6E, the angioinhibitory activity of CCN11–3 was associated with a significant reduction of VEGF, MMP-2, αv integrin, and TNF-α transcript levels as compared with the activity of the intact CCN1 protein. Conversely, VEGF, fibronectin, and TNF-α transcript levels in CCN11–2-treated cells were the same as those induced by CCN1. Only COL4A1 and MMP-2 levels were reduced in cells exposed to CCN11–2. VEGF and MMP-2 transcript levels were reduced by 31% in CCN11-treated cells as compared with CCN1-treated cells. This suggests that CCN1 signaling may be defined by the interactions among its modular domains.
Three-dimensional Structure of the CCN1-truncated Variants and It Potential Impact on Their Biological Activity
We examined the changes of the spatial organization of the CCN1 protein as its C-terminal domains were serially truncated, and we assessed the organization of the remaining domains in the predicted three-dimensional structure of those variant forms. Fig. 7 shows the predicted three-dimensional ribbon structure of CCN1, CCN11–3, CCN11–2, and CCN11 with the highest confidence score given by I-TASSER software. Overall, the three-dimensional structure of the CCN1 protein has a globular appearance with a “W”-shaped arrangement of its domains brought about by the proximity of the two consecutive domains at the N- and C-terminal ends and the long hinge region in the middle (Fig. 7A). The relatively long linker region between the vWC and TSP1 domains and the shorter linkers between IGFBP and vWC and between TSP1 and CT provide both an overall flexibility of the full-length molecule and a local rigidity within the molecule with the IGFBP and CT domain (at the extreme N- and C-terminal ends, respectively) cocked in almost opposite directions. The vWC and TSP1 domains that contain stretches of amino acid residues involved in integrin binding form a synergy region located on the same face of the molecule. This region contains potential binding sites for growth factors (e.g. VEGF and TGF-β), suggesting that the simultaneous interaction of growth factors and integrins with this region may be sterically hindered and thus, mutually exclusive. The CT domain at the extreme C-terminal end is well exposed, which may facilitate the oligomerization of the CCN1 proteins and/or its interaction with other ECM proteins. Of particular interest is that the removal of the CT domain unmasked a short peptide sequence within the TSP1 domain, TSWSQCSKTCGTGISTRV, that computationally based peptidomics approaches have previously identified as cyrostatin, a potential suppressor of endothelial cell growth and migration (45). Moreover, further removal of the TSP1 domain resulted in a more compact molecule in which most residues were exposed and readily accessible to receptors and interacting partners (Fig. 7C). Finally, conformational change of the IGFBP domain alone may not be sufficient to unmask new activity of this domain given the paucity of information contained within its primary sequence. Many of the functional and structural features of all truncated forms of CCN1 will become apparent with the availability of experimentally determined conformational changes.
FIGURE 7.

Three-dimensional models of CCN1 and its truncated variants. A, schematic representations of the three-dimensional structure of the CCN1 protein (panel a) and the truncated isoforms CCN11–3 (panel b), CCN11–2 (panel c), and CCN11 (panel d). Coiled chains are α-helices. Parallel and antiparallel β-sheets are represented with wide green arrows and disulfide bonds are shown with yellow bars. B, primary structure of the CCN1 protein. The amino acid residues of the modular domains are annotated in blue, and the amino acid residues of the linker peptides are annotated in black. Proteins interacting with the stretches of amino acid in each domain are highlighted.
DISCUSSION
This study provided new insights into the structure-to-function relationship of the CCN1 protein and its potential relevance in pathological angiogenesis associated with proliferative retinopathy. The large majority of vitreal fluid samples from PDR patients contained CCN1 fragments recognizable by specific antibodies to IGFBP and vWC domains, and only a few samples exhibited an additional product that was also detectable by TSP1-specific antibodies. The truncated form containing either partially or completely the two-module form IGFBP-vWC was predominantly represented in PDR fluids along with other proangiogenic and permeability factors such as VEGF and MMP-2. Whether clinical parameters such as type of diabetes, extent of neovascularization, fibrosis/pre-retinal fibrotic membrane grading, and degree of hemorrhage correlate with the qualitative and quantitative changes of the CCN1 protein in vitreal fluids is unknown and will be investigated using a larger cohort of patients.
The contribution of MMPs to the generation of the CCN1-truncated forms is supported by several observations. First, MMP-2 proteolytic activity was four times higher in PDR fluids than in controls. Second, the CCN1 primary sequence contains MMP cleavage sites precisely located in the large cysteine residue-free sterically unhindered hinge region separating the N-terminal region composed of the IGFBP and vWC domains and the C-terminal region composed of the TSP1 and CT domains. The latter contain numerous cleavage sites for proteases that potentially increased their sensitivity to proteolytic degradation, whereas the N-terminal region contains fewer cleavage sites that may account for its resistance to digestion. Third, proteolytic cleavage of CCN1 by MMPs in a cell culture environment generated truncated forms of CCN1, some of which contained completely or partially the two-module form IGFBP-vWC further supporting a cause-effect relationship.
However, when interpreting the profile of CCN1 degradome products in vivo, it is important to consider that the CCN1 protein may be targeted by multiple proteases, although the identity and specific roles of each protease in such a complex environment are difficult to glean. Indeed, the primary sequence of CCN1 contains numerous cleavage sites for aspartic, cysteine, and serine proteases. Concordantly, Guillon-Munos et al. (46) have validated CCN1 as a substrate of kallikrein-related peptidases, an emerging group of secreted serine proteases that cleave CCN1 at different sites and regulate angiogenesis. Similarly, plasmin, a serine protease that promotes fibrinolysis, cleaves CCN1 either directly or indirectly via activation of MMPs (47). These and other proteases do not operate alone but in linear and amplification cascades. The MMPs, in particular, play an important role in the process of interconnecting different types of proteases creating a dynamic “protease web” (48). Thus, expression of MMPs (e.g. MMP-2 and MMP-14) may activate other proteases from all classes of the degradome and/or inactivate new protease inhibitors leading to either a more severe degradation of the CCN1 protein and/or generating a mixture of degradome products. The loss/total proteolysis of CCN1 upon co-expression of both MMP-2 and MMP-14 in a cell culture environment suggests that an initial processing by MMPs may render their substrates susceptible to further processing by the same proteases and/or other proteases. In this context, d'Ortho et al. (49) showed that MMP-14 induced TNF-α processing and release of mature TNF-α, which furthered its degradation into smaller fragments. Future utilization of degradomic approaches will provide insights into the relative contribution of specific proteases to the formation of a diverse range of CCN1 degradome products.
Another interesting outcome of our study design is the functional implication of truncated variants of the CCN1 protein in the OIR mouse model that simulates the final neovascular stage common to different human ischemic retinopathies. Conceptually, it is difficult to determine the true biological activities of the degradome products of CCN1 because the truncated variants predominantly present in PDR samples or in culture media may not necessarily contain the full amino acid sequences of the constituent modules revealed by domain-specific antibodies. If cryptic or protein-binding sites are to be exposed or modified as a result of CCN1 processing, their function may be altered accordingly. However, we surmised that the activity of CCN1 variants containing the complete sequence of IGFBP, vWC, or TSP1 domain may provide insights into the functional significance of the breakdown products predominantly found in PDR fluids and/or culture media. Our data clearly demonstrated an in vivo angiostimulatory activity of the truncated variant, CCN11–2, containing the complete sequence of the IGFBP and vWC domains and an angioinhibitory activity of the variant CCN11–3 containing the additional TSP1 domain. Clearly, the main structural information contained within the vWC and TSP1 modules is critical for the overall activity of the CCN1-truncated variants. We showed that the CCN11–2 variant was able to induce retinal endothelial cell growth, adhesion, and capillary-like tube formation to the same extent as the full-length protein. Many of these activities derive from CCN1 interaction with integrin receptors localized in the vWC domain that contains binding sites bearing structural homologies to those involved in the interaction with αvβ3, αvβ5, and α2β3 (20). The interaction between αvβ3 integrin and the vWC domain was shown to be largely responsible for CCN1-mediated endothelial cell adhesion, migration, survival, and angiogenic activity (19). Surprisingly, even though the CCN11–2 variant lacks the heparan sulfate proteoglycan-binding site, which has been mapped in the TSP1 and CT domains, it still conveyed a potent angiogenic activity in vivo and strongly increased proangiogenic factor gene expression such as VEGF. Thus, exposure of the vWC domain at the C-terminal end of the CCN11–2 molecule likely produced conformational changes that modified its functional potential.
Most domains in multimodular ECM proteins are quite rigid, and the function of the parent protein is defined by the relative spatial arrangement of adjacent domains. Truncation of one or two domains of the CCN1 molecule was accompanied by structural changes that are propagated to the remaining domains at the opposite pole. In the CCN1 protein, vWC consists of an upper section comprising β-sheets and a less structured lower fibronectin-like domain held together by disulfide bonds (Fig. 7). The fact that the vWC and fibronectin-like domains are phylogenetically linked and bear sequence homologies with one another raises the possibility that they may, at least in part, be functionally similar as well. VEGF has been shown to bind to fibronectin via the fibronectin-like domain and promotes mitogen-activated protein kinase activation increasing endothelial cell growth and migration (50). Therefore, the neovascular effects of the vWC domain may depend on interaction with and/or potentiation of VEGF activity.
The variant containing completely or partially the IGFBP, vWC, and TSP1 domains was seldom found in vitreal fluids from PDR patients suggesting that the TSP1 domain is highly susceptible to proteolytic degradation, which is consistent with previous reports (51). Interestingly, delivery into the eyes of the three-module form IGFBP-vWC-TSP1 (i.e. CCN11–3) significantly reduced the formation of neovascular tufts in OIR mice. Our studies in cultured endothelial cells further showed that CCN11–3 promoted neither cell growth nor capillary tube formation. There are a number of potential explanations to account for the ability of CCN11–3 to exhibit an antiangiogenic activity via its TSP1 module. First, the antiangiogenic function of TSP1, which is present in more than 100 different proteins, occurs through either direct inhibition of endothelial cell migration and induction of cell arrest as evidenced by our in vitro data or indirectly by inhibiting expression of growth factors, cytokines, and proteases. CCN11–3 did not promote endothelial cell apoptosis (data not shown), but it significantly reduced VEGF gene expression (Fig. 7). These data are consistent with those of Huwiler et al. (52), who showed that at nanomolar concentrations, the TSP1 repeat alone inhibited growth factor-induced endothelial cell growth and migration. Furthermore, a study by Karagiannis and Popel (45) has used computational bioinformatics analyses and identified specific TSP1 sequences with an antiangiogenic potential in proteins traditionally known to be proangiogenic, including the CCN proteins. The TSP1 domain of CCN1 contains a short peptide, referred to as cyrostatin, with experimentally validated antiproliferative and antimigratory activities. Utilization of neutralizing antibodies to CD36, the main TSP1 repeat receptor, abolished the peptide's activity. Therefore, because only an isolated fragment of the TSP1 domain was needed to inhibit proliferation and migration of endothelial cells, it is conceivable that conformational changes alone are sufficient to expose the angioinhibitory activity of the CN11–3 variant. Such observations are in line with the inhibitory effects of CCN11–3 on abnormal vessel outgrowth in OIR mice. Second, the functional and/or physical interaction between the TSP1 domain of CCN11–3 and VEGF alters the bioavailability and function of VEGF. Indeed, in the ischemic environment of the retina, the severely metabolically deprived neurons and astrocytes produce excessive amounts of VEGF in an attempt to restore their nutrient supply by triggering blood vessel growth. Under these conditions, the TSP1 domain binds preferentially to the heparin-binding VEGF isoform in an antiangiogenic mode of action (53). Such interaction hinders VEGF binding to its receptor and reduced VEGF-mediated angiogenesis (54). Third, the biological activities of the CCN1 protein depend on the bioavailability of many other interacting partners that recognize each of the constitutive domains of the protein. Therefore, truncation of one, two, or three domains suppresses the biological properties conveyed by the missing domain(s). The CT domain that is missing in the truncated variant CCN11–3 has been considered as a functionally critical component of the full-length CCN1 protein by virtue of its ability to physically bind and/or functionally modulate integrin, heparin, growth factor signaling, as well as components of the Wnt and Notch signaling pathways (25, 55, 56). Notably, the CT domain allows dimerization and oligomerization of the CCN1 proteins bringing several identical domains together in the same proximity that further increase their affinity to their interacting partners (57). Thus, the absence of the CT domains simply deprive the truncated CCN1 variant of these biological properties. The truncated variant CCN11–3 may act as a dominant negative form of the full-length protein. A similar finding was reported for CCN2, a family member structurally related to CCN1. Deletion of the CT domain of the CCN2 protein generated a variant that antagonized the activity of the native CCN2 molecule, which further supports the function of CCN11–3 as a potential dominant negative form (32).
Overall, our findings suggest that CCN1 variants containing completely or partially the IGFBP and vWC domains may convey angiostimulatory effects that potentially exacerbate neovascularization in proliferative retinopathy such as PDR. Conversely, truncated variants containing completely or partially the IGFBP, vWC, and TSP1 domains, rarely found in PDR fluids, may convey angioinhibitory effects. This putative dominant form of CCN1 offers an alternative treatment option to suppress retinal neovascularization associated with PDR or retinopathy of prematurity.
Mechanistically, how proteolytic events of CCN1 translate into defined cellular response have yet to be elucidated. During blood vessel growth, the amount and phenotype of endothelial tip cells forming the front of sprouts may be guided by, among other signals, degradome products of ECM proteins, including those of CCN1. In both normal and pathological angiogenesis, sprouts are headed by migrating endothelial tip cells, which dynamically extend long filopodial protrusions in a polarized way, migrate into the ECM, and sense their environment for attractive and repulsive signals for guidance. The number and extent of filipodial protrusions of tip cells have been found to be abnormally high in blood vessel sprouts of human pathological specimens of tumor neovascularization and in neovascular tufts of OIR retinas (58, 59). A study by Koziol et al. (60) has shown that the ability of endothelial cells to acquire a tip cell phenotype is associated with MMP-14-induced proteolytic degradation of several proteins, including CCN1. The absence of MMP-14 in stimulated endothelial cells resulted in impaired processing and accumulation of the substrate dampening the capillary sprouting process and favoring quiescence of the vasculature. These observations reinforce the notion that degradome products of CCN1 are potential contributing factors to the increased number and activation state of tip cells during tissue neovascularization. Further studies investigating such activity and their implications in pathological angiogenesis are warranted.
Acknowledgments
We thank David Little, Kit Poon, Sunna Vyatra Hutagalung, Iyesha Williams, and Rahul Parmar for their help and technical assistance with the experiments and critical discussion and review of this work. We also thank Dr. Jane Sottile (University of Rochester, Rochester, NY) for sharing the MMP-14 adenoviral vector.
This work was supported, in whole or in part, by National Institutes of Health Grants EY022091-01 and EY019387 -01A1 (to B. C.) and EY01260-12 and EY007739-23 from NEI (to M. B. G.).
- ECM
- extracellular matrix
- PDR
- proliferative diabetic retinopathy
- IGFBP
- insulin-like growth factor-binding protein
- vWC
- von Willebrand factor type C
- TSP1
- thrombospondin type 1
- MMP
- matrix metalloproteinase
- qPCR
- quantitative real time PCR
- CM
- conditioned medium
- OIR
- oxygen-induced retinopathy
- REC
- retinal endothelial cell
- P
- postnatal day
- CT
- C-terminal cysteine knot.
REFERENCES
- 1. Bradley J., Ju M., Robinson G. S. (2007) Combination therapy for the treatment of ocular neovascularization. Angiogenesis 10, 141–148 [DOI] [PubMed] [Google Scholar]
- 2. Rivera J. C., Sapieha P., Joyal J. S., Duhamel F., Shao Z., Sitaras N., Picard E., Zhou E., Lachapelle P., Chemtob S. (2011) Understanding retinopathy of prematurity: update on pathogenesis. Neonatology 100, 343–353 [DOI] [PubMed] [Google Scholar]
- 3. Lu P., Takai K., Weaver V. M., Werb Z. (2011) Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harbor Perspect. Biol. 3, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Senger D. R., Davis G. E. (2011) Angiogenesis. Cold Spring Harbor Perspect. Biol. 3, a005090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boosani C. S., Sudhakar Y. A. (2011) Proteolytically derived endogenous angioinhibitors originating from the extracellular matrix. Pharmaceuticals 4, 1551–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jun J. I., Lau L. F. (2011) Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 10, 945–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hasan A., Pokeza N., Shaw L., Lee H. S., Lazzaro D., Chintala H., Rosenbaum D., Grant M. B., Chaqour B. (2011) The matricellular protein cysteine-rich protein 61 (CCN1/Cyr61) enhances physiological adaptation of retinal vessels and reduces pathological neovascularization associated with ischemic retinopathy. J. Biol. Chem. 286, 9542–9554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hilfiker-Kleiner D., Kaminski K., Kaminska A., Fuchs M., Klein G., Podewski E., Grote K., Kiian I., Wollert K. C., Hilfiker A., Drexler H. (2004) Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation 109, 2227–2233 [DOI] [PubMed] [Google Scholar]
- 9. Jin Y., Kim H. P., Ifedigbo E., Lau L. F., Choi A. M. (2005) Cyr61 protects against hyperoxia-induced cell death via Akt pathway in pulmonary epithelial cells. Am. J. Respir. Cell Mol. Biol. 33, 297–302 [DOI] [PubMed] [Google Scholar]
- 10. Kubota S., Takigawa M. (2007) CCN family proteins and angiogenesis: from embryo to adulthood. Angiogenesis 10, 1–11 [DOI] [PubMed] [Google Scholar]
- 11. Hughes J. M., Kuiper E. J., Klaassen I., Canning P., Stitt A. W., Van Bezu J., Schalkwijk C. G., Van Noorden C. J., Schlingemann R. O. (2007) Advanced glycation end products cause increased CCN family and extracellular matrix gene expression in the diabetic rodent retina. Diabetologia 50, 1089–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan L., Chaqour B. (2013) Cysteine-rich protein 61 (CCN1) and connective tissue growth factor (CCN2) at the crosshairs of ocular neovascular and fibrovascular disease therapy. J. Cell Commun. Signal. 10.1007/s12079-013-0206-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kivelä R., Silvennoinen M., Lehti M., Jalava S., Vihko V., Kainulainen H. (2008) Exercise-induced expression of angiogenic growth factors in skeletal muscle and in capillaries of healthy and diabetic mice. Cardiovasc. Diabetol. 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X., Yu W., Dong F. (2012) Cysteine-rich 61 (CYR61) is up-regulated in proliferative diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 250, 661–668 [DOI] [PubMed] [Google Scholar]
- 15. O'Leary J. M., Hamilton J. M., Deane C. M., Valeyev N. V., Sandell L. J., Downing A. K. (2004) Solution structure and dynamics of a prototypical chordin-like cysteine-rich repeat (von Willebrand factor type C module) from collagen IIA. J. Biol. Chem. 279, 53857–53866 [DOI] [PubMed] [Google Scholar]
- 16. Chen C. C., Lau L. F. (2009) Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 41, 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao R., Brigstock D. R. (2003) Low density lipoprotein receptor-related protein (LRP) is a heparin-dependent adhesion receptor for connective tissue growth factor (CTGF) in rat activated hepatic stellate cells. Hepatol. Res. 27, 214–220 [DOI] [PubMed] [Google Scholar]
- 18. Leask A., Abraham D. J. (2006) All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 119, 4803–4810 [DOI] [PubMed] [Google Scholar]
- 19. Leu S. J., Lam S. C., Lau L. F. (2002) Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins αvβ3 and α6β1 in human umbilical vein endothelial cells. J. Biol. Chem. 277, 46248–46255 [DOI] [PubMed] [Google Scholar]
- 20. Leu S. J., Liu Y., Chen N., Chen C. C., Lam S. C., Lau L. F. (2003) Identification of a novel integrin α6β1-binding site in the angiogenic inducer CCN1 (CYR61). J. Biol. Chem. 278, 33801–33808 [DOI] [PubMed] [Google Scholar]
- 21. Schober J. M., Chen N., Grzeszkiewicz T. M., Jovanovic I., Emeson E. E., Ugarova T. P., Ye R. D., Lau L. F., Lam S. C. (2002) Identification of integrin αMβ2 as an adhesion receptor on peripheral blood monocytes for Cyr61 (CCN1) and connective tissue growth factor (CCN2): immediate-early gene products expressed in atherosclerotic lesions. Blood 99, 4457–4465 [DOI] [PubMed] [Google Scholar]
- 22. Segarini P. R., Nesbitt J. E., Li D., Hays L. G., Yates J. R., 3rd, Carmichael D. F. (2001) The low density lipoprotein receptor-related protein/α2-macroglobulin receptor is a receptor for connective tissue growth factor. J. Biol. Chem. 276, 40659–40667 [DOI] [PubMed] [Google Scholar]
- 23. Liu H., Yang R., Tinner B., Choudhry A., Schutze N., Chaqour B. (2008) Cysteine-rich protein 61 and connective tissue growth factor induce deadhesion and anoikis of retinal pericytes. Endocrinology 149, 1666–1677 [DOI] [PubMed] [Google Scholar]
- 24. Athanasopoulos A. N., Schneider D., Keiper T., Alt V., Pendurthi U. R., Liegibel U. M., Sommer U., Nawroth P. P., Kasperk C., Chavakis T. (2007) Vascular endothelial growth factor (VEGF)-induced up-regulation of CCN1 in osteoblasts mediates proangiogenic activities in endothelial cells and promotes fracture healing. J. Biol. Chem. 282, 26746–26753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Latinkic B. V., Mercurio S., Bennett B., Hirst E. M., Xu Q., Lau L. F., Mohun T. J., Smith J. C. (2003) Xenopus Cyr61 regulates gastrulation movements and modulates Wnt signalling. Development 130, 2429–2441 [DOI] [PubMed] [Google Scholar]
- 26. Zhou D., Herrick D. J., Rosenbloom J., Chaqour B. (2005) Cyr61 mediates the expression of VEGF, αv-integrin, and α-actin genes through cytoskeletally based mechanotransduction mechanisms in bladder smooth muscle cells. J. Appl. Physiol. 98, 2344–2354 [DOI] [PubMed] [Google Scholar]
- 27. Grote K., Salguero G., Ballmaier M., Dangers M., Drexler H., Schieffer B. (2007) The angiogenic factor CCN1 promotes adhesion and migration of circulating CD34+ progenitor cells: potential role in angiogenesis and endothelial regeneration. Blood 110, 877–885 [DOI] [PubMed] [Google Scholar]
- 28. Schütze N., Schenk R., Fiedler J., Mattes T., Jakob F., Brenner R. E. (2007) CYR61/CCN1 and WISP3/CCN6 are chemoattractive ligands for human multipotent mesenchymal stroma cells. BMC Cell Biol. 8, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Butler J. M., Guthrie S. M., Koc M., Afzal A., Caballero S., Brooks H. L., Mames R. N., Segal M. S., Grant M. B., Scott E. W. (2005) SDF-1 is both necessary and sufficient to promote proliferative retinopathy. J. Clin. Invest. 115, 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi F., Sottile J. (2011) MT1-MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J. Cell Sci. 124, 4039–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. MacKenzie T. C., Kobinger G. P., Kootstra N. A., Radu A., Sena-Esteves M., Bouchard S., Wilson J. M., Verma I. M., Flake A. W. (2002) Efficient transduction of liver and muscle after in utero injection of lentiviral vectors with different pseudotypes. Mol. Ther. 6, 349–358 [DOI] [PubMed] [Google Scholar]
- 32. Chintala H., Liu H., Parmar R., Kamalska M., Kim Y. J., Lovett D., Grant M. B., Chaqour B. (2012) Connective tissue growth factor regulates retinal neovascularization through p53 protein-dependent transactivation of the matrix metalloproteinase (MMP)-2 gene. J. Biol. Chem. 287, 40570–40585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D'Amato R., Sullivan R., D'Amore P. A. (1994) Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 [PubMed] [Google Scholar]
- 34. Zudaire E., Gambardella L., Kurcz C., Vermeren S. (2011) A computational tool for quantitative analysis of vascular networks. PLoS One 6, e27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tamura I., Rosenbloom J., Macarak E., Chaqour B. (2001) Regulation of Cyr61 gene expression by mechanical stretch through multiple signaling pathways. Am. J. Physiol. Cell Physiol. 281, C1524–C1532 [DOI] [PubMed] [Google Scholar]
- 36. van Wijngaarden P., Brereton H. M., Coster D. J., Williams K. A. (2007) Stability of housekeeping gene expression in the rat retina during exposure to cyclic hyperoxia. Mol. Vis. 13, 1508–1515 [PubMed] [Google Scholar]
- 37. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Feng L., Hu J., Xie C., Wang F. (2013) Differentiating vitreous proteomes in proliferative diabetic retinopathy using high-performance liquid chromatography coupled to tandem mass spectrometry. Exp. Eye Res. 108, 110–119 [DOI] [PubMed] [Google Scholar]
- 39. Dean R. A., Butler G. S., Hamma-Kourbali Y., Delbé J., Brigstock D. R., Courty J., Overall C. M. (2007) Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol. Cell. Biol. 27, 8454–8465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Butler G. S., Dean R. A., Tam E. M., Overall C. M. (2008) Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol. 28, 4896–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song J., Tan H., Perry A. J., Akutsu T., Webb G. I., Whisstock J. C., Pike R. N. (2012) PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato H., Takino T., Okada Y., Cao J., Shinagawa A., Yamamoto E., Seiki M. (1994) A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 370, 61–65 [DOI] [PubMed] [Google Scholar]
- 43. Perbal B. (2009) Alternative splicing of CCN mRNAs. It has been upon us. J. Cell Commun. Signal. 3, 153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott A., Fruttiger M. (2010) Oxygen-induced retinopathy: a model for vascular pathology in the retina. Eye 24, 416–421 [DOI] [PubMed] [Google Scholar]
- 45. Karagiannis E. D., Popel A. S. (2008) A systematic methodology for proteome-wide identification of peptides inhibiting the proliferation and migration of endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 105, 13775–13780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guillon-Munos A., Oikonomopoulou K., Michel N., Smith C. R., Petit-Courty A., Canepa S., Reverdiau P., Heuzé-Vourc'h N., Diamandis E. P., Courty Y. (2011) Kallikrein-related peptidase 12 hydrolyzes matricellular proteins of the CCN family and modifies interactions of CCN1 and CCN5 with growth factors. J. Biol. Chem. 286, 25505–25518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pendurthi U. R., Tran T. T., Post M., Rao L. V. (2005) Proteolysis of CCN1 by plasmin: functional implications. Cancer Res. 65, 9705–9711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Overall C. M., Blobel C. P. (2007) In search of partners: linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 8, 245–257 [DOI] [PubMed] [Google Scholar]
- 49. d'Ortho M. P., Will H., Atkinson S., Butler G., Messent A., Gavrilovic J., Smith B., Timpl R., Zardi L., Murphy G. (1997) Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable with many matrix metalloproteinases. Eur. J. Biochem. 250, 751–757 [DOI] [PubMed] [Google Scholar]
- 50. Martino M. M., Hubbell J. A. (2010) The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 24, 4711–4721 [DOI] [PubMed] [Google Scholar]
- 51. Elzie C. A., Murphy-Ullrich J. E. (2004) The N terminus of thrombospondin: the domain stands apart. Int. J. Biochem. Cell Biol. 36, 1090–1101 [DOI] [PubMed] [Google Scholar]
- 52. Huwiler K. G., Vestling M. M., Annis D. S., Mosher D. F. (2002) Biophysical characterization, including disulfide bond assignments, of the anti-angiogenic type 1 domains of human thrombospondin-1. Biochemistry 41, 14329–14339 [DOI] [PubMed] [Google Scholar]
- 53. Inoki I., Shiomi T., Hashimoto G., Enomoto H., Nakamura H., Makino K., Ikeda E., Takata S., Kobayashi K., Okada Y. (2002) Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 16, 219–221 [DOI] [PubMed] [Google Scholar]
- 54. Ishida S., Usui T., Yamashiro K., Kaji Y., Amano S., Ogura Y., Hida T., Oguchi Y., Ambati J., Miller J. W., Gragoudas E. S., Ng Y. S., D'Amore P. A., Shima D. T., Adamis A. P. (2003) VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J. Exp. Med. 198, 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bork P. (1993) The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 327, 125–130 [DOI] [PubMed] [Google Scholar]
- 56. Brigstock D. R. (2003) The CCN family: a new stimulus package. J. Endocrinol. 178, 169–175 [DOI] [PubMed] [Google Scholar]
- 57. Holbourn K. P., Acharya K. R., Perbal B. (2008) The CCN family of proteins: structure-function relationships. Trends Biochem. Sci. 33, 461–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fukushima Y., Okada M., Kataoka H., Hirashima M., Yoshida Y., Mann F., Gomi F., Nishida K., Nishikawa S., Uemura A. (2011) Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J. Clin. Invest. 121, 1974–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schlingemann R. O., Rietveld F. J., Kwaspen F., van de Kerkhof P. C., de Waal R. M., Ruiter D. J. (1991) Differential expression of markers for endothelial cells, pericytes, and basal lamina in the microvasculature of tumors and granulation tissue. Am. J. Pathol. 138, 1335–1347 [PMC free article] [PubMed] [Google Scholar]
- 60. Koziol A., Gonzalo P., Mota A., Pollán Á., Lorenzo C., Colomé N., Montaner D., Dopazo J., Arribas J., Canals F., Arroyo A. G. (2012) The protease MT1-MMP drives a combinatorial proteolytic program in activated endothelial cells. FASEB J. 26, 4481–4494 [DOI] [PubMed] [Google Scholar]