

Background: Small heterodimer partner (SHP) is a key mediator of bile acid signaling.
Results: Bile acid signal-induced phosphorylation at Thr-55 by protein kinase Cζ is important for SHP-mediated recruitment of chromatin modifiers and histone modifications.
Conclusion: Thr-55 phosphorylation is critical for epigenomic regulation of liver metabolic genes.
Significance: SHP Thr-55 phosphorylation may potentially provide a therapeutic target for bile acid-related diseases.
Keywords: Bile Acid, Fibroblast Growth Factor (FGF), Histone Modification, Liver Metabolism, Post-translational Modification, CDCA, Cyp7a1, FGF19, Epigenomics
Abstract
Bile acids (BAs) are recently recognized key signaling molecules that control integrative metabolism and energy expenditure. BAs activate multiple signaling pathways, including those of nuclear receptors, primarily farnesoid X receptor (FXR), membrane BA receptors, and FXR-induced FGF19 to regulate the fed-state metabolism. Small heterodimer partner (SHP) has been implicated as a key mediator of these BA signaling pathways by recruitment of chromatin modifying proteins, but the key question of how SHP transduces BA signaling into repressive histone modifications at liver metabolic genes remains unknown. Here we show that protein kinase Cζ (PKCζ) is activated by BA or FGF19 and phosphorylates SHP at Thr-55 and that Thr-55 phosphorylation is critical for the epigenomic coordinator functions of SHP. PKCζ is coimmunopreciptitated with SHP and both are recruited to SHP target genes after bile acid or FGF19 treatment. Activated phosphorylated PKCζ and phosphorylated SHP are predominantly located in the nucleus after FGF19 treatment. Phosphorylation at Thr-55 is required for subsequent methylation at Arg-57, a naturally occurring mutation site in metabolic syndrome patients. Thr-55 phosphorylation increases interaction of SHP with chromatin modifiers and their occupancy at selective BA-responsive genes. This molecular cascade leads to repressive modifications of histones at metabolic target genes, and consequently, decreased BA pools and hepatic triglyceride levels. Remarkably, mutation of Thr-55 attenuates these SHP-mediated epigenomic and metabolic effects. This study identifies PKCζ as a novel key upstream regulator of BA-regulated SHP function, revealing the role of Thr-55 phosphorylation in epigenomic regulation of liver metabolism.
Introduction
The function of bile acids (BAs)2 to facilitate digestion of lipid nutrients is well known, but only recently have BAs been recognized as key signaling molecules that control integrative metabolism and energy expenditure (1–4). BAs are endogenous ligands for nuclear BA receptors, primarily FXR, and membrane BA G-protein coupled receptors, including TGR5 (1–3). Furthermore, BA-activated farnesoid X receptor (FXR) induces expression of intestinal fibroblast growth factor 19 (FGF19, FGF15 is the mouse ortholog) that is secreted and acts at the liver (5). After a meal, in addition to activation of FXR, binding of BAs or BA-induced FGF19 to their hepatic membrane receptors triggers signaling cascades that mediate postprandial responses, such as inhibition of BA and glucose synthesis and stimulation of glycogen synthesis (6). However, little is known about how BA signaling is transmitted to control transcriptional programs of fed-state metabolism.
Accumulating evidence indicates that small heterodimer partner (SHP) is a key mediator of BA signaling (5, 7–10). SHP is an orphan nuclear receptor lacking a DNA binding domain and acts as a transcriptional corepressor by inhibiting numerous transcription factors, such as HNF-4, LRH-1, ERRγ, p53, and NF-κB, in diverse biological pathways including metabolism, proliferation, and inflammation (11–14). Of these functions, the role of SHP in maintaining BA homeostasis by inhibiting hepatic BA synthetic Cyp7a1 and Cyp8b1 genes is well established (9, 10, 15). Our group has shown that in response to elevated hepatic BA levels, SHP represses the Cyp7a1 gene by coordinately recruiting repressive chromatin modifying cofactors, HDAC-containing mSin3A and NcoR corepressor complexes, G9a methyltransferase, and the Brm-containing Swi/Snf remodeling complex, resulting in sequential epigenomic modifications at the promoter (16–18). However, the key question of how BAs trigger increased recruitment of SHP-associated chromatin modifiers and consequently epigenomic regulation is not known.
Post-translational modifications (PTMs) of regulatory proteins like SHP profoundly modulate their cellular function by altering protein interactions, nuclear localization, and protein stability. For more than a decade, the prevailing mechanism for increased hepatic SHP activity was its transcriptional induction by FXR (9, 10). We have recently shown that repression activity and protein stability are also regulated by BA signal-induced PTMs of SHP (8, 19). Relevant to this study, SHP is methylated at Arg-57 by PRMT5 methyltransferase upon BA treatment and this methylation enhances SHP activity by selectively increasing its interaction with repressive chromatin modifiers (19). Remarkably, a naturally occurring mutation of Arg-57 to Trp in SHP is associated with metabolic syndrome (20). Here we show that an atypical protein kinase Cζ (PKCζ) is activated by BA or FGF19 signaling and phosphorylates SHP at Thr-55 and that Thr-55 phosphorylation of SHP is important for the epigenomic coordinator function of SHP in the regulation of fed-state liver metabolism.
EXPERIMENTAL PROCEDURES
Reagents
Antibodies were purchased for SHP (sc-30169), lamin A (sc-20680), tubulin (sc-8035), HDAC1 (sc-7872), Brm (sc-6450), LRH-1 (sc-5995X), HNF-4 (sc-8987), PGC1α (sc-13067), RNA pol II (sc-9001), GFP, (sc-8334), β-tubulin (sc-5274), and actin (sc-1616) from Santa Cruz Biotechnology; for Foxa-2 (number 3143), phospho-Thr (number 9381), phospho-PKCζ (number 9378), and PKCζ (number 9368) from Cell Signaling; for H3K9/K14-Ac (number 06-599), H3K9-me2 (number 07-030), H3K4-me3 (number 07-521), and G9a from Millipore; and M2 from Sigma. The siRNA for PKCζ was purchased from Ambion and siRNA for ERK and control siRNA were purchased from Applied Biosystems. A phospho-Thr-55-specific SHP antibody was produced commercially (Abmart, Inc.).
Animal Experiments
Male BALB/c male mice (8–12 weeks old) were injected via the tail vein with Ad-FLAG-SHP WT or T55A mutant (0.5–1.0 × 109 active viral particles in 200 μl of PBS), which resulted in SHP levels about 10-fold greater than endogenous SHP (data not shown). This is comparable with 5–6-fold increases in SHP compared with endogenous levels in SHP liver-specific transgenic mice and to hepatic SHP levels in response to BA signaling (21). The construction of Ad-FLAG-SHP WT has been described previously (19). The adenovirus selectively infects the liver so that little or no expression occurs in other tissues (22). Five to 7 days after infection, the mice were fasted for 5–6 h and treated for 15 min with 1 mg/kg of FGF19 injected i.v. or fed 0.5% CA-chow (Harlan Teklad TD05271) for 3 h. BA pools were measured by colorimetric analysis (Trinity Biotechnology). Liver triglyceride (TG) levels were measured using a kit (TR0100, Sigma). For glucose tolerance test, mice were fasted for 6 h and injected intraperitoneally with glucose solution (Sigma, 2 g/kg of body weight), and blood glucose levels were measured using a glucometer (Roche Applied Science). All animal use and adenoviral protocols were approved by the Animal Care and Use and Biosafety Committees at the University of Illinois at Urbana-Champaign.
Tandem Mass Spectrometry Analysis
FLAG-human SHP was adenovirally expressed in HepG2 cells and 48 h later, the cells were treated with 5 μm MG132 for 4 h to inhibit proteasomal degradation of SHP (8) and then treated with chenodeoxycholic acid (CDCA) for 1 h. FLAG-SHP was isolated using M2-agarose, proteins were separated and visualized by colloidal staining; and bands containing FLAG-SHP were subjected to MS/MS analysis.
IP/IB-based Phosphorylation Assay
Endogenous SHP or FLAG-SHP was immunoprecipitated with 1 μg of SHP antibody or M2 antibody, respectively, for 30 min in IP buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 0.5% Nonidet P-40, 2 mm EDTA, and 5% glycerol). Then, 35 μl of 25% protein G-agarose was added, and 2 h later samples were washed with IP buffer, and SHP phosphorylated at Thr was detected by IB using phospho-Thr antibody or specific phospho-Thr-55 antibody.
In Vitro PKCζ Assay
HepG2 cells were infected with Ad-FLAG-SHP WT or T55A, and 24 h later, cell extracts were prepared in lysis buffer (1% Nonidet P-40, 50 mm Tris-HCl, pH 8.0, 150 mm NaCl). FLAG-SHP was isolated by affinity to M2-agarose beads and the beads were washed with the lysis buffer and then with kinase buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 mm DTT). FLAG-SHP bound to M2-agarose was incubated with 20 μm ATP and 20 ng of purified PKCζ (Millipore) in kinase buffer at 30 °C for 30 min. To determine the effect of FGF19 or CDCA treatment on PKCζ activity, HepG2 cells were infected with Ad-PKCζ (23) and treated with vehicle or FGF19 or CDCA for 15 min, PKCζ was immunoprecipitated, and kinase activity was determined in in vitro assays with purified FLAG-SHP as substrate.
Co-IP, GST Pull-down, and Reporter Assays
Cell extracts were prepared in co-IP buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 2 mm EDTA, 0.3% Nonidet P-40, 10% glycerol) and incubated with 1–2 μg of antibodies for 30 min, and 35 μl of 25% protein G-agarose was added. Two h later, samples were washed with the co-IP buffer and interacting proteins were detected by IB. GST pull-down and reporter assays were performed as described previously (19, 24, 25).
Immunofluorescence Cellular Localization Assays
HepG2 cells were incubated with serum-free media overnight and treated with vehicle or FGF19 for 15 min. After fixation with 4% paraformaldehyde for 25 min, cells were treated with 0.5% Triton X-100/PBS for 15 min, incubated with primary antibody (dilution 1:200 in PBS) for 2 h and secondary antibody Texas Red goat anti-rabbit IgG (dilution 1:200 in PBS) for 1 h. Nuclei were labeled with DAPI (dilution 1:1000) for 5 min. Images were taken and processed using ZEN software (Zeiss LSM700).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed as previously described (17–19, 25, 26). Briefly, livers were finely minced and incubated in PBS containing 1% formaldehyde for 10 min, and glycine was added to stop the reaction. Cells were resuspended in hypotonic buffer and lysed by homogenization. Nuclei were pelleted and sonicated. The chromatin sample was precleared and chromatin was immunoprecipitated using 1–1.5 μg of antibody. For re-ChIP assays, FLAG-SHP was immunoprecipitated first by M2 antibody, eluted by adding 50 μl of 10 mm DTT at 37 °C for 30 min. Then, chromatin samples were diluted (20-fold) with buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100) and re-precipitated using antibodies as indicated. Chromatin was extensively washed, eluted, and the amounts of genomic DNA were determined by PCR. Primer sequences for semi-qPCR have been previously described (19) and qPCR primer sequences are available upon request.
RT-qPCR
Total RNA was isolated and cDNA was synthesized using a reverse transcriptase kit (Promega), and RT-qPCR was performed. Primer sequences are available upon request.
RESULTS
SHP Is Phosphorylated at Thr-55 in Response to BA or FGF19 Signaling
To identify BA signal-induced phosphorylation site(s) in SHP, purified SHP was analyzed by tandem mass spectrometry. SHP was phosphorylated at Thr-55 after treatment of HepG2 cells with a primary BA, CDCA (Fig. 1A). Treatment with CDCA or FGF19 resulted in increased Thr phosphorylation of FLAG-SHP (Fig. 1B), but these effects were not observed with GW4064, a synthetic agonist for FXR (Fig. 1C). Thr phosphorylation of endogenous SHP was also increased in HepG2 cells after CDCA or FGF19 treatments (Fig. 1D) and in mouse liver after FGF19 injection (Fig. 1E). A large fraction, roughly 70%, of total hepatic SHP proteins was phosphorylated after FGF19 treatment (data not shown). Importantly, mutation of Thr-55 (T55A) abolished phosphorylation of SHP, identifying Thr-55 as the site of BA-induced Thr phosphorylation (Fig. 1F).
FIGURE 1.

Phosphorylation of SHP at Thr-55 in response to BA or FGF19 signaling. A, tandem mass (MS/MS) spectrum of the SHP peptide showing phosphorylation of Thr-55 (T∧, phosphorylated Thr-55; T#, Thr-55 with loss of water; R*, methylated Arg-57). FLAG-SHP expressed in HepG2 cells treated with CDCA was isolated, visualized by colloidal staining, and excised for MS/MS analysis. B and C, phospho-Thr levels of FLAG-SHP in HepG2 cells treated with FGF19, CDCA, or GW4064 for 1 h were detected by IP of FLAG-SHP followed by IB using phospho-Thr antibody. D and E, phospho-Thr levels of endogenous SHP in mouse liver or HepG2 cells treated with FGF19 or CDCA are shown. In B and E, bands were quantified and phosphor-Thr SHP levels relative to untreated cells (CTL) shown (right panel) and statistical significance was determined by the Student's t test, * indicates p < 0.05, S.E., n = 3. F, FLAG-SHP WT or T55A was expressed in HepG2 cells and phospho-Thr SHP was detected by IP/IB analysis. G and H, the experimental outline is shown at the top. Phospho-Thr-55 of SHP in liver extracts was detected by IP/IB analysis using general phosphor-Thr antibody (G) or phosphor-Thr-55-specific SHP antibody (H). FLAG-SHP and control GFP from the adenoviral infection are shown at the bottom. I, phosphorylation of endogenous SHP at Thr-55 in mouse liver extracts was detected by IB using a phosphor-Thr-55-specific SHP antibody. J, alignment of the SHP region containing Thr-55 (underlined) from various species.
To determine whether SHP is phosphorylated in vivo at Thr-55, a phospho-Thr-55-specific SHP antibody was developed. To assess the specificity of antibody, mice were injected with adenoviral vectors expressing SHP wild type (WT) or a phosphorylation-defective T55A mutant and phosphorylation of SHP was detected with a phospho-Thr antibody or with the phospho-Thr-55-specific SHP antibody. Robust Thr phosphorylation was detected in FGF19-treated mice expressing FLAG-SHP WT, but was not detectable in mice expressing the T55A mutant (Fig. 1G). Importantly, phosphorylation in WT, but not in the T55A mutant, was detected by the phosphor-Thr-55-specific SHP antibody, demonstrating the specificity of this antibody (Fig. 1H) and confirming that Thr-55 is the major site of phosphorylation in exogenously expressed FLAG-SHP in vivo. Using this site-specific antibody, we also confirmed that endogenous SHP in mouse liver is phosphorylated at Thr-55 after FGF19 treatment (Fig. 1I) and that Thr-55 phosphorylation of FLAG-SHP was increased in response to FGF19 or CDCA in HepG2 cells (data not shown). These results demonstrate that SHP is phosphorylated at Thr-55 in response to BA or FGF19. Notably, the Thr-55 is well conserved in vertebrates (Fig. 1J), suggesting that phosphorylation at Thr-55 of SHP may be functionally important.
Thr-55 in SHP Is Phosphorylated by PKCζ upon BA/FGF19 Signaling
To identify the kinase(s) catalyzing the Thr-55 phosphorylation of SHP, we utilized both pharmacological inhibitors and siRNA. We first used inhibitors of kinases potentially involved in BA/FGF19 signaling pathways (4, 6, 27). Treatment with inhibitors of PI3K or PKCζ decreased Thr phosphorylation of SHP induced by FGF19 or CDCA (Fig. 2A). In contrast, treatment with inhibitors of PKC (Fig. 2A), ERK, JNK, PKB (Fig. 2B), or p38 kinase (data not shown) did not reduce the phosphorylation levels. Because some of these inhibitors are not specific and ERK has been shown to be activated by BA or FGF19 treatment (6, 27), we confirmed our findings using siRNA for PKCζ or ERK. Down-regulation of PKCζ abolished Thr phosphorylation by FGF19, whereas phosphorylation of SHP was not inhibited by down-regulation of ERK (Fig. 2C). Consistently, levels of the active phosphorylated form of PKCζ were elevated in HepG2 cells or in mice after BA or FGF19 treatment (Fig. 2D). These results suggest that the PI3K/PKCζ pathway is activated upon BA signaling and that PKCζ phosphorylates SHP at Thr-55.
FIGURE 2.

PKCζ phosphorylates SHP at Thr-55 upon BA/FGF19 signaling. A and B, HepG2 cells infected with Ad-FLAG-SHP were treated with each kinase inhibitor as indicated, for PKC (G06983, 1 mm), PI3K (wortmannin, 100 nm), PKCζ (myristoylated PKCζ pseudo-substrate, 20 μm; or PKCζ pseudo-substrate, 20 mm), p38 (SB203580, 5 μm), ERK (PD98059, 20 μm), or JNK (SP600125, 20 μm) for 30 min and then further treated with FGF19 or CDCA for 15 min. Phospho-Thr levels of SHP were detected by IP/IB analysis. Consistent results were observed in 2–4 experiments. C, HepG2 cells were transfected with siRNA as indicated and 24 h later, cells were infected with Ad-FLAG-SHP, and then 24 h later treated with FGF19 for 15 min. Phospho-Thr levels of SHP were detected by IP/IB analysis. In the right panel, phospho-Thr SHP relative to the untreated control siRNA in lane 1 are shown and statistical significance determined by the Student's t test, * and **, and NS indicate p < 0.05, p < 0.01, and statistically nonsignificant, respectively, S.E., n = 3. D, HepG2 cells of mice were treated with FGF19 or CDCA for 15 min and phospho- or total PKCζ levels were detected. E, co-IP, the interaction of FLAG-SHP with PKCζ was detected by co-IP assays in HepG2 cells. F, GST pull-down, schematic diagrams of domains of SHP are shown at the top. Amounts of input GST or GST-SHP proteins are shown (middle) and PKCζ proteins bound to GST-SHP proteins were detected (bottom). G, experimental outline for the in vitro kinase assay is shown (left panel). Phospho-Thr-55 levels of FLAG-SHP WT were detected by IP/IB or IB analysis (right panel). H, experimental outline for the in vitro kinase assay is shown (top) and phospho-Thr levels of FLAG-SHP WT or T55A mutant were detected by IP/IB analysis (upper panel). Total PKCζ and SHP levels are shown below.
To further support this conclusion, the interaction of SHP with PKCζ was studied. In co-IP studies, FGF19 treatment increased the interaction of SHP with PKCζ and this interaction was not observed for the phosphorylation-defective T55A mutant (Fig. 2E). In vitro GST pulldown assays showed that SHP directly interacted with PKCζ through the N-terminal domain of SHP (Fig. 2F).
To determine whether PKCζ is activated by BA or FGF19 treatment, PKCζ was overexpressed in HepG2 cells and cells were treated with vehicle, FGF19, or CDCA. PKCζ was immunoprecipitated and its activity was assessed in in vitro kinase assays (Fig. 2G, left panel). In response to either FGF19 or CDCA treatment, phosphorylation of both PKCζ and SHP, detected by the phosphor-Thr-55-specific antibody, was substantially increased (Fig. 2G, right panel). Moreover, Thr phosphorylation of SHP was observed for FLAG-SHP WT, but not the T55A mutant (Fig. 2H). These results demonstrate that BA signaling results in activation of PKCζ and phosphorylation of SHP at Thr-55.
Effects of FGF19 or BA on Nuclear Localization of PKCζ and SHP
We next examined the effect of FGF19 or BA treatment on the cellular localization of phosphorylated PKCζ and SHP by immunofluorescence studies. PKCζ and phosphorylated PKCζ were localized exclusively in the cytoplasm in untreated cells and FGF19 treatment resulted in a dramatic increase in the levels of both forms of PKCζ in the nucleus (Fig. 3, A and B). SHP was localized in both the cytoplasm and nucleus and FGF19 treatment resulted in modest increases in nuclear abundance (Fig. 3C). Strikingly, however, low levels of SHP phosphorylated at Thr-55 were detected in the cytoplasm in untreated cells, but levels were dramatically increased and nearly exclusively localized in the nucleus after FGF19 treatment (Fig. 3D). These results confirm that PKCζ is activated by FGF19 signaling and suggest that SHP is phosphorylated by PKCζ in the nucleus. Consistent with these results, in biochemical fractionation studies in mice in vivo, SHP WT was localized in both the cytoplasm and nucleus, but the T55A phosphorylation-defective SHP mutant was predominantly located in the cytoplasm (data not shown). These results suggest that the phosphorylation of SHP at Thr-55 is likely important for retention of SHP in the nucleus.
FIGURE 3.

Effects of FGF19 or BA signaling on nuclear localization of PKCζ and SHP. A–D, immunofluorescence cellular localization study. HepG2 cells were treated with vehicle (Veh) or FGF19 for 15 min and localization of PKCζ and SHP was detected by antibody against phosphor-PKCζ, PKCζ, phosphor-SHP, or SHP. Nuclei were stained with DAPI. Merged images are shown below. E and F, ChIP: mice were treated with FGF19 (15 min) or fed CA chow (3 h), livers were pooled from 3 mice, and ChIP assays were performed.
The nuclear localization of activated PKCζ suggested the possibility that PKCζ might be co-recruited with SHP to its target genes. To test this possibility, liver ChIP assays were done in mice treated with FGF19 or CA. Recruitment of PKCζ was compared with that of ERK and PRMT5, which modulate PTM of SHP in response to BA or FGF19 signaling (8, 19). In mice treated with FGF19 or fed CA chow, occupancies of SHP, PKCζ, PRMT5, and ERK (8, 19) were increased at the Cyp7a1 promoter (Fig. 3, E and F). Conversely, occupancy of RNA polymerase II was decreased, consistent with the suppression of Cyp7a1 expression by SHP. These results, collectively with cellular localization studies and co-IP protein interaction studies above, suggest that PKCζ is activated by BA or FGF19 signaling and phosphorylates SHP at Thr-55, possibly by co-localizing with SHP at its target genes.
Thr-55 Phosphorylation Is Essential for Arg-57 Methylation
We showed recently that SHP is methylated at Arg-57 by PRMT5 in response to BA signaling and that this methylation is important for SHP repression activity (19). We, therefore, further examined possible functional cross-talk between Arg-57 methylation and Thr-55 phosphorylation. Mutation of Thr-55 (T55A) largely abolished methylation of Arg-57 after FGF19 treatment, whereas mutation of Arg-57 (R57W) did not affect Thr-55 phosphorylation (Fig. 4A). Similar results were observed when the cells were treated with CDCA (Fig. 4B). Consistent with these results, the interaction of SHP with PRMT5, which catalyzes methylation at Arg-57, was impaired by mutation of Thr-55, as detected in co-IP studies (Fig. 4, C and D). These results indicate that PKCζ-mediated Thr-55 phosphorylation is likely a key upstream event that is required for Arg-57 methylation upon BA signaling.
FIGURE 4.
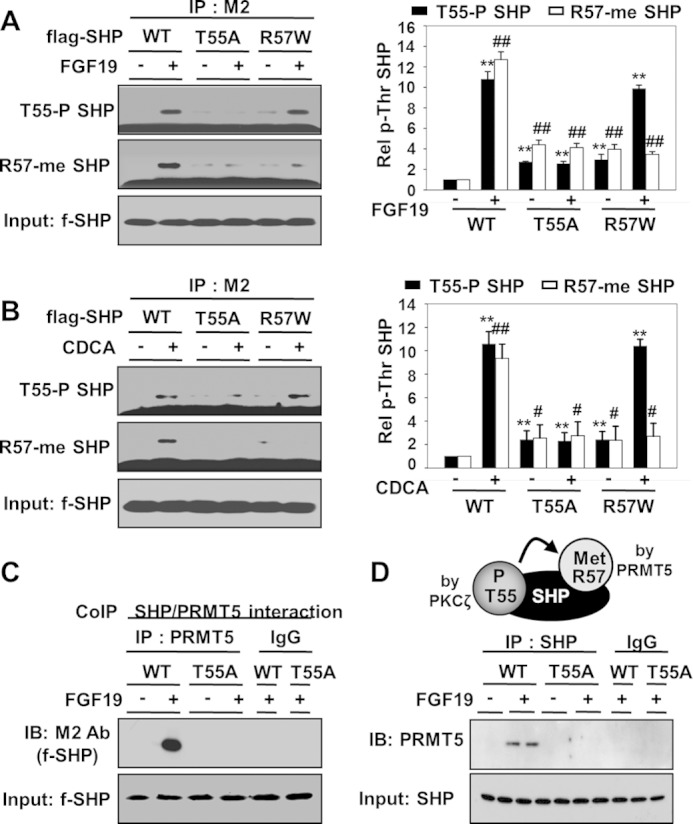
Thr-55 phosphorylation of SHP is required for Arg-57 methylation. A and B, HepG2 cells were infected with Ad-FLAG-SHP WT, T55A, or R57W, and treated with FGF19 or CDCA for 15 min, and phosphorylated Thr or methylated Arg levels were detected by IP/IB analysis. In the right panels, levels of phospho-Thr SHP or methyl-Arg SHP levels were quantified relative to WT are shown and statistical significance was determined by the Student's t test, *, indicates p < 0.05, S.E., n = 3. C and D, co-IP: HepG2 cells were transfected with an expression plasmid for PRMT5 and FLAG-SHP WT or T55A mutant and then treated with FGF19. Interaction of SHP with PRMT5 was detected by co-IP. On the top of D is a diagram illustrating the dependence of Arg-57 methylation by PRMT5 on Thr-55 phosphorylation by PKCζ.
Thr-55 Phosphorylation Is Important for SHP Repression Activity
To determine the functional relevance of Thr-55 phosphorylation, cell-based reporter assays were performed. Expression of SHP WT decreased HNF-4-mediated transactivation in HepG2 cells treated with either FGF19 (Fig. 5A) or CDCA (Fig. 5B). These SHP-mediated repression effects were substantially impaired for the phosphorylation-defective T55A mutant, but, in contrast, enhanced with the phosphorylation mimic T55D mutant. Similar results were observed with a promoter-luciferase reporter for Cyp7a1 (Fig. 5, C and D). Because Thr-55 phosphorylation is required for Arg-57 methylation (Fig. 4), the effects of the Thr mutation were compared with Arg-57 mutation. Increasing amounts of SHP WT decreased LRH-1 transactivation and this repression was reversed by the R57K mutation similar to, but to a slightly less extent than, the reversal by the T55A mutation (Fig. 5, E and F). Similar amounts of SHP WT and mutant proteins were expressed in cell-based reporter assays (Fig. 5G). These results suggest that Thr-55 phosphorylation augments the SHP repression activity and this may be, at least in part, mediated by methylation at Arg-57 that is dependent on Thr-55 phosphorylation.
FIGURE 5.
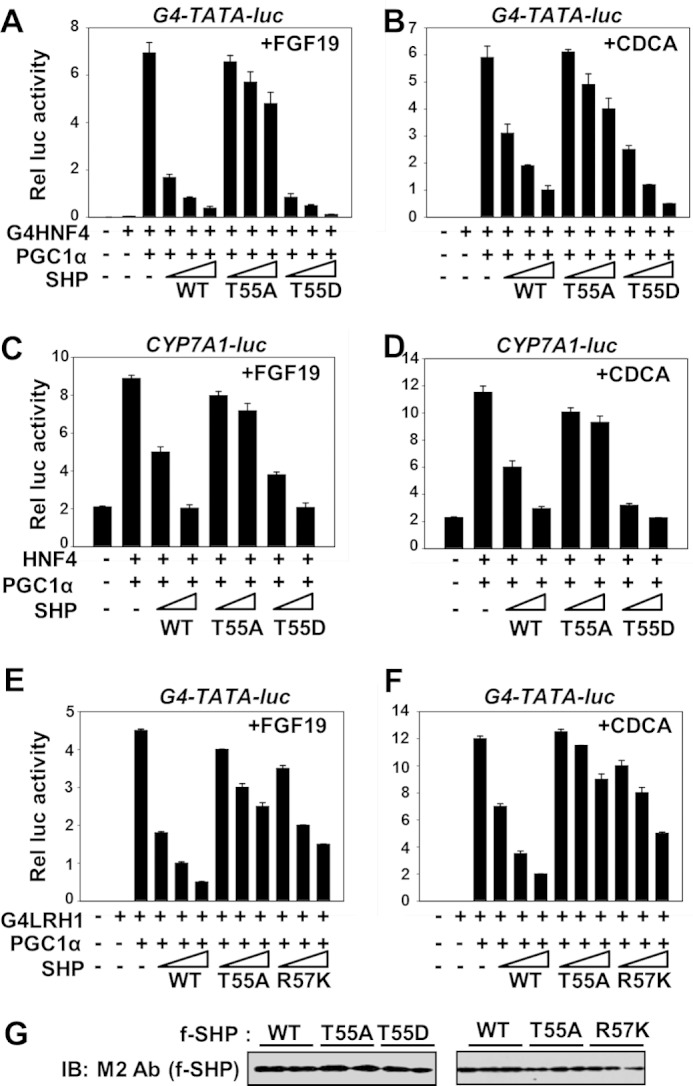
Thr-55 phosphorylation is important for SHP repression activity. A–F, HepG2 cells were transfected with plasmids as indicated and treated with either FGF19 (8 h) or CDCA overnight and reporter assays were performed. The triangle indicates increasing amounts of the SHP expression plasmids. The values for firefly luciferase activities were normalized by β-galactosidase activities. The mean ± S.E. are plotted (n = 3). G, expression levels of SHP WT and the mutants from cell extracts are shown.
Thr-55 Phosphorylation Selectively Increases Occupancy of SHP-interacting Chromatin Modifiers at BA-responsive Target Genes
To elucidate the molecular basis underlying the enhanced SHP repression activity by Thr-55 phosphorylation, we first examined the effect of mutation of Thr-55 of SHP on interaction with its known transcription regulators. FGF19 treatment dramatically increased the interaction with known SHP-interacting transcriptional factors LRH-1 and HNF-4, coactivator PGC-1α, and chromatin modifiers G9a and Brm (Fig. 6A). With the exception of G9a, SHP interaction with these proteins was nearly abolished by mutation of Thr-55 (Fig. 6A). These results suggest that Thr-55 phosphorylation is critical for interaction of SHP with transcription factors and some of its associated chromatin modifying proteins.
FIGURE 6.

Thr-55 phosphorylation increases SHP interaction with repressive chromatin modifiers and their recruitment to BA-responsive target genes. A, co-IP: transcriptional regulators were immunoprecipitated and the presence of FLAG-SHP was detected by M2 antibody. B–F, sequential liver chromatin IP. B, experimental outline. C–F, chromatin was immunoprecipitated with M2 antibody, eluted, and re-precipitated with antibody as indicated. Precipitated genomic DNA was quantified by semi-qPCR.
Because the interaction of SHP with LRH-1/HNF-4, which should increase SHP recruitment at its target genes, was decreased by mutation of Thr-55 (data not shown), we next determined the effects of mutation of Thr-55 on promoter occupancy of SHP (Fig. 6B). Occupancies of FLAG-SHP were examined at BA-responsive SHP target genes, Cyp7a1, Cyp8b1, and Shp genes, which contain LRH-1/HNF-4 sites at their promoters (15, 18, 19). FLAG-SHP was immunoprecipitated with M2 antibody first and then re-immunoprecipitated with SHP antibody. Treatment with FGF19 or CA feeding increased SHP occupancy at the Cyp7a1 and Cyp8b1 genes, which was decreased with the T55A mutant (Fig. 6, C and D). In contrast, mutation of Thr-55 did not substantially decrease SHP occupancy at the Shp promoter. These results indicate that Thr-55 phosphorylation is important for recruitment of SHP in a gene-specific manner.
We next determined the effects of mutation of Thr-55 on recruitment of SHP-associated transcriptional regulators. Although treatment with FGF19 or CA did not change HNF-4 occupancy, the occupancy of HDAC1/2, G9a, and Brm was increased with FLAG-SHP WT, but recruitment of HDAC1/2 and Brm was reduced in mice expressing the T55A mutant (Fig. 6, E and F). In contrast, the mutation did not affect recruitment of G9a, which is consistent with the co-IP studies presented above (Fig. 6A). SHP inhibits its own expression by recruiting these repressive histone modifiers, including Brm, to its own promoter (18). Indeed, FGF19 treatment or CA feeding increased occupancy of G9a and Brm, at the Shp promoter and the Thr-55 mutation had little effect on these changes (Fig. 6, E and F). These results collectively demonstrate that Thr-55 phosphorylation of SHP is important for recruitment of chromatin modifiers to a subset of BA target genes.
Thr-55 Phosphorylation of SHP Is Important for Epigenomic Events at a Subset of BA-responsive Genes
We further investigated in vivo the effects of mutation of Thr-55 on histone modifications at SHP target genes. BA-responsive SHP target genes involved in BA, fatty acid, and glucose metabolism, Cyp7a1, Cyp8b1, Srebp-1c, and Pepck, were examined (15, 18, 19). Upon FGF19 treatment, levels of histone H3 acetylated at K9/14 and tri-methylated at K4, two well known gene activation histone marks (28), were decreased at all of these genes in mice expressing SHP WT, but not in mice expressing the T55A mutant (Fig. 7A). Interestingly, di-methylated histone H3K9 (K9-me2), a known gene repression mark catalyzed by G9a (17, 28, 29), was increased after FGF19 treatment in both SHP WT and T55A (Fig. 7A). In contrast, at the Shp promoter, levels of gene activation histone marks, H3K9/14-Ac and H3K4-me3, were increased and levels of a gene repression mark, H3K9-me2, were decreased and the T55A mutant did not have marked effects on histone modifications (Fig. 7A). Strikingly, the genes overall epigenomic events for mice fed CA chow (Fig. 7B) were similar to those observed after FGF19 treatment. The occupancy of SHP and chromatin modifying factors and histone modifications were confirmed at the Cyp7a1 promoter using ChIP-qPCR analysis (Fig. 7C, D). Collectively, these findings indicate that Thr-55 phosphorylation of SHP is important for BA signal-induced epigenomic regulation of a subset of liver metabolic genes.
FIGURE 7.

Thr-55 phosphorylation of SHP is critical for epigenomic events at liver metabolic genes. Mice were tail vein injected with Ad-FLAG-SHP WT or T55A and 7 days later, mice were treated with FGF19 or fed CA chow, and liver ChIP assays were performed. Chromatin was immunoprecipitated with M2 antibody first and re-precipitated with antibody as indicated. Precipitated DNA was quantified by semi-qPCR (A and B) and qPCR (C and D).
Functional Consequences of Thr-55 Phosphorylation of SHP in Vivo
Finally, we determined the functional consequences of mutation of Thr-55 in SHP in vivo (Fig. 8A). Acute hepatic overexpression of SHP WT or T55A resulted in repression of the BA-responsive target genes involved in BA synthesis and transport, lipogenesis, and gluconeogenesis, whereas expression increased for genes involved in FA transport and β-oxidation (Fig. 8B). Remarkably, the effects of mutation of Thr-55 were gene-dependent. For most genes, the T55A mutation at least partially reversed the induction or repression mediated by SHP WT (Fig. 8B). For repressed BA synthetic and lipogenic genes, expression with T55A was greater than with the Ad-empty control, suggesting that T55A acts in a dominant negative manner to inhibit endogenous SHP activity. In contrast, for a subset of genes, Bsep, CytC, CD36, and Srb1, the SHP effects were not significantly affected by mutation of Thr-55. Consistent with these patterns of gene expression, acute overexpression of SHP WT decreased the BA pool sizes and hepatic TG levels, whereas these effects were not observed for the Thr-55 mutant (Fig. 8C). Notably in contrast, the effects of the Thr-55 mutation on expression of gluconeogenic genes, Pepck and Glc-6-pase, were absent or relatively modest compared with the genes involved in BA and FA synthesis. In line with these results, glucose tolerance was improved in mice overexpressing SHP WT and was still markedly improved with the T55A mutant (Fig. 8D). These findings, together with the in vitro and in vivo studies, indicate that Thr-55 phosphorylation of SHP by PKCζ is important for transmitting BA signals, resulting in selective transcriptional and metabolic responses in liver.
FIGURE 8.

Functional consequences of Thr-55 phosphorylation of SHP in vivo. A, experimental outline is shown at the top. B, the mRNA levels of the indicated genes were determined by RT-qPCR. C, effects of mutation of Thr-55 on BA pools and hepatic TG levels are shown. D, a glucose tolerance test was performed and serum glucose levels were detected at the indicated times. Statistical significance was determined by the Student's t test, *, **, and NS indicate p < 0.05, p < 0.01, and statistically nonsignificant, respectively, S.E. (n = 5).
DISCUSSION
This study demonstrates the critical role of phosphorylation of SHP by PKCζ at a single site, Thr-55, in epigenomic regulation of the fed-state hepatic metabolism. As summarized in Fig. 9, binding of BA or FGF19 to their respective membrane receptors triggers signaling cascades in hepatocytes, which results in the activation of downstream signaling pathways including the PI3K/PKCζ pathway. Although activated PKCζ is present in the cytoplasm as well as the nucleus, it is not clear whether PKCζ is activated before or after translocation into the nucleus in response to BA signaling. BA also binds to FXR and induces BA-responsive genes, including Shp. Activated PKCζ, localized in the nucleus, phosphorylates SHP at Thr-55 and possibly other regulatory proteins as well. Thr-55 phosphorylation increases the SHP interaction with PRMT5, which is required for subsequent Arg-57 methylation. These sequential PTMs selectively increase the SHP interaction with repressive chromatin modifying cofactors, HDACs and Brm, and recruitment of these proteins to a subset of BA-responsive genes, resulting in histone modifications and gene repression (16–18). Phosphorylated PKCζ, as well as PRMT5, is also recruited to SHP target genes and may be in the same SHP complex that contains chromatin modifiers at BA-responsive SHP target genes but this has not been established. PKCζ and PRMT5 may catalyze PTMs of histones in addition to regulator factors. Epigenomic events mediated by signal-induced PTMs of SHP contribute to postprandial responses including decreased BA and TG levels.
FIGURE 9.
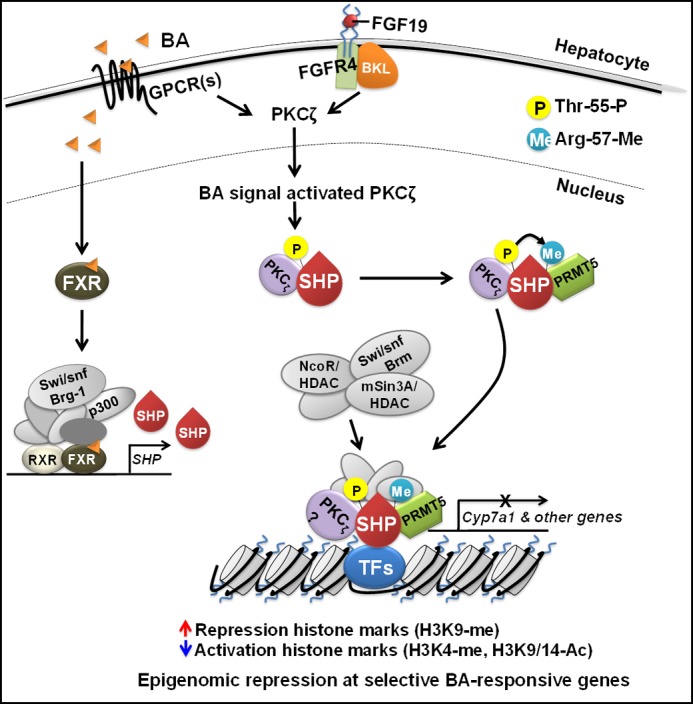
Proposed model. Phosphorylation of SHP by PKCζ at a single site is important for translating BA or FGF19 signaling into epigenomic events at a subset of liver metabolic genes. Binding of BA or FGF19 to their respective membrane receptors triggers signaling cascades in hepatocytes, which results in nuclear translocation and activation of PKCζ. BA also binds to FXR and induces BA-responsive genes including Shp. Activated PKCζ phosphorylates SHP at Thr-55. Thr-55 phosphorylation increases the SHP interaction with PRMT5, which is required for subsequent Arg-57 methylation. These sequential PTMs selectively increase the SHP interaction with repressive chromatin modifying cofactors, HDACs and Brm, and the recruitment of these proteins to a subset of BA-responsive genes, resulting in histone modifications and gene repression. PKCζ is also recruited to SHP target genes, possibly in the same complex that contains PRMT5 and the chromatin modifiers (16–18).
This study reveals several intriguing features of BA signaling pathways and the PTMs that modulate SHP activity. First, BA and FGF19 signaling both resulted in Thr-55 phosphorylation by PKCζ, although different hepatic membrane receptors initiate their signaling cascades, which suggests that these two pathways converge before PKCζ phosphorylation of Thr-55. In addition, epigenomic events triggered by BA or FGF19 signaling at the selected SHP target genes tested were strikingly similar. Second, Thr-55 phosphorylation is essential for Arg-57 methylation. We recently showed that Arg-57 methylation by PRMT5 methyltransferase is important for enhancing SHP repression activity (19). Interestingly, a naturally occurring mutation of Arg-57 to Trp in SHP is present in human metabolic syndrome patients (20). Third, a recent study of transgenic mice has shown that PKCζ functions as a negative regulator of obesity-induced inflammation and counteracts liver steatosis (30). This study is in line with our study showing that PKCζ-mediated phosphorylation of SHP is important for reduction of hepatic TG levels. Fourth, PKCζ was identified for the first time as a key regulator of FGF19 as well as BA signaling in hepatocytes. Our findings indicated that PKCζ is localized in the cytoplasm and FGF19 treatment dramatically increases phosphorylation and nuclear abundance of PKCζ. Consistent with these results, phosphorylated PKCζ was predominantly located in the nucleus. Moreover, SHP phosphorylated at Thr-55 was also largely detected in the nucleus. These findings, together with the increased occupancy of PKCζ at the Cyp7a1 promoter, suggest that phosphorylation of SHP by activated PKCζ likely occurs in the nucleus at the SHP target genes. With regard to PKCζ in BA-regulated hepatic function, a recent study has shown that overexpression of a membrane ATPase, ATP8B1, results in PKCζ-mediated phosphorylation of FXR with enhanced activity and nuclear localization upon BA signaling (31). Our findings, together with this previous study, suggest that PKCζ may coordinately regulate the activity of two key mediators of BA signaling, FXR and SHP. It will be interesting to see whether PKCζ and PKCζ-mediated PTMs of these regulators play a role in the development of BA-related diseases including cholestasis, steatosis, and cancer.
The reported studies on the role of SHP in hepatic lipid metabolism have been inconsistent. Our current study shows that adenoviral-mediated acute liver-specific overexpression of SHP WT decreased hepatic TG levels and BA pools and that PKCζ-mediated phosphorylation of SHP is important for these effects. In line with our findings, it was shown that BAs reduce TG levels by activating the FXR/SHP pathway and that SHP is essential for BA-mediated down-regulation of hepatic lipogenesis by inhibiting expression of Srebp-1c, a key lipogenic activator (7). In contrast, chronic elevation of hepatic SHP levels in transgenic mice resulted in decreased BA pools and increased liver TG levels (21), whereas SHP-KO mice were lean and had increased (14) or unchanged insulin sensitivity (32). These differences may be due to acute versus chronic manipulation of SHP levels in mice, to mixed genetic backgrounds of transgenic mice, and also to effects from non-hepatic tissues on the overall energy balance and metabolic outcomes in the whole body SHP-KO mice. Inducible liver-specific mouse models will be valuable to precisely assess these important issues.
An unexpected effect of expression in the liver of the either WT or SHP mutated at Thr-55 was improved glucose tolerance. Glucose tolerance is primarily mediated by glucose uptake in peripheral tissues, so these results suggest that the several days of altered metabolism in the liver indirectly affects the glucose sensitivity of peripheral tissues. Further studies will be required to determine the mechanism of this effect.
Consistent with the role of SHP in recruiting chromatin-modifying cofactors (16–18, 33), occupancy of HDACs and Brm at the tested BA-responsive genes was indeed substantially decreased by mutation of Thr-55. However, it was surprising that the occupancy of G9a was not affected by the Thr-55 mutation. The underlying mechanisms are not clear but other BA signal-induced PTMs of SHP may be important for G9a recruitment. PTMs of regulatory proteins like SHP change the conformation of the protein interface and thus may selectively alter interaction with transcriptional regulatory proteins, which then may contribute to gene-specific regulation. If the regulation of a subset of BA-responsive SHP target genes is more dependent on promoter occupancy of HDACs and Brm, compared with G9a, then these genes would be selectively dependent on Thr-55 phosphorylation of SHP. Future studies will be required to determine the molecular basis of gene-specific regulation by BA signal-induced PTMs of SHP and other key transcriptional regulators.
Epigenomics has emerged as an important aspect of regulation of many biological processes including liver BA metabolism (33). In addition to our studies (16–18) described above, other recent studies have demonstrated that chromatin-modifying cofactors have a critical role in maintaining BA/cholesterol homeostasis. MLL3, a histone lysine methyltransferase, functions as a critical coactivator of FXR and catalyzes trimethylation of K4 of histone H3 at FXR target genes, including the Shp gene and remarkably, inactivation of MLL3 resulted in disrupted BA homeostasis in vivo (34, 35). Consistent with these findings, occupancies of MLL3 and H3K4 methylation at BA transporter genes were decreased in a mouse model of liver cholestasis (36). Also, the importance of HDAC7 in the feedback regulation of CYP7A1 has been reported, revealing HDAC7 as a potential target for treating hypercholesterolemia (37). Although BAs have beneficial roles in removing excess cholesterol and counteracting obesity by reducing TG levels, abnormally elevated hepatic BA levels are associated with hepatobiliary disease, like cholestasis, as recently demonstrated in SHP-KO (38) and SHP/FXR-KO mouse studies (39). In this work, we have shown the crucial role of phosphorylation of SHP at a single site by PKCζ in BA signal-induced epigenomic regulation of liver metabolic genes. Our findings thus suggest that Thr-55 phosphorylation of SHP may provide a novel potential target for the treatment of BA-related diseases associated with SHP dysfunction.
Acknowledgments
We are grateful to Jie Chen, University of Illinois at Urbana-Champaign, for providing expert advice on cell signaling. Ad-PKCζ was kindly provided by Drs. R. Farese and M. Sajan, University of South Florida. We thank Johan Auwerx, Ecole Polytechnique, Switzerland, for the GST-SHP constructs. We also thank Byron Kemper for helpful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK71662 (to H. E. X.), DK66202 (to H. E. X.), DK62777 (to J. K. K.), and DK95842 (to J. K. K.) and federal funds from National Institutes of Health NCI Contract HHSN261200800001E.
- BA
- bile acid
- SHP
- small heterodimer partner
- FXR
- farnesoid X receptor
- CDCA
- chenodeoxycholic acid
- CA
- cholic acid
- FGF15/19
- fibroblast growth factor 15/19
- PTM
- post-translational modifications
- PRMT5
- protein arginine methyltransferase 5
- ERK
- extracellular signal-regulated kinase
- TG
- triglyceride
- IP
- immunoprecipitation
- IB
- immunoblot
- qPCR
- quantitative PCR
- HDAC
- histone deacetylase.
REFERENCES
- 1. Chiang J. Y. (2009) Bile acids. Regulation of synthesis. J. Lipid Res. 50, 1955–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas C., Pellicciari R., Pruzanski M., Auwerx J., Schoonjans K. (2008) Targeting bile acid signalling for metabolic diseases. Nat. Rev. Drug. Discov. 7, 678–693 [DOI] [PubMed] [Google Scholar]
- 3. Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. (2009) Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 89, 147–191 [DOI] [PubMed] [Google Scholar]
- 4. Hylemon P. B., Zhou H., Pandak W. M., Ren S., Gil G., Dent P. (2009) Bile acids as regulatory molecules. J. Lipid Res. 50, 1509–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., Gerard R. D., Repa J. J., Mangelsdorf D. J., Kliewer S. A. (2005) Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2, 217–225 [DOI] [PubMed] [Google Scholar]
- 6. Kir S., Beddow S. A., Samuel V. T., Miller P., Previs S. F., Suino-Powell K., Xu H. E., Shulman G. I., Kliewer S. A., Mangelsdorf D. J. (2011) FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 331, 1621–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113, 1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miao J., Xiao Z., Kanamaluru D., Min G., Yau P. M., Veenstra T. D., Ellis E., Strom S., Suino-Powell K., Xu H. E., Kemper J. K. (2009) Bile acid signaling pathways increase stability of small heterodimer partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 23, 986–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., Mangelsdorf D. J. (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell. 6, 507–515 [DOI] [PubMed] [Google Scholar]
- 10. Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., Maloney P. R., Willson T. M., Kliewer S. A. (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6, 517–526 [DOI] [PubMed] [Google Scholar]
- 11. Lee Y. K., Moore D. D. (2002) Dual mechanism for repression of the monomeric orphan receptor liver receptor homologous protein-1 (LRH-1) by the orphan small heterodimer partner (SHP). J. Biol. Chem. 277, 2463–2467 [DOI] [PubMed] [Google Scholar]
- 12. Lee J., Padhye A., Sharma A., Song G., Miao J., Mo Y. Y., Wang L., Kemper J. K. (2010) A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J. Biol. Chem. 285, 12604–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuk J. M., Shin D. M., Lee H. M., Kim J. J., Kim S. W., Jin H. S., Yang C. S., Park K. A., Chanda D., Kim D. K., Huang S. M., Lee S. K., Lee C. H., Kim J. M., Song C. H., Lee S. Y., Hur G. M., Moore D. D., Choi H. S., Jo E. K. (2011) The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 12, 742–751 [DOI] [PubMed] [Google Scholar]
- 14. Wang L., Liu J., Saha P., Huang J., Chan L., Spiegelman B., Moore D. D. (2005) The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab. 2, 227–238 [DOI] [PubMed] [Google Scholar]
- 15. Chiang J. Y. L. (2002) Bile acid regulation of gene expression. Roles of nuclear hormone receptors. Endocr. Rev. 23, 443–463 [DOI] [PubMed] [Google Scholar]
- 16. Kemper J. K., Kim H., Miao J., Bhalla S., Bae Y. (2004) Role of a mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Mol. Cell Biol. 24, 7707–7719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fang S., Miao J., Xiang L., Ponugoti B., Treuter E., Kemper J. K. (2007) Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol. Cell Biol. 27, 1407–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miao J., Fang S., Lee J., Comstock C., Knudsen K. E., Kemper J. K. (2009) Functional specificities of Brm and Brg-1 Swi/Snf ATPases in the feedback regulation of hepatic bile acid biosynthesis. Mol. Cell Biol. 29, 6170–6181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanamaluru D., Xiao Z., Fang S., Choi S. E., Kim D. H., Veenstra T. D., Kemper J. K. (2011) Arginine methylation by PRMT5 at a naturally-occurring mutation site is critical for liver metabolic regulation by Small Heterodimer Partner. Mol. Cell Biol. 31, 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nishigori H., Tomura H., Tonooka N., Kanamori M., Yamada S., Sho K., Inoue I., Kikuchi N., Onigata K., Kojima I., Kohama T., Yamagata K., Yang Q., Matsuzawa Y., Miki T., Seino S., Kim M. Y., Choi H. S., Lee Y. K., Moore D. D., Takeda J. (2001) Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc. Natl. Acad. Sci. U.S.A. 98, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boulias K., Katrakili N., Bamberg K., Underhill P., Greenfield A., Talianidis I. (2005) Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO J. 24, 2624–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Connelly S., Mech C. (2004) Delivery of adenoviral DNA to mouse liver. Methods Mol. Biol. 246, 37–52 [DOI] [PubMed] [Google Scholar]
- 23. Sajan M. P., Rivas J., Li P., Standaert M. L., Farese R. V. (2006) Repletion of atypical protein kinase C following RNA interference-mediated depletion restores insulin-stimulated glucose transport. J. Biol. Chem. 281, 17466–17473 [DOI] [PubMed] [Google Scholar]
- 24. Kemper J. K., Xiao Z., Ponugoti B., Miao J., Fang S., Kanamaluru D., Tsang S., Wu S. Y., Chiang C. M., Veenstra T. D. (2009) FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 10, 392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ponugoti B., Kim D. H., Xiao Z., Smith Z., Miao J., Zang M., Wu S. Y., Chiang C. M., Veenstra T. D., Kemper J. K. (2010) SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 285, 33959–33970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee J., Seok S., Yu P., Kim K., Smith Z., Rivas-Astroza M., Zhong S., Kemper J. K. (2012) Genomic analysis of hepatic Farnesoid X receptor (FXR) binding sites reveals altered binding in obesity and direct gene repression by FXR. Hepatology 56, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu T., Choi S. E., Kim D. H., Seok S., Suino-Powell K. M., Xu H. E., Kemper J. K. (2012) Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor β-Klotho. Proc. Natl. Acad. Sci. U.S.A. 109, 16137–16142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 29. Boulias K., Talianidis I. (2004) Functional role of G9a-induced histone methylation in small heterodimer partner-mediated transcriptional repression. Nucleic Acids Res. 32, 6096–6103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee S. J., Kim J. Y., Nogueiras R., Linares J. F., Perez-Tilve D., Jung D. Y., Ko H. J., Hofmann S. M., Drew A., Leitges M., Kim J. K., Tschöp M. H., Diaz-Meco M. T., Moscat J. (2010) PKCζ-regulated inflammation in the nonhematopoietic compartment is critical for obesity-induced glucose intolerance. Cell Metab. 12, 65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frankenberg T., Miloh T., Chen F. Y., Ananthanarayanan M., Sun A. Q., Balasubramaniyan N., Arias I., Setchell K. D., Suchy F. J., Shneider B. L. (2008) The membrane protein ATPase class I type 8B member 1 signals through protein kinase Cζ to activate the farnesoid X receptor. Hepatology 48, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park Y. J., Kim S. C., Kim J., Anakk S., Lee J. M., Tseng H. T., Yechoor V., Park J., Choi J. S., Jang H. C., Lee K. U., Novak C. M., Moore D. D., Lee Y. K. (2011) Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J. Lipid Res. 52, 2234–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith Z., Ryerson D., Kemper J. K. (2013) Epigenomic regulation of bile acid metabolism. Emerging role of transcriptional cofactors. Mol. Cell Endocrinol. 368, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim D. H., Lee J., Lee B., Lee J. W. (2009) ASCOM controls farnesoid X receptor transactivation through its associated histone H3 lysine 4 methyltransferase activity. Mol. Endocrinol. 23, 1556–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim D. H., Kim J., Lee J. W. (2011) Requirement for MLL3 in p53 regulation of hepatic expression of small heterodimer partner and bile acid homeostasis. Mol. Endocrinol. 25, 2076–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ananthanarayanan M., Li Y., Surapureddi S., Balasubramaniyan N., Ahn J., Goldstein J. A., Suchy F. J. (2011) Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is down-regulated in cholestasis. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mitro N., Godio C., De Fabiani E., Scotti E., Galmozzi A., Gilardi F., Caruso D., Vigil Chacon A. B., Crestani M. (2007) Insights in the regulation of cholesterol 7α-hydroxylase gene reveal a target for modulating bile acid synthesis. Hepatology 46, 885–897 [DOI] [PubMed] [Google Scholar]
- 38. Park Y. J., Qatanani M., Chua S. S., LaRey J. L., Johnson S. A., Watanabe M., Moore D. D., Lee Y. K. (2008) Loss of orphan receptor small heterodimer partner sensitizes mice to liver injury from obstructive cholestasis. Hepatology 47, 1578–1586 [DOI] [PubMed] [Google Scholar]
- 39. Anakk S., Watanabe M., Ochsner S. A., McKenna N. J., Finegold M. J., Moore D. D. (2011) Combined deletion of FXR and SHP in mice induces Cyp17a1 and results in juvenile onset cholestasis. J. Clin. Invest. 121, 86–95 [DOI] [PMC free article] [PubMed] [Google Scholar]