

Background: Phosphorylation of 4E-BP1 plays a critical role in controlling its ability to inhibit protein translation.
Results: PPM1G is identified as a protein phosphatase that dephosphorylates 4E-BP1 in vitro and in cells.
Conclusion: PPM1G negatively regulates protein translation by controlling the phosphorylation of 4E-BP1.
Significance: PPM1G-mediated dephosphorylation of 4E-BP1 provides a novel mechanism in the regulation of protein translation.
Keywords: Cell Growth, eIF4E, Gene Silencing, Phosphatase, Translation, 4E-BP1, PPM1G, Dephosphorylation
Abstract
Protein translation initiation is a tightly controlled process responding to nutrient availability and mitogen stimulation. Serving as one of the most important negative regulators of protein translation, 4E binding protein 1 (4E-BP1) binds to translation initiation factor 4E and inhibits cap-dependent translation in a phosphorylation-dependent manner. Although it has been demonstrated previously that the phosphorylation of 4E-BP1 is controlled by mammalian target of rapamycin in the mammalian target of rapamycin complex 1, the mechanism underlying the dephosphorylation of 4E-BP1 remains elusive. Here, we report the identification of PPM1G as the phosphatase of 4E-BP1. A coimmunoprecipitation experiment reveals that PPM1G binds to 4E-BP1 in cells and that purified PPM1G dephosphorylates 4E-BP1 in vitro. Knockdown of PPM1G in 293E and colon cancer HCT116 cells results in an increase in the phosphorylation of 4E-BP1 at both the Thr-37/46 and Ser-65 sites. Furthermore, the time course of 4E-BP1 dephosphorylation induced by amino acid starvation or mammalian target of rapamycin inhibition is slowed down significantly in PPM1G knockdown cells. Functionally, the amount of 4E-BP1 bound to the cap-dependent translation initiation complex is decreased when the expression of PPM1G is depleted. As a result, the rate of cap-dependent translation, cell size, and protein content are increased in PPM1G knockdown cells. Taken together, our study has identified protein phosphatase PPM1G as a novel regulator of cap-dependent protein translation by negatively controlling the phosphorylation of 4E-BP1.
Introduction
Protein translation is a fundamentally important process that plays an essential role in maintaining normal homeostasis in cells. Given the fact that protein synthesis is a highly energy-consuming process, it is tightly regulated according to the growth and proliferation needs of the organism. Among the steps of protein translation, which include initiation, elongation, and termination, translation initiation is considered the rate-limiting step because it has the capacity to integrate and respond to various signaling inputs (1). Disruption of precise controls over protein translation is often associated with human diseases. For example, recent studies have linked up-regulation of protein synthesis to increased cancer malignancy and resistance to therapy (2, 3). Defects in protein synthesis resulting from mutations in genes involved in translation are found to be associated with inherited human diseases (4).
Numerous studies have demonstrated that mammalian target of rapamycin (mTOR)3 plays a critical role in controlling the translation initiation step in protein synthesis (5). mTOR is a serine/threonine kinase that exists in two distinct functional complexes: mTORC1 and mTORC2 (6). The mTORC1 complex is responsible for controlling protein translation downstream of growth factors, nutrients, and stress signals. Serving as one the major substrates of mTOR, 4E-BP1 directly regulates the rate of translation by affecting the assembly of the translation initiation complex (20). During the translation initiation step in mammalian cells, the cap structure of the mRNA is recognized by the eIF4F complex, which is comprised of eIF4A, eIF4G, and eIF4E proteins. The hypophosphorylated form of 4E-BP1 binds to the cap-binding protein eIF4E and prevents it from interacting with the scaffolding protein eIF4G, thus suppressing cap-dependent translation. Activation of mTOR leads to phosphorylation of 4E-BP1 and disruption of the binding between 4E-BP1 and eIF4E. As a result, 4E-BP1 is released from the cap structure, which allows the association of eIF4G with eIF4E to form the initiation complex and protein translation to proceed (1, 7, 8). Given its role in controlling protein translation, mTOR-mediated phosphorylation of 4E-BP1 has been studied extensively (5, 9, 10). Specifically, two sets of phosphorylation sites have been identified in 4E-BP1 upon mTOR activation, in which mTOR is responsible for directly phosphorylating Thr-37 and Thr-46 and priming for additional phosphorylation at Ser-65 and Thr-70. Furthermore, the phosphorylation status of 4E-BP1 has been identified as a biomarker to indicate the efficacy of anticancer treatments because a complete dephosphorylation of 4E-BP1 is required to effectively inhibit cancer cell growth in vitro and in vivo (11). However, how 4E-BP1 is dephosphorylated in cells is currently unknown.
In this study, we report the identification of PPM1G as the phosphatase that directly dephosphorylates multiple phosphorylation sites in 4E-BP1 both in vitro and in cells. In addition, we examine the role of PPM1G in regulating cap-dependent protein translation by controlling the binding of 4E-BP1 with the cap structure.
EXPERIMENTAL PROCEDURES
Reagents
The expression plasmid for HA-tagged 4E-BP1/4A mutant (pBabe-HA-4E-BP1/4A) was a gift from Dr. Qingbai She (University of Kentucky), and the dual Renilla firefly luciferase pcDNA3-rLuc-PolioIRES-fLuc reporter was provided by Dr. John Blenis (Harvard Medical School). Amino acid-free minimum Eagle's medium was purchased from US Biological. The following antibodies were purchased from the following commercial sources: polyclonal antibodies for PPM1G from Bethyl Laboratory; polyclonal antibodies against 4E-BP1, p70S6K, Akt, S6, phospho-4E-BP1 (pThr-37/46, pSer-65, and pThr-70), phospho-Akt (pThr-308), phospho-p70S6K (pThr-389), and phospho-S6 (Ser-240/244) from Cell Signaling Technology; and anti-γ-tubulin mAb from Sigma. Calyculin A, okadaic acid, and PP242 were obtained from EMD.
Cells
Human colon cancer HCT116 cells were cultured in McCoy's 5A medium, and 293E cells were cultured in DMEM. All media were supplemented with 10% FBS (Sigma) and 1% penicillin/streptomycin. Stable HCT116 cells overexpressing HA-4E-BP1 or HA-4E-BP1/4A were provided by Dr. Qingbai She. Transient transfections of these cells were performed using polyethylenimine. To generate a collection of PPM knockdown 293E cell lines, shRNA lentivirus-targeting constructs in the pLKO.1-puro vector for human PPM1A, PPM1B, PPM1D, PPM1E, PPM1F, PPM1G, PPP2CA, and PPP2CB were purchased from Sigma-Aldrich. There are four shRNA targeting sequences for each phosphatase. Lentivirus-mediated delivery of shRNA and selection for stable knockdown cells were carried out as described previously (12). Two different stable cell lines were created for each phosphatase by combining lentiviruses derived from two different shRNA targeting constructs.
Generation of Expression Constructs
To express PPM1G in mammalian cells, the cDNA of human PPM1G was obtained by using aSuperScript III one-step RT-PCR kit (Invitrogen) and total RNA isolated from 293E cells as the template. The entire coding sequence of PPM1G was confirmed by sequencing and subsequently subcloned in-frame into the p3XFLAG-CMV vector (Sigma-Aldrich). To generate GST-tagged fusion protein in bacteria, the coding sequence of PPM1G was amplified using PCR and subcloned in-frame into the pGEX-6P3 vector.
Immunoprecipitation and Immunoblotting
To detect the level of protein expression and phosphorylation, cells were lysed in lysis buffer (50 mm Na2HPO4, 1 mm sodium pyrophosphate, 20 mm NaF, 2 mm EDTA, 2 mm EGTA, 1% Triton X-100, 1 mm DTT, 200 mm benzamidine, 40 mg/ml leupeptin, 200 mm PMSF), and the detergent-solubilized cell lysates were separated by SDS-PAGE and analyzed by immunoblotting (13). For immunoprecipitation experiments, cell lysates were incubated with anti-HA affinity matrix (Roche) or protein A/G beads coupled to specific antibodies (14). For cap binding assays, cell lysates were incubated with 7-methyl-GTP (m7GTP)-Sepharose at 4 °C for 2 h as described previously (15, 16). Immunoprecipitates were washed with lysis buffer, and bound proteins were analyzed by SDS-PAGE and immunoblotting. The density of Western blot signals was obtained and quantified using a FluorChem digital imaging system (Alpha Innotech).
In Vitro Dephosphorylation
The GST-tagged PPM1G fusion proteins were expressed and purified from bacteria by following procedures described previously (13). The GST-PPM1G was treated with PreScission protease to release PPM1G recombinant proteins. The presence of PPM1G phosphatase activity was verified by using para-nitrophenylphosphate as the substrate (17). To obtain phosphorylated 4E-BP1 proteins as a substrate, stable HCT116 cells overexpressing wild-type HA-4E-BP1 were lysed in lysis buffer. Cell lysates were incubated with anti-HA affinity matrix, and the immunoprecipitates were washed twice with lysis buffer and twice with phosphatase buffer (0.1 m sodium acetate, 0.05 m bis-Tris, 0.05 m Tris (pH 7.5)). The dephosphorylation reactions were carried out by incubating HA-4E-BP1-bound beads with purified PPM1G (0.1 μg) at room temperature for 0–30 min in the phosphatase buffer supplemented with MnCl2 (2 mm) and DTT (10 mm) (13).
Analysis of 4E-BP1 Dephosphorylation in Cells
Amino acid starvation or mTOR inhibitor treatment were used to induce dephosphorylation of 4E-BP1 in cells. For amino acid starvation, cells grown in regular growth medium were rinsed with PBS and then incubated in amino acid-free and serum-free minimum Eagle's medium (supplemented with 584 μg/ml l-glutamine) for 0–45 min. For PP242 treatment, cells grown to ∼60–70% confluency were treated with PP242 (100 nm) for the indicated time in regular growth medium. At the end of each treatment, cell lysates were prepared in lysis buffer and analyzed by immunoblotting.
Dual Luciferase Assay
The cap-dependent translation was measured using a dual luciferase reporter in which the Renilla luciferase was linked with the 5′ UTR sequence of HIF-1α and the firefly luciferase was driven by the polio virus internal ribosome entry site (16, 18). Cells transfected with the dual luciferase reporter were treated as specified and the luciferase activities were measured using a Dual-Luciferase reporter assay system (Promega). The rate of cap-dependent translation was defined as the ratio of Renilla/firefly luciferase activities.
RESULTS
Identification of PPM1G as the Phosphatase for 4E-BP1
In our studies to examine the function of mTOR in regulating protein translation, we became interested in determining which protein phosphatases may be involved in dephosphorylating 4E-BP1, the key downstream effector of mTOR. The serine/threonine phosphatases are divided into two families: phosphoprotein phosphatases (PPPs) and metal-dependent protein phosphatases (PPMs). Phosphatase inhibitors such as okadaic acid (OA) and calyculin A are known to inhibit the PPP family of protein phosphatases such as PP1 and PP2A but not PPM family members (19). To begin our search for 4E-BP1 phosphatase, we first treated cells with OA and calyculin A to determine whether the phosphorylation of 4E-BP1 is sensitive to the inhibition of the PPP family of phosphatases. Interestingly, the level of phosphorylation at both Thr-37/Thr-46 and Ser-65 sites were not altered significantly in cells treated with OA or calyculin A (Fig. 1A). As a positive control, the Thr-308 site in Akt, a phosphorylation site known to be regulated by PP2A (13), was largely increased in cells treated with either phosphatase inhibitor (Fig. 1A). These results suggest that the phosphorylation of 4E-BP1 at both sets of phosphorylation sites is likely regulated by the PPM family of phosphatases, which is known to be resistant to the phosphatase inhibitors tested here. To screen for specific phosphatases of 4E-BP1, we generated a small library of knockdown 293E cell lines using shRNAs targeting different PPM phosphatases, including PPM1A, PPM1B, PPM1D, PPM1E, PPM1F, and PPM1G. Among them, knockdown of PPM1G resulted in the most significant increase in 4E-BP1 phosphorylation (Fig. 1B). Confirming our results obtained with phosphatase inhibitors of PP2A, knockdown of the catalytic subunits of PP2A, including PPP2CA and PPP2CB, did not increase the phosphorylation of 4E-BP1 under normal growth conditions (Fig. 1B). Following this initial observation, we generated additional PPM1G knockdown cell lines using 293E and HCT116 cells to further characterize the function of PPM1G in regulating 4E-BP1 phosphorylation. The results showed that the level of phosphorylation at the Thr-37/Thr-46 and Ser-65 sites in 4E-BP1 was increased by ∼2- to 3-fold in both PPM1G knockdown cell lines (Fig. 1, C and D). In contrast, the phosphorylation of p70S6K at Thr-398, another mTOR-dependent phosphorylation site, was not affected by silencing PPM1G expression (Fig. 1C). This suggests that PPM1G functions at the level of 4E-BP1 and downstream of mTOR. All four phosphorylation sites on 4E-BP1 (including Thr-37/Thr-46, Ser-65, and Thr-70) seemed to be affected by knocking down PPM1G. Our subsequent experiments mainly focused on the Thr-37/Thr-46 and Ser-65 sites. Taken together, we have identified PPM1G as a potential phosphatase of 4E-BP1.
FIGURE 1.

Identification of PPM1G as a phosphatase of 4E-BP1. A, phosphorylation of 4E-BP1 is not sensitive to the treatment of the phosphatase inhibitors OA and calyculin A. 293E cells grown in regular growth medium containing 10% FBS were treated with dimethyl sulfoxide (DMSO), OA (100 nm), or calyculin A (10 nm) for 15 min (lanes 1–3, respectively), and cell lysates were analyzed using immunoblotting. The phosphorylation status of 4E-BP1 at the Thr-37/46 and Ser-65 sites was detected using the p37/46 and p65 antibodies, respectively. The phosphorylation of Akt at the Thr-308 site was used as a control. The total 4E-BP1, Akt, and tubulin proteins were detected using respective antibodies. B, to screen for phosphatases of 4E-BP1, a small library of knockdown 293E cell lines were generated using shRNAs targeting different PPM phosphatases including PPM1A, PPM1B, PPM1D, PPM1E, PPM1F, and PPM1G as well as PPP2CA and PPP2CB. Cell lysates were prepared and analyzed for phosphorylation of 4E-BP1 at the Thr-37/Thr-46, Ser-65, and Thr-70 sites using corresponding phosphospecific antibodies. The total protein expression of 4E-BP1 was detected by the 4E-BP1 antibody. β-actin was used as the loading control. C, knockdown of PPM1G promotes 4E-BP1 phosphorylation. Stable control (sh-Con) and two different PPM1G knockdown (sh-PPM1G#1 and sh-PPM1G#2) 293E and HCT116 cell lines (lanes 1–3 and 4-6, respectively) were cultured in regular growth medium containing 10% FBS until they reached ∼60–70% confluency. Cell lysates were prepared and analyzed for the phosphorylation status of 4E-BP1 by immunoblotting. The phosphorylation of 4E-BP1 at the Thr-37/46 and Ser-65 sites was detected by the corresponding phosphospecific antibodies. The phosphorylation of p70S6K at the Thr-389 site (p-S6K) was shown as a negative control. D, the relative phosphorylation of 4E-BP1 was obtained by normalizing ECL signals generated by the p37/46 or p65 antibody to that of total 4E-BP1 in both 293E and HCT116 cells. The value of control cells was set to 1, and data shown in the bar graphs represent the mean ± S.D. (n = 3). *, p < 0.05 as determined by two-sample Student's t tests compared with the control cells. Note that the quantified results were obtained from one of the stable PPM1G knockdown cell lines (Sh-PPM1G#1) and that this knockdown cell line was used in all subsequent experiments in our study. Similar results were obtained using the other knockdown cell line (Sh-PPM1G#2).
Next, we examined the interaction between PPM1G and 4E-BP1 because protein phosphatases usually have relatively high affinity for their substrates. HCT116 stable cells overexpressing HA-4E-BP1 were transfected with FLAG-PPM1G and subsequently treated with PP242 to inhibit mTOR activity. Cell lysates were immunoprecipitated with the anti-FLAG antibody to determine whether PPM1G is associated with 4E-BP1. As shown in Fig. 2A, PPM1G was found coimmunoprecipitated with 4E-BP1 regardless of the phosphorylation status of 4E-BP1. To further confirm that the interaction between PPM1G and 4E-BP1 is independent of the phosphorylation status of 4E-BP1, we performed the coimmunoprecipitation experiment using HCT116 cells expressing either WT 4E-BP1 or the phosphorylation-deficient 4E-BP1/4A mutant. The results showed that PPM1G interacted with both forms of 4E-BP1 equally well (Fig. 4B). Finally, we examined the interaction between the endogenous PPM1G and 4E-BP1. Coimmunoprecipitation of these two endogenous proteins was readily detected in 293E cells (Fig. 2C).
FIGURE 2.
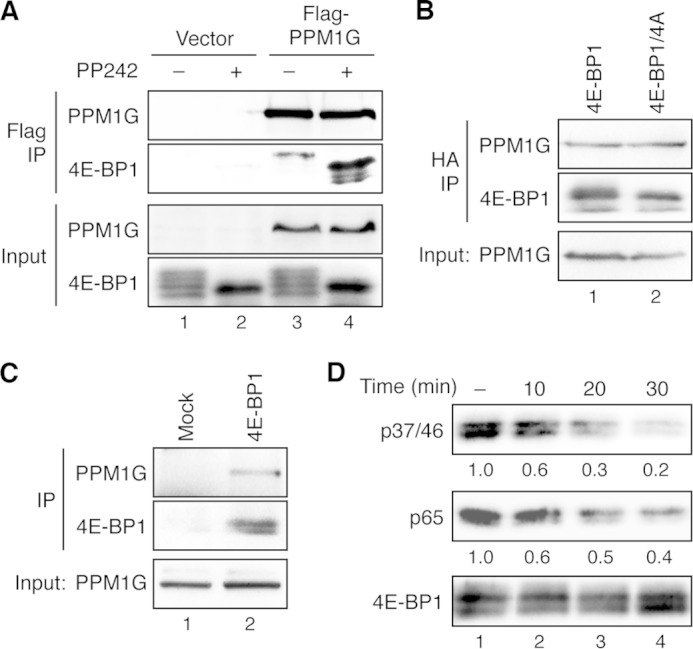
4E-BP1 is a direct substrate of PPM1G. A, PPM1G interacts with 4E-BP1 in cells. Stable HCT116 cells overexpressing HA-4E-BP1 were transfected with vector (lanes 1–2) or FLAG-PPM1G (lanes 3–4), and the transfected cells were treated with dimethyl sulfoxide or PP242 for 2 h. Cell lysates were prepared and immunoprecipitated (IP) with the anti-FLAG affinity gel. The presence of PPM1G in immunoprecipitates and cell lysates (10% input) was detected using the PPM1G antibody, whereas 4E-BP1 proteins were detected by the 4E-BP1 antibody. B, PPM1G interacts with both the WT and phosphorylation-deficient mutant 4E-BP1 in cells. Cell lysates were prepared from stable HCT116 cells overexpressing either WT HA-4E-BP1 or the HA-4E-BP1/4A mutant and subjected to immunoprecipitation using the anti-HA affinity matrix. The presence of PPM1G in immunoprecipitates and cell lysates (10% input) was detected using the PPM1G antibody, whereas 4E-BP1 proteins were detected by the 4E-BP1 antibody. C, the endogenous PPM1G and 4E-BP1 interact. Equal amounts of 293E cell lysates were incubated with protein A/G beads alone (Mock) or beads plus the 4E-BP1 antibody. The presence of PPM1G and 4E-BP1 in the immunoprecipitates and input were detected with the PPM1G and 4E-BP1 antibodies, respectively. D, dephosphorylation of 4E-BP1 in vitro. The HA-tagged 4E-BP1 was immunoprecipitated from stable HCT116/4E-BP1 cells using the anti-HA affinity matrix. Dephosphorylation reactions were carried out by incubating the immunoprecipitates with the purified PPM1G for the indicated time (lanes 2–4). As control for nonspecific dephosphorylation, HA-4E-BP1-bound beads were incubated in the dephosphorylation buffer without adding purified PPM1G for 30 min at room temperature (lane 1). Phosphorylation of 4E-BP1 at the Thr-37/46 and Ser-65 sites was detected using the phosphospecific antibodies specific for these sites. The relative phosphorylation of 4E-BP1 was quantified by normalizing ECL signals generated by phosphospecific antibodies to that of total 4E-BP1 and shown below the corresponding blots.
FIGURE 4.

PPM1G regulates the amount of 4E-BP1 associated with the cap structure. A, knockdown of PPM1G decreases the amount of 4E-BP1 bound with the m7GTP cap complex. The stable control and PPM1G knockdown 293E cells were subjected to one of the following treatments: kept in serum-containing medium (+S), serum-starved overnight (−S), serum-starved overnight and subsequently treated with insulin (+Ins), and pretreated with rapamycin before adding insulin (+Rapa/Ins). Cell lysates were incubated with m7GTP-beads to pull down cap-associated proteins. The amount of proteins and phosphoproteins in precipitates and lysates were detected by immunoblotting analysis. B, the amount of 4E-BP1 associated with the m7GTP-beads was quantified and normalized to the amount of cap-bound eIF4E. This number was set to 1 for cells grown in serum-containing medium. Data shown in the graph represent the mean ± S.E. (n = 4). *, p < 0.01; #, p < 0.05 as determined by two-sample Student's t tests comparing PPM1G knockdown (Sh-PPM1G) HCT116 cells with stable control (sh-Con) cells under each condition.
To further assess whether 4E-BP1 is dephosphorylated by PPM1G directly, we performed in vitro dephosphorylation experiments using purified recombinant PPM1G proteins. Phosphorylated 4E-BP1 isolated from stable HCT116 cells expressing HA-4E-BP1 was incubated with purified PPM1G protein. The time course of 4E-BP1 dephosphorylation revealed that both the Thr-37/Thr-46 and Ser-65 sites were dephosphorylated readily by PPM1G in vitro. At the end of 30 min, the phosphorylation of Thr-37/Thr-46 and Ser-65 was decreased by 80 and 60%, respectively (Fig. 2D). As a control for specificity, we found that HA-S6K isolated from cells was not a substrate of purified PPM1G in vitro under the condition where HA-4E-BP1 was dephosphorylated (data not shown). Collectively, these findings suggest that PPM1G binds to 4E-BP1 basally in cells and that 4E-BP1 is a direct substrate of PPM1G in vitro.
Knockdown of PPM1G Prolongs the Phosphorylation of 4E-BP1 in cells
We next determined whether PPM1G controls the dephosphorylation time course of 4E-BP1 in cells. To induce 4E-BP1 dephosphorylation, control and PPM1G knockdown cells were treated with PP242, a potent ATP-competitive kinase inhibitor of mTOR, for 0–60 min and 18 h. As shown in Fig. 1, the phosphorylation at both the Thr-37/Thr-46 and Ser-65 sites was elevated in PPM1G knockdown HCT116 cells (Fig. 3A, compare lanes 1 and 7). More significantly, the rate of PP242-induced dephosphorylation was largely slowed down in PPM1G knockdown cells (Fig. 3, A and B). At the end of 60 min, phosphorylation was detected in only ∼20% of endogenous 4E-BP1 protein, whereas more than 50% of 4E-BP1 remained phosphorylated at both the Thr-37/Thr-46 and Ser-65 sites in cells with decreased expression of PPM1G. Similarly, when cells were incubated in amino acid-free medium to inactivate mTOR, 4E-BP1 became rapidly dephosphorylated within 45 min in control 293E cells (Fig. 3C, lanes 1–6). However, the dephosphorylation time course of 4E-BP1 at both the Thr-37/Thr-46 and Ser-65 sites were delayed significantly in PPM1G knockdown cells (Fig. 3C, lanes 7–12). After nutrient deprivation for 45 min, 4E-BP1 was almost completely dephosphorylated in control cells, whereas about 30–40% of 4E-BP1 proteins remained phosphorylated in PPM1G knockdown cells (Fig. 3D). As a negative control, the dephosphorylation of p70S6K induced by PP242 or amino acid starvation was not altered by silencing PPM1G in cells (Fig. 3, A and C). These results confirm that the level of PPM1G expression controls the dephosphorylation time course of 4E-BP1.
FIGURE 3.
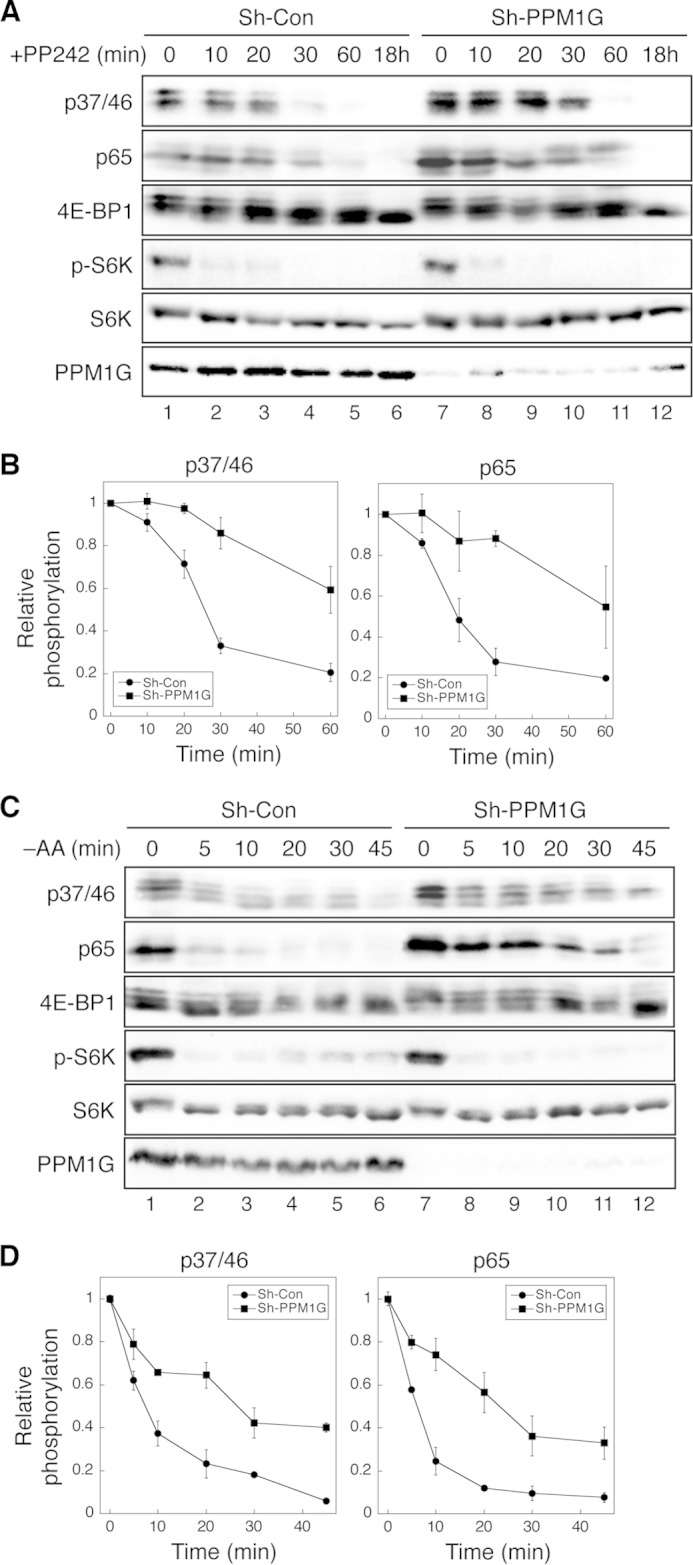
Knockdown of PPM1G delays the dephosphorylation of 4E-BP1 induced by mTOR inhibition. A, the time course of 4E-BP1 dephosphorylation upon inhibition of mTOR kinase activity. Stable control (Sh-Con) and PPM1G knockdown (Sh-PPM1G) HCT116 cells were treated with PP242 (100 nm) for 0–60 min or 18 h. Cell lysates were analyzed using immunoblotting. The phosphorylation of 4E-BP1 at Thr-37/46 and Ser-65 and p70S6K at Thr-398 was detected using phosphospecific antibodies. B, Western blot analyses as shown in A were quantified, the relative phosphorylation of 4E-BP1 was obtained by normalizing ECL signals generated by the p37/46 or p65 antibody to that of total 4E-BP1, and the quantitative results were expressed graphically. Data shown in the graph represent mean ± S.D. (n = 3). C, time course of 4E-BP1 dephosphorylation upon amino acid starvation (−AA). Stable control and PPM1G knockdown 293E cells were incubated in amino acid-free medium for 0–45 min. Cell lysates were analyzed using immunoblotting. The phosphorylation of 4E-BP1 at Thr-37/46 and Ser-65 and p70S6K at Thr-398 was detected using phosphospecific antibodies. D, Western blot analyses as shown in C were quantified, the relative phosphorylation of 4E-BP1 was obtained by normalizing ECL signals generated by the p37/46 or p65 antibody to that of total 4E-BP1, and the quantitative results were expressed graphically. Data shown in the graph represent mean ± S.D. (n = 3).
PPM1G Controls the Association of 4E-BP1 with the Cap Structure
It has been shown previously that the level of 4E-BP1 phosphorylation regulates protein translation by controlling the amount of 4E-BP1 associated with eIF4E and the cap structure (1, 18). We first determined the effect of knocking down PPM1G on the association of 4E-BP1 with the cap structure. Control and PPM1G knockdown 293E cells were cultured under different conditions to alter the phosphorylation status of 4E-BP1, and cap structure-associated proteins were pulled down from cell lysates using m7GTP beads. Consistent with previous studies, the amount of 4E-BP1 found associated with the cap structure was conversely correlated with the phosphorylation status of 4E-BP1 (Fig. 4). Specifically, in control cells, when the phosphorylation of 4E-BP1 was decreased under serum-starved and rapamycin-treated conditions, total 4E-BP1 bound to the cap complex was ∼50% and 1.2-fold higher than that in cells grown in serum-containing medium (Fig. 4, A and B, compare lanes 3 and 7 with lane 1). On the other hand, increased phosphorylation of 4E-BP1 reduced the amount of 4E-BP1 bound to the cap structure in insulin-treated control cells (Fig. 4, A and B, compare lanes 3 and 5). Furthermore, silencing PPM1G resulted in a decrease in the amount of 4E-BP1 associated with the cap structure under each condition and an increase in 4E-BP1 phosphorylation, as detected in cell lysates compared with control cells (Fig. 4A, compare lanes 2, 4, 6, and 8 with lanes 1, 3, 5, and 7, respectively). As a control, the phosphorylation of cap-bound ribosomal S6 protein (p-S6) was not altered by knocking down PPM1G expression (Fig. 4A). Taken together, we showed that PPM1G-mediated dephosphorylation of 4E-BP1 promotes the association of 4E-BP1 with the cap complex.
PPM1G Negatively Regulates the Cap-dependent Translation and Cell Growth
Because 4E-BP1 is a key negative regulator of cap-dependent protein translation, we next determined the rate of protein translation in control and PPM1G knockdown cells. A dual luciferase reporter system was used to measure the cap-dependent translation under different conditions (15). As shown in Fig. 5, A and B, knockdown of PPM1G resulted in an increase in the cap-dependent translation in both 293E and HCT116 cells basally (+S condition). Under the serum-starved condition, the rate of cap-dependent translation was decreased as expected. However, the translation remained significantly higher in PPM1G knockdown cells (Fig. 5, A and B, −S). Furthermore, insulin-stimulated translation was enhanced further by depleting PPM1G expression in both cell lines (Fig. 5, A and B, +Ins). Therefore, PPM1G negatively regulates the cap-dependent translation in cells. Consistent with the finding that PPM1G-mediated dephosphorylation of 4E-BP1 suppressed the cap-dependent translation (Fig. 5, A and B), knockdown of PPM1G in 293E and HCT116 cells resulted in increased protein contents (C). Specifically, for the same numbers of cells, the protein content was significantly higher in PPM1G knockdown cells, suggesting enhanced protein synthesis in these cells (Fig. 5C).
FIGURE 5.
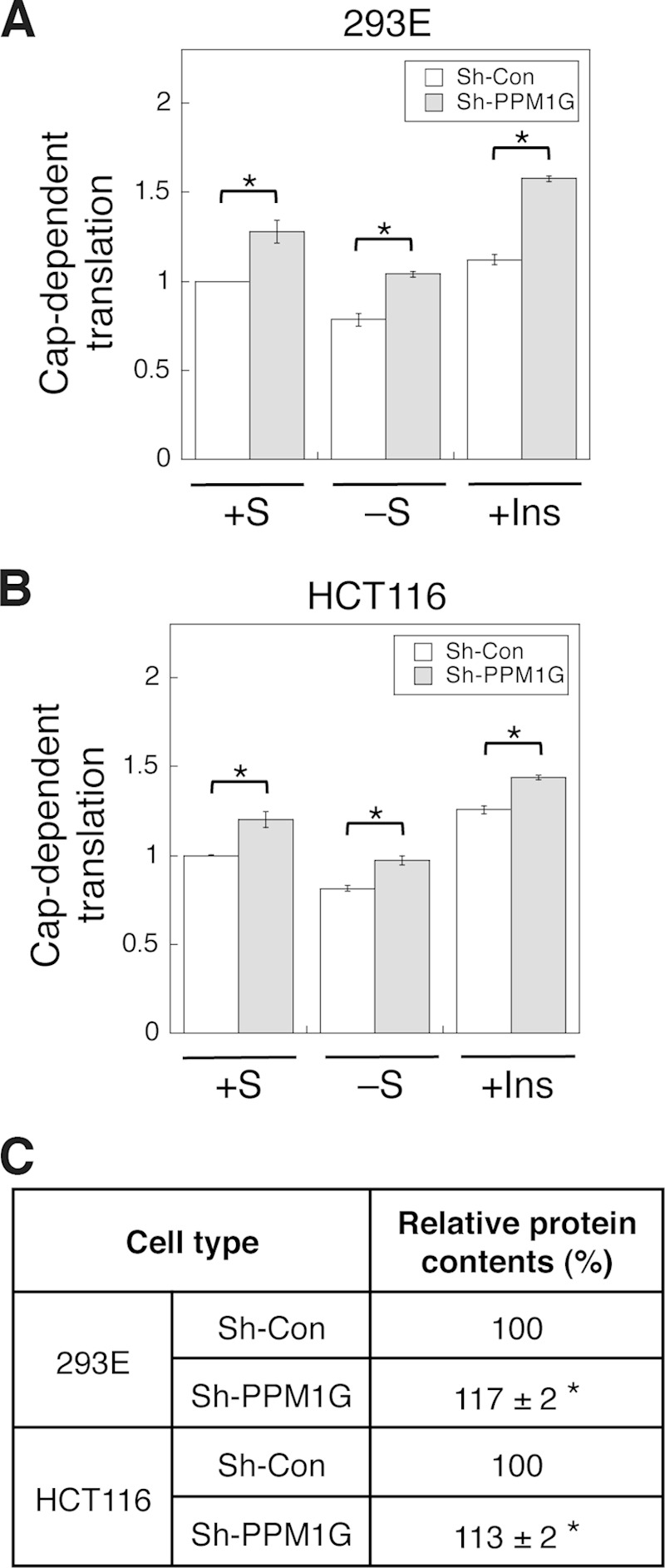
Knockdown of PPM1G increases protein translation and cell growth. A and B, stable control (Sh-Con) and PPM1G (Sh-PPM1G) knockdown 293E and HCT116 cells were transfected with a dual luciferase reporter. The transfected cells were subjected to one of the following treatments: kept in serum-containing medium (+S), serum-starved overnight (−S), and serum-starved overnight followed by treating with insulin for an additional 24 h (+Ins). Cells were then collected at the same time, and luciferase activities were measured using dual luciferase assays. The cap-dependent translation was determined using the ratio of Renilla/firefly luciferase light units. The value of control cells in the serum-containing medium was set to 1. Data shown in the graph represent the mean ± S.D. (n = 3). *, p < 0.01 as determined by two-sample Student's t tests compared with the control cells. C, stable control and PPM1G knockdown 293E and HCT116 cells were cultured in regular growth medium to ∼50% confluence. For protein content measurement, equal numbers of control and PPM1G knockdown cells were lysed in lysis buffer, and protein concentrations were determined using Bradford assays. Each experiment was done in duplicate, and three independent experiments were averaged. The average protein contents shown in the table represent mean ± S.D. *, p < 0.05 as determined by two-sample Student's t tests compared with the control cells.
To further verify the specificity of the PPM1G-mediated effect on protein translation, we examined whether a phosphorylation-deficient 4E-BP1 can suppress the cap-dependent translation in PPM1G knockdown cells. Prior studies have indicated that a 4E-BP1–4A mutant that carries alanine substitutions at all four major phosphorylation sites serves as a constitutively active inhibitor of eIF4E and impairs the assembly of the eIF4F complex (11, 21, 22). Consistent with the results shown in Fig. 4A, the cap-dependent translation was significantly higher in PPM1G knockdown cells compared with control cells under all three conditions tested (Fig. 6A). As expected, overexpression of 4E-BP1–4A resulted in a decrease in the rate of translation in control cells (Fig. 6A). Most importantly, 4E-BP1–4A was capable of inhibiting protein translation in PPM1G knockdown cells, and no statistical significance was observed between control and PPM1G knockdown cells transfected with 4E-BP1–4A (Fig. 6A). These results suggested that the PPM1G-mediated effect on protein translation is largely dependent on its ability to dephosphorylate 4E-BP1. The expression of HA-4E-BP1–4A in control and PPM1G knockdown cells was confirmed using Western blotting (Fig. 6B). Taken together, our results demonstrate that PPM1G inhibits protein translation and cell growth by directly suppressing the phosphorylation of 4E-BP1.
FIGURE 6.
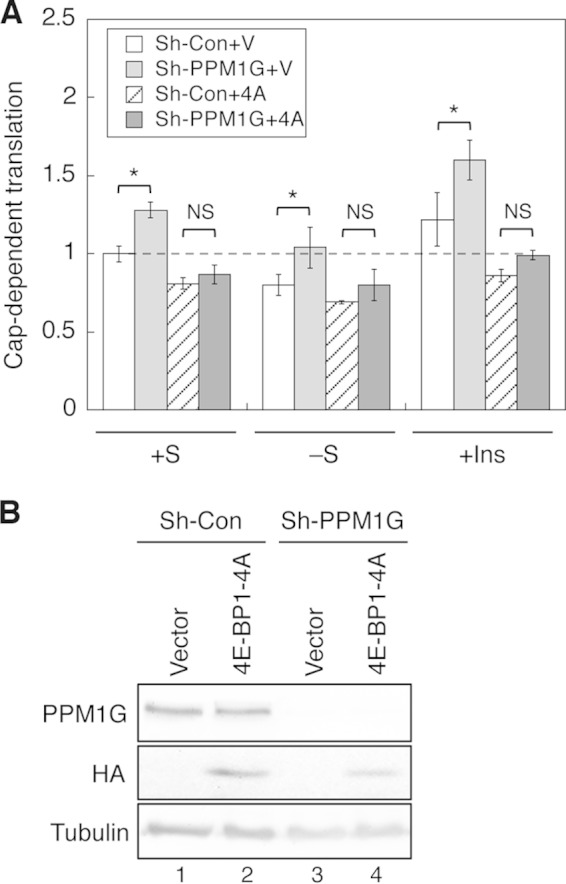
PPM1G-mediated regulation of cap-dependent translation is 4E-BP1-dependent. A, stable control (sh-Con) and PPM1G knockdown (sh-PPM1G) 293E cells were transiently cotransfected with the dual luciferase reporter together with either the pBabe vector or pBabe-HA-4EBP1/4A plasmid. Approximately 24 h post-transfection, cells were subjected to one of the following treatments: kept in serum-containing medium (+S), serum-starved overnight (−S), and serum-starved overnight and subsequently treated with insulin for another 24 h (+Ins). Cells were collected at the same time, and luciferase activities were measured using dual luciferase assays. The cap-dependent translation was determined using the ratio of Renilla/firefly luciferase light units. The value of control cells in the serum-containing medium was set to 1. Data shown in the graph represent the mean ± S.D. (n = 3). *, p < 0.05 as determined by two-sample Student's t tests comparing sh-PPM1G with sh-Con cells. NS, no statistical significance). The dashed line indicates the value of relative cap-dependent translation at 1 for better comparison. B, cell lysates were prepared from transfected cells and analyzed using immunoblotting to detect the expression of HA-4E-BP1/4A protein.
In summary, we identified PPM1G as a novel phosphatase that controls the phosphorylation of 4E-BP1. Loss of PPM1G expression leads to sustained phosphorylation of 4E-BP1 and promotes cap-dependent translation and cell growth.
DISCUSSION
An increasing amount of evidence has linked dysregulation of protein translation with human diseases (2–4). In various types of tumors, protein translation is often up-regulated as the result of hyperactivation of signaling pathways that control cell growth and proliferation (6, 7, 22). For example, increased levels of 4E-BP1 phosphorylation and expression of eIF4E have been found to be associated with higher tumor grade and decreased survival in breast cancer (23). Therefore, loss of proper control over 4E-BP1 phosphorylation may play an important role in promoting tumorigenesis. In this study, we showed that a PPM family protein phosphatase, PPM1G, functions to dephosphorylate 4E-BP1 in vitro and in cells. Specifically, knockdown of PPM1G results in an increase in 4E-BP1 phosphorylation basally and a decrease in the time course of 4E-BP1 dephosphorylation upon inhibiting mTOR activity. Moreover, by controlling the association of 4E-BP1 with the cap structure and the assembly of the initiation complex, PPM1G plays a negative role in regulating cap-dependent protein translation and cell growth.
Protein phosphorylation is one of the most commonly used schemes in signal transduction. It is estimated that one-third of cellular proteins are phosphorylated and that the majority of the phosphorylation occurs on Ser and Thr residues (24). The number of Ser/Thr phosphatases is very limited compared with the vast pool of phosphorylated proteins as potential substrates. Facing the challenge of controlling a large number of substrates, many Ser/Thr phosphatases are found to regulate multiple substrates (19). In the case of PPM1G, a limited numbers of studies have identified several other substrates of PPM1G, which include USP7, coilin, survival motor neuron complex proteins, and histones H2A and H2B (25–28). By controlling the dephosphorylation of these substrates, the function of PPM1G has been implicated in regulating p53-dependent DNA damage response, RNA splicing, and chromatin assembly (25–28). Interestingly, it has been shown that PPM1G functions in the nucleus to regulate RNA splicing and to dephosphorylate histones (27, 28). We have found that PPM1G is localized in both cytoplasmic and nuclear compartments in 293E and several colon cancer cell lines (data not shown). Thus, it is likely that PPM1G is capable of shuffling between the cytoplasm and nucleus to dephosphorylate different substrates. Future studies are needed to determine whether altered nuclear localization of PPM1G affects its ability to dephosphorylate 4E-BP1. On the flip side, although we found that PPM1G controls the basal phosphorylation of 4E-BP1 under normal growth conditions, it has been shown previously that up-regulation of PP2A/PP1 phosphatase activity as the result of oxidative stress promotes the dephosphorylation of 4E-BP1 (29, 30). Thus, 4E-BP1 as a substrate can potentially be dephosphorylated by multiple phosphatases in a cell context-dependent manner.
A recent study has shown that the phosphorylation of 4E-BP1 not only controls the interaction between eIF4E and eIF4G but also plays an important role in regulating the expression level of 4E-BP1 because hyperphosphorylated 4E-BP1 is resistant to ubiquitation-dependent degradation (31). In addition, a transcriptome-scale ribosome profiling study has revealed that mTORC1 specifically regulates the translation of mRNAs containing so called TOP (5′ terminal oligopyrimidine) or TOP-like motifs and that the 4E-BP family of translational repressors is identified as a master effector responsible for controlling the majority of mTORC1-dependent translational activity (22). In this study, we have demonstrated that the interaction between PPM1G and 4E-BP1 is not altered by the phosphorylation status of 4E-BP1. Thus, it is likely that the level of PPM1G in cells controls the basal amount of 4E-BP1 bound to the cap structure. Because it has been suggested that 4E-BP1 affects the translation of TOP and TOP-like mRNAs much more than other mRNAs by regulating the interaction between the cap-binding protein eIF4E and eIF4G (22), it will be interesting to determine whether the translation of specific TOP or TOP-like mRNAs (such as EEF2 and VIM) are affected by PPM1G in future studies.
Previous studies have shown that 4E-BP1 serves as a master regulator of cap-dependent translation downstream of mTOR and that dephosphorylation of 4E-BP1 is required to achieve therapeutic effects when using inhibitors targeting mTOR or other signaling molecules upstream of mTOR (11, 32). We found in this study that loss of PPM1G expression delays the dephosphorylation time course of 4E-BP1. Thus, the expression and activity of PPM1G may play an important role in regulating the effectiveness of mTOR inhibitors in cancer treatment. Taken together, our study has identified a novel role of PPM1G in negatively regulating protein translation by directly dephosphorylating 4E-BP1. This finding may help to better understand the complexity of inhibiting mTOR signaling in cancer.
Acknowledgments
We thank Dr. John Blenis for providing the pcDNA3-rLuc-PolioIRES-fLuc reporter plasmid and Dr. Qingbai She (University of Kentucky) for providing the HA-4E-BP1/4A expression plasmid and stable HCT116 cells expressing HA-4E-BP1 and HA-4E-BP1/4A.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA133429-01A1 (to T. G.). This work was also supported by American Cancer Society Grant RSG0822001TBE (to T. G.).
- mTOR
- mammalian target of rapamycin
- mTORC1
- mammalian target of rapamycin complex 1
- m7GTP
- 7-methyl-GTP
- PPP
- phosphoprotein phosphatase
- PPM
- metal-dependent protein phosphatase
- OA
- okadaic acid
- TOP
- 5′ terminal oligopyrimidine.
REFERENCES
- 1. Mahoney S. J., Dempsey J. M., Blenis J. (2009) Cell signaling in protein synthesis: ribosome biogenesis and translation initiation and elongation. Prog. Mol. Biol. Transl. Sci. 90, 53–107 [DOI] [PubMed] [Google Scholar]
- 2. Silvera D., Formenti S. C., Schneider R. J. (2010) Translational control in cancer. Nat. Rev. Cancer 10, 254–266 [DOI] [PubMed] [Google Scholar]
- 3. Grzmil M., Hemmings B. A. (2012) Translation regulation as a therapeutic target in cancer. Cancer Res. 72, 3891–3900 [DOI] [PubMed] [Google Scholar]
- 4. Scheper G. C., van der Knaap M. S., Proud C. G. (2007) Translation matters. Protein synthesis defects in inherited disease. Nat. Rev. Genet. 8, 711–723 [DOI] [PubMed] [Google Scholar]
- 5. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 6. Guertin D. A., Sabatini D. M. (2007) Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 [DOI] [PubMed] [Google Scholar]
- 7. Sonenberg N., Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes. Mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fraser C. S. (2009) The molecular basis of translational control. Prog. Mol. Biol. Transl. Sci. 90, 1–51 [DOI] [PubMed] [Google Scholar]
- 9. Fadden P., Haystead T. A., Lawrence J. C., Jr. (1997) Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. J. Biol. Chem. 272, 10240–10247 [DOI] [PubMed] [Google Scholar]
- 10. Gingras A. C., Gygi S. P., Raught B., Polakiewicz R. D., Abraham R. T., Hoekstra M. F., Aebersold R., Sonenberg N. (1999) Regulation of 4E-BP1 phosphorylation. A novel two-step mechanism. Genes Dev. 13, 1422–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. She Q. B., Halilovic E., Ye Q., Zhen W., Shirasawa S., Sasazuki T., Solit D. B., Rosen N. (2010) 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 18, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu J., Weiss H. L., Rychahou P., Jackson L. N., Evers B. M., Gao T. (2009) Loss of PHLPP expression in colon cancer. Role in proliferation and tumorigenesis. Oncogene 28, 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao T., Furnari F., Newton A. C. (2005) PHLPP. A phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 [DOI] [PubMed] [Google Scholar]
- 14. Li X., Liu J., Gao T. (2009) β-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol. Cell. Biol. 29, 6192–6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holz M. K., Ballif B. A., Gygi S. P., Blenis J. (2005) mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580 [DOI] [PubMed] [Google Scholar]
- 16. Liu J., Stevens P. D., Li X., Schmidt M. D., Gao T. (2011) PHLPP-mediated dephosphorylation of S6K1 inhibits protein translation and cell growth. Mol. Cell. Biol. 31, 4917–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brognard J., Sierecki E., Gao T., Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 [DOI] [PubMed] [Google Scholar]
- 18. Choo A. Y., Yoon S. O., Kim S. G., Roux P. P., Blenis J. (2008) Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. U.S.A. 105, 17414–17419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi Y. (2009) Serine/threonine phosphatases. Mechanism through structure. Cell 139, 468–484 [DOI] [PubMed] [Google Scholar]
- 20. Schalm S. S., Fingar D. C., Sabatini D. M., Blenis J. (2003) TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 13, 797–806 [DOI] [PubMed] [Google Scholar]
- 21. Fingar D. C., Salama S., Tsou C., Harlow E., Blenis J. (2002) Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16, 1472–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., Sabatini D. M. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coleman L. J., Peter M. B., Teall T. J., Brannan R. A., Hanby A. M., Honarpisheh H., Shaaban A. M., Smith L., Speirs V., Verghese E. T., McElwaine J. N., Hughes T. A. (2009) Combined analysis of eIF4E and 4E-binding protein expression predicts breast cancer survival and estimates eIF4E activity. Br. J. Cancer 100, 1393–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Virshup D. M., Shenolikar S. (2009) From promiscuity to precision. Protein phosphatases get a makeover. Mol. Cell 33, 537–545 [DOI] [PubMed] [Google Scholar]
- 25. Khoronenkova S. V., Dianova I. I., Ternette N., Kessler B. M., Parsons J. L., Dianov G. L. (2012) ATM-dependent down-regulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol. Cell 45, 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hearst S. M., Gilder A. S., Negi S. S., Davis M. D., George E. M., Whittom A. A., Toyota C. G., Husedzinovic A., Gruss O. J., Hebert M. D. (2009) Cajal-body formation correlates with differential coilin phosphorylation in primary and transformed cell lines. J. Cell Sci. 122, 1872–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Petri S., Grimmler M., Over S., Fischer U., Gruss O. J. (2007) Dephosphorylation of survival motor neurons (SMN) by PPM1G/PP2Cγ governs Cajal body localization and stability of the SMN complex. J. Cell Biol. 179, 451–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kimura H., Takizawa N., Allemand E., Hori T., Iborra F. J., Nozaki N., Muraki M., Hagiwara M., Krainer A. R., Fukagawa T., Okawa K. (2006) A novel histone exchange factor, protein phosphatase 2Cγ, mediates the exchange and dephosphorylation of H2A-H2B. J. Cell Biol. 175, 389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guan L., Song K., Pysz M. A., Curry K. J., Hizli A. A., Danielpour D., Black A. R., Black J. D. (2007) Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. J. Biol. Chem. 282, 14213–14225 [DOI] [PubMed] [Google Scholar]
- 30. Pham F. H., Sugden P. H., Clerk A. (2000) Regulation of protein kinase B and 4E-BP1 by oxidative stress in cardiac myocytes. Circ. Res. 86, 1252–1258 [DOI] [PubMed] [Google Scholar]
- 31. Yanagiya A., Suyama E., Adachi H., Svitkin Y. V., Aza-Blanc P., Imataka H., Mikami S., Martineau Y., Ronai Z. A., Sonenberg N. (2012) Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol. Cell 46, 847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guertin D. A., Sabatini D. M. (2009) The pharmacology of mTOR inhibition. Sci. Signal 2, pe24. [DOI] [PubMed] [Google Scholar]