

Background: LacCer is known to regulate PLA2 activity in cells, but the precise mechanisms have not been elucidated.
Results: LacCer binds to cPLA2α and increases its enzymatic activity.
Conclusion: LacCer is identified as a novel and direct activator of cPLA2α.
Significance: This research provides new insights into the regulatory mechanisms of cPLA2α and the physiological functions of LacCer as a signaling molecule.
Keywords: Arachidonic acid, Glycosphingolipid, Phospholipase A, Sphingolipid, Tumor Necrosis Factor (TNF)
Abstract
Lactosylceramide (LacCer) is a member of the glycosphingolipid family and is known to be a bioactive lipid in various cell physiological processes. However, the direct targets of LacCer and cellular events mediated by LacCer are largely unknown. In this study, we examined the effect of LacCer on the release of arachidonic acid (AA) and the activity of cytosolic phospholipase A2α (cPLA2α). In CHO-W11A cells, treatment with 1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP), an inhibitor of glucosylceramide synthase, reduced the glycosphingolipid level, and the release of AA induced by A23187 or platelet-activating factor was inhibited. The addition of LacCer reversed the PPMP effect on the stimulus-induced AA release. Exogenous LacCer stimulated the release of AA, which was decreased by treatment with an inhibitor of cPLA2α or silencing of the enzyme. Treatment of CHO-W11A cells with LacCer induced the translocation of full-length cPLA2α and its C2 domain from the cytosol to the Golgi apparatus. LacCer also induced the translocation of the D43N mutant of cPLA2α. Treatment of L929 cells with TNF-α induced LacCer generation and mediated the translocation of cPLA2α and AA release, which was attenuated by treatment with PPMP. In vitro studies were then conducted to test whether LacCer interacts directly with cPLA2α. Phosphatidylcholine vesicles containing LacCer increased cPLA2α activity. LacCer bound to cPLA2α and its C2 domain in a Ca2+-independent manner. Thus, we propose that LacCer is a direct activator of cPLA2α.
Introduction
The production of the eicosanoids, proinflammatory lipid mediators that include prostaglandins, thromboxanes, and leukotrienes, is dependent on the availability of their precursor, free arachidonic acid (AA).3 The release of AA from the sn-2 position of glycerophospholipids is a highly regulated process that occurs in response to a wide variety of stimuli, such as cytokines, growth factors, and neurotransmitters (1, 2). The 85-kDa group IVA cytosolic phospholipase A2α (cPLA2α) hydrolyzes glycerophospholipids at the sn-2 position to liberate AA. cPLA2α is regulated mainly by binding of Ca2+, phosphorylation at serine residues, and interaction with lipids. The binding of Ca2+ to the C2 domain of cPLA2α triggers translocation of cPLA2α from the cytosol to the perinuclear region, including the Golgi apparatus, endoplasmic reticulum, and nuclear envelope (3). The phosphorylation of Ser505 by MAPK appears to increase enzymatic activity (4). Anionic phospholipids, particularly phosphatidylinositol 4,5-bisphosphate (PIP2) and ceramide 1-phosphate (C1P), promote binding to lipid vesicles and increase the activity of cPLA2α (1, 2). PIP2 activates the enzyme by increasing catalytic efficiency through increased penetration of the membrane (5). C1P enhances the activity of cPLA2α by increasing the resident time of the enzyme in the membrane through electrostatic interactions with cationic residues in the C2 domain (6).
Glycosphingolipids were largely known as components of the cell membrane. However, recently, these lipids have been accorded functional roles in multiple signal transduction pathways that lead to critical phenotypic changes in cells, such as cell proliferation, adhesion, and apoptosis (7–9). Lactosylceramide (LacCer) is a member of the glycosphingolipid family and plays pivotal roles as a precursor in the biosynthesis of complex glycosphingolipids. LacCer has also been shown to be a bioactive lipid involved in various cell signaling cascades. It has been reported that LacCer stimulates PECAM-1 (platelet endothelial cell adhesion molecule-1) in the adhesion and diapedesis of monocytes/lymphocytes via Ca2+-independent PLA2 and cPLA2α (10). However, the role of LacCer in the activity of PLA2 and AA release remains unclear.
Previously, we examined the role of sphingolipids in the activity of cPLA2α and the release of AA and demonstrated that sphingomyelin disturbs the binding to glycerophospholipids and so reduces the activity of cPLA2α (11). We also found that C1P is a direct activator of cPLA2α via the C2 domain (12). Sphingosine was shown to be a direct inhibitor of cPLA2α (13). Thus, several sphingolipids regulate the activity of cPLA2α, but the role of glycosphingolipids in the activity of the enzyme is unknown. In this study, we investigated the mechanisms by which glycosphingolipids, especially LacCer, activate cPLA2α. LacCer was found to bind cPLA2α directly and to be an inducer of cPLA2α activation in vitro and in cells. Furthermore, we show that the activation of cPLA2α in response to TNF-α was mediated through the generation of LacCer.
EXPERIMENTAL PROCEDURES
Materials
[5,6,8,9,11,12,14,15-3H]AA (215 Ci/mmol, 7.96 TBq/mmol) was purchased from Amersham Biosciences; 1-palmitoyl-2-[14C]arachidonylphosphatidylcholine (PAPC; 48 mCi/mmol, 1776 MBq/mmol) from PerkinElmer Life Sciences; A23187 from Calbiochem; platelet-activating factor (PAF) and sphingosine 1-phosphate from Cayman Chemical (Ann Arbor, MI); U0126 from Promega; 1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP), bovine glucosylceramide (GlcCer), bovine LacCer, and bovine GM1 from Matreya, LLC (Pleasant Gap, PA); C16-C1P, 1-palmitoyl-2-oleoylphosphatidylinositol, 1-palmitoyl-2-oleoylphosphatidylcholine, and 1-palmitoyl-2-oleoylphosphatidylethanolamine from Avanti Polar Lipids (Alabaster, AL); BODIPY-C5-LacCer and Oregon Green 488 BAPTA-1/AM from Invitrogen; and TNF-α from PeproTech (Rocky Hill, NJ). Pyrrophenone was generously provided by Dr. Kohji Hanasaki (Shionogi Co. Ltd., Osaka, Japan).
Cells and Cell Cultures
The CHO-W11A cell line stably expressing the guinea pig PAF receptor was cultured in Ham's F-12 medium supplemented with 10% FBS, 100 units/ml penicillin G sodium, and 100 μg/ml streptomycin sulfate at 37 °C and 5% CO2. L929 mouse fibrosarcoma cells and a stable clone of L929 cells lacking cPLA2α (L929-cPLA2α-shRNA, clone 49) established previously (14) were cultured in the same manner except for the use of DMEM containing 5% FBS instead of Ham's F-12 medium. The HEK293T cell line was cultured in DMEM containing 10% FBS.
AA Release Assay
Cells (2 × 104 cells) on 24-well plates were labeled by incubation for 18 h in 0.5 ml of medium containing 33 nCi of [3H]AA and 0.1% BSA. The cells were washed and stimulated with reagents in medium containing 0.1% BSA and 10 mm HEPES (pH 7.4) for specific periods at 37 °C. The radioactivity of supernatants and cell lysates (in 1% Triton X-100) was measured by liquid scintillation counting. The amount of radioactivity released into the supernatant is expressed as a percentage of the total amount of radioactivity incorporated.
Plasmid Construction, Transfection, and Confocal Microscopy
The plasmids for chimeric proteins containing GFP or DsRed at the N terminus of cPLA2α (GFP-cPLA2α and DsRed-cPLA2α), the C2 domain (GFP-C2), and the D43N mutant (GFP-D43N-cPLA2α) were kindly donated by Dr. Tetsuya Hirabayashi (The Tokyo Metropolitan Institute of Medical Science). For protein expression, cells were seeded at a density of 2 × 105 cells/60-mm dish and transiently transfected with 2 μg of expression vector with Lipofectamine PLUS (Invitrogen) according to the manufacturer's protocol. After 3 h of incubation, transfected cells were seeded on coverslips (12 mm in diameter) of glass-bottomed dishes (Iwaki) at a density of 1 × 104. After another 48 h of incubation, the culture medium was replaced, and the cells were washed with Hanks' balanced salt solution containing 10 mm HEPES (pH 7.4) and 0.1% BSA and stimulated with reagents in the same buffer. Fluorescent images were taken with a FluoView confocal laser scanning microscope system (Olympus).
PLA2 Assay
HEK293T cells were transfected with an expression vector for human cPLA2α (pcDNA4/HisMaxA-hcPLA2α) using Lipofectamine PLUS. Following transfection, the cells were homogenized with a Potter homogenizer in lysis buffer (0.34 m sucrose, 100 μm dithiothreitol, 10 mm HEPES (pH 7.4), 0.2% CHAPS, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 100 μm phenylmethylsulfonyl fluoride). PLA2 activity was measured using mixed liposomes, each containing PAPC, LacCer, and Triton X-100 as a substrate. The mixed lipids in the solvent (1:1 chloroform/methanol) were dried under nitrogen. A solution of 0.00125% Triton X-100 was added, and the lipid was vortexed vigorously for 2 min and then sonicated for 5 min in a water bath. The assay buffer contained 100 mm HEPES (pH 7.4), 1 mg/ml BSA, 1 mm CaCl2, and 10 mm dithiothreitol. The reaction was started by the addition of enzyme sources (12.5 μg), and the reaction mixture was incubated at 37 °C for 30 min. The reaction was terminated with Dole's reagent, and silica gel powder was used to recover free fatty acid in an n-heptane layer. Radioactivity was measured with a liquid scintillation counter.
Western Blotting
Cells were scraped and homogenized with ice-cold buffer containing 10 mm HEPES, 250 mm sucrose, 1 mm EDTA, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 100 μm phenylmethylsulfonyl fluoride. The lysates were centrifuged at 1000 × g for 10 min. Soluble fractions were then centrifuged at 100,000 × g for 30 min, and equal amounts of proteins from membrane and cytosolic fractions were separated by 7.5% SDS-PAGE and electrotransferred onto polyvinyl difluoride membranes (Bio-Rad). cPLA2α was detected using an anti-cPLA2α monoclonal antibody (Santa Cruz Biotechnology) followed by an anti-mouse horseradish peroxidase antibody (Amersham Biosciences). The immunoreactive bands were visualized by enhanced chemiluminescence. Results were analyzed using a LAS-1000plus system equipped with Science Lab software (Fujifilm, Tokyo, Japan). The intensity of chemiluminescence was measured using NIH ImageJ software.
Measurement of Glycosphingolipids
CHO-W11A cells were washed three times with PBS. Lipids were extracted by the Bligh and Dyer method (15). The organic phase was then subjected to Iatrobead column chromatography. The mixture of glycosphingolipids was eluted with 9:1 acetone/methanol and dried under nitrogen. The lipids were spotted onto a Silica Gel 60 TLC plate. The plate was sprayed with 47% sulfuric acid and then heated at 150 °C on a hot plate and imaged using the LAS1000plus system.
RNA Interference
L929 cells were transfected with mouse GlcCer synthase siRNA (Santa Cruz Biotechnology) using Lipofectamine RNAiMAX (Invitrogen) as recommended by the manufacturer. Following 30 h of transfection, cells were labeled with [3H]AA for 18 h and then washed and stimulated with TNF-α for 6 h.
Measurement of LacCer Synthesis
L929 cells were incubated in culture medium containing [14C]serine for 18 h. The cells were washed and incubated with or without TNF-α for 2 h. The total lipid extract was then subjected to Iatrobead column chromatography. The mixture of glycosphingolipids was eluted with 9:1 acetone/methanol and dried under nitrogen. Glycosphingolipids were separated onto a Silica Gel 60 TLC plate using 100:42:6 chloroform/methanol/water. The TLC plate was dried, exposed to an imaging plate (BAS-IP MS 2025, Fujifilm), and visualized using a Typhoon FLA 9000 system (GE Healthcare).
Lipid-Protein Overlay Assay
Lipids were spotted onto a Hybond C membrane (Amersham Biosciences) and dried under nitrogen. The membrane was rewetted in water and blocked for 1 h in 2% BSA/TBS-T (TBS containing Tween 20). It was then exposed overnight at 4 °C to 0.5 μg/ml purified GST-cPLA2α or GST-C2. The membrane was washed with TBS-T and exposed overnight at 4 °C to a 1:1000 dilution of anti-GST antibody in 2% BSA/TBS-T. It was washed with TBS-T and exposed to a 1:3000 dilution of horseradish peroxidase-conjugated anti-goat IgG antibody in 2% BSA/TBS-T for 1 h at room temperature. The immunoreactive spots were visualized by enhanced chemiluminescence. Purified recombinant GST did not bind any of the lipids using this protocol (data not shown).
Large Multilamellar Vesicle Binding Assay
Large multilamellar vesicles for LacCer were produced by drying 68.3 μl of a 1 mg/ml solution (100 μl) of 50 mm Tris-HCl (pH 7.4) and 150 mm NaCl for each reaction, and the lipid was vortexed vigorously for 2 min. It was then mixed with 200 μl of buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 4 mm EGTA) and, for the calcium-containing reaction, mixed with 4.4 mm CaCl2. The binding reaction was initiated by the addition of 100 μl of 50 mm Tris-HCl (pH 7.4) and 150 mm NaCl containing 0.5 μg of GST-cPLA2α. After 5 min at room temperature, the reaction was centrifuged at 10,000 × g for 10 min, and the supernatant was removed. 100 μl of 1× Laemmli buffer was added to the lipid pellet, and 50 μl of the lipid pellet was subjected to SDS-PAGE. Purified recombinant GST did not bind LacCer using this protocol (data not shown).
Calcium Imaging
CHO-W11A cells were seeded on glass-bottomed dishes and incubated for 48 h. The cells were washed and incubated with 5 μm Oregon Green 488 BAPTA-1/AM for 1 h at 37 °C. Cells were washed three times and then imaged using the FluoView confocal laser scanning microscope system.
Statistics
Values are the means ± S.E. for three to four independent experiments performed in triplicate. In the case of multiple comparisons, the significance of differences was determined using a one-way analysis of variance by Dunnett's or Tukey's test. For pairwise comparisons, Student's two-tailed t test was used. p values <0.05 were considered to be significant.
RESULTS
LacCer Induces cPLA2α-dependent AA Release from Cells
To analyze the role of glycosphingolipids in cPLA2α-dependent AA release from cells, glycosphingolipid biosynthesis was inhibited by treatment with PPMP, a competitive inhibitor of GlcCer synthase. As shown in Fig. 1A, treatment of CHO-W11A cells with 1 μm PPMP for 48 h led to a significant decrease in cellular glycosphingolipid levels. We confirmed that the treatment of cells with PPMP at the concentrations used did not cause cytotoxicity and alteration of cPLA2α expression levels during the test period (data not shown). Using this culture system, we determined whether the reduction in the cellular glycosphingolipid level affected the release of AA from cells. We previously reported that stimulation of CHO-W11A cells with A23187 or PAF induces cPLA2α-dependent AA release (11). As shown in Fig. 1B, the release of AA induced by 1 μm A23187 or 100 nm PAF was decreased in PPMP-treated cells. The reduced release of AA from PPMP-treated cells was rescued by supplementation of the culture medium with 30 μm GlcCer or LacCer. We next tested the effect of exogenous glycosphingolipids on the release of AA from CHO-W11A cells. As shown in Fig. 1C, treatment with LacCer increased the release of AA in a concentration-dependent manner. In addition, 30 μm LacCer induced the release of AA in a time-dependent manner, and the release at 30 min following stimulation was 2.5-fold of that upon treatment with vehicle (Fig. 1D). Other glycosphingolipids, such as GlcCer, galactosylceramide, and GM1, did not increase the release of AA from CHO-W11A cells (data not shown). Treatment of cells with 2 μm pyrrophenone, a selective inhibitor of cPLA2α, reduced the LacCer-induced release of AA (Fig. 1E). We next investigated the effect of LacCer on another cell type, murine L929 fibroblast cells. As shown in Fig. 1F, stimulation for 30 min with LacCer significantly increased the release of AA from L929 cells, which was inhibited by pyrrophenone. Furthermore, in stable cPLA2α knockdown L929 cells (L929-cPLA2α-shRNA), LacCer did not induce the release of AA. These results suggest that glycosphingolipids, especially LacCer, play an important role in the regulation of cPLA2α-dependent release of AA in cells.
FIGURE 1.

LacCer induces cPLA2α-dependent AA release from cells. A, CHO-W11A cells were cultured for 48 h in culture medium supplemented with vehicle or 1 μm PPMP. After the cells were washed, glycosphingolipids were extracted and spotted onto a TLC plate. B, CHO-W11A cells were cultured for 30 h in culture medium supplemented with or without 1 μm PPMP, 10 μm GlcCer, and/or 10 μm LacCer. The cells were then labeled by incubation for 18 h in 0.1% BSA- and [3H]AA-containing medium supplemented with the same reagents. The labeled cells were washed and stimulated with 1 μm A23187 or 100 nm PAF for 30 min at 37 °C. a, p < 0.05, significantly different from the control without PPMP. C, [3H]AA-labeled cells were stimulated with the indicated concentrations of LacCer for 30 min at 37 °C. D, [3H]AA-labeled cells were stimulated with vehicle or 30 μm LacCer for the indicated time periods at 37 °C. ○, vehicle; ●, LacCer. E, the labeled cells were pretreated with vehicle (Control) or 2 μm pyrrophenone for 30 min prior to stimulation with vehicle (Vehi) or 30 μm LacCer for 30 min at 37 °C. a, p < 0.05, significantly different from the control without LacCer; b, p < 0.05, significantly different from the control without pyrrophenone. F, [3H]AA-labeled L929 and L929-cPLA2α-shRNA cells were pretreated with 2 μm pyrrophenone (Pyrro) for 30 min and then stimulated with 30 μm LacCer for 30 min at 37 °C. The expression levels of cPLA2α in both cells are shown. a, p < 0.05, significantly different from the control without LacCer. The data shown are the means ± S.E. for three experiments.
Effect of LacCer on cPLA2α Translocation in Cells
To examine whether LacCer induces translocation of cPLA2α, we monitored the localization of GFP-cPLA2α in living cells by confocal laser fluorescence microscopy. GFP-cPLA2α was almost homogeneously present in the cytosol in CHO-W11A cells in the resting state. Treatment with LacCer triggered the translocation of GFP-cPLA2α (but not GFP) to the membrane in a pattern consistent with the Golgi apparatus, endoplasmic reticulum, and nuclear envelope (Fig. 2A). To demonstrate that LacCer-activated cPLA2α was translocated to Golgi membranes, the fluorescent signal of cPLA2α was overlaid with a marker of the Golgi, red fluorescent protein-Golgi. As shown in Fig. 2B, GFP-cPLA2α activated by LacCer was co-localized with red fluorescent protein-Golgi. The fluorescent LacCer BODIPY-C5-LacCer is internalized into endosomes and then transferred to the Golgi (16). Significant co-localization was observed between BODIPY-C5-LacCer and DsRed-cPLA2α (Fig. 2C). Because the C2 domain of cPLA2α is sufficient for translocation and can interact with lipids, we monitored the cellular localization of the C2 domain of cPLA2α fused to GFP (GFP-C2). As shown in Fig. 2D, LacCer induced the translocation of GFP-C2 to the Golgi. These results suggest that cPLA2α is translocated mainly to Golgi membranes through the C2 domain in response to LacCer.
FIGURE 2.
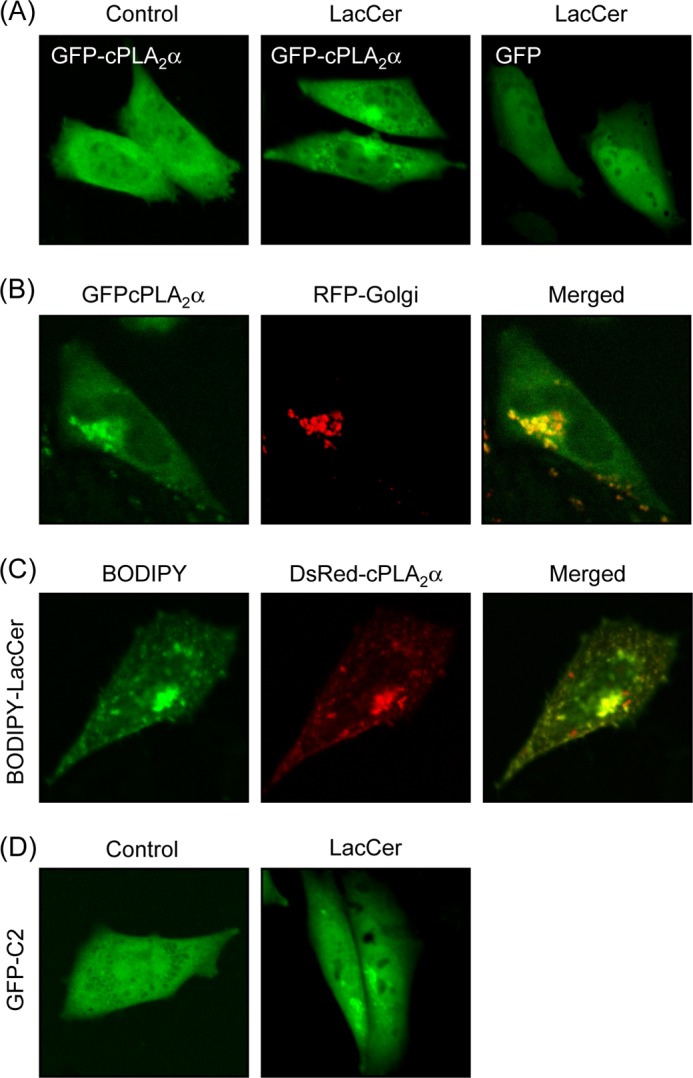
LacCer induces cPLA2α translocation in cells. A, CHO-W11A cells transiently transfected with expression vectors for GFP-cPLA2α and GFP were treated with vehicle (Control) or 30 μm LacCer for 1 h. B, CHO-W11A cells transiently transfected with expression vectors for GFP-cPLA2α and red fluorescent protein (RFP)-Golgi were treated with 30 μm LacCer. C, DsRed-cPLA2α-expressing CHO-W11A cells were treated with 100 nm BODIPY-C5-LacCer for 1 h. D, GFP-C2-expressing CHO-W11A cells were treated with 30 μm LacCer for 1 h. In A–D, data are representative of three independent experiments.
LacCer Activates cPLA2α without Intracellular Ca2+ Mobilization in Cells
To determine the Ca2+ requirement for LacCer-induced cPLA2α translocation, the cellular localization of GFP-cPLA2α in the presence of EGTA was examined. As shown in Fig. 3A, LacCer triggered the translocation of GFP-cPLA2α in the presence of EGTA. The single substitution D43N in the C2 domain abrogates the Ca2+-dependent translocation of cPLA2α to membranes (17). The translocation of GFP-D43N-cPLA2α was induced by treatment with LacCer, but not with A23187 (Fig. 3B). We next investigated whether LacCer would induce a rise in intracellular Ca2+ concentrations in CHO-W11A cells. Treatment with LacCer did not increase intracellular Ca2+ concentrations (Fig. 3C). Next, we determined the effect of EGTA on the release of AA induced by LacCer. As shown in Fig. 3D, LacCer induced the release of AA even in the presence of EGTA. Thus, LacCer triggers the translocation of cPLA2α and then induces the release of AA without intracellular Ca2+ mobilization in CHO-W11A cells.
FIGURE 3.

LacCer activates cPLA2α without intracellular Ca2+ mobilization. A, CHO-W11A cells transiently transfected with expression vector for GFP-cPLA2α were treated with vehicle (Control) or 30 μm LacCer for 1 h in the presence of 2 mm EGTA. B, GFP-D43N-cPLA2α-expressing CHO-W11A cells were treated with 30 μm LacCer or 5 μm A23187 for 1 h. C, CHO-W11A cells labeled with Oregon Green 488 were washed and then treated with 30 μm LacCer for 10 or 60 min or with 5 μm A23187 for 5 min. In A–C, data are representative of three independent experiments. D, [3H]AA-labeled CHO-W11A cells were stimulated with vehicle (Vehi) or 30 μm LacCer for 30 min at 37 °C in the presence or absence of 2 mm EGTA. a, p < 0.05, significantly different from the control without LacCer.
LacCer Directly Binds and Activates cPLA2α in Vitro
We next determined whether LacCer would bind directly to cPLA2α using a lipid-protein overlay assay. As shown in Fig. 4A, cPLA2α bound as little as 80 nmol of LacCer. cPLA2α was also found to bind to 1-palmitoyl-2-oleoylphosphatidylinositol, although to a lesser extent compared with LacCer (Fig. 4B). In contrast, cPLA2α did not bind to 1-palmitoyl-2-oleoylphosphatidylcholine, 1-palmitoyl-2-oleoylphosphatidylethanolamine, GM1, or sphingosine 1-phosphate when 100 nmol was bound to the membrane. The binding between LacCer and cPLA2α could be detected even in the presence of EGTA (Fig. 4C), indicating that LacCer binds to cPLA2α in a Ca2+-independent manner. C1P is known to bind cPLA2α in a calcium-dependent manner. As shown in Fig. 4C, C1P bound cPLA2α in the presence of Ca2+, which was inhibited in the presence of EGTA. The region of cPLA2α that binds LacCer was also determined using the recombinantly expressed C2 domain of cPLA2α. As shown in Fig. 4D, LacCer bound to the C2 domain of cPLA2α. Next, we used a large multilamellar vesicle binding assay for cPLA2α. cPLA2α bound to LacCer in both the presence and absence of Ca2+ (Fig. 4E). Thus, cPLA2α interacts with LacCer via the C2 domain in a Ca2+-independent manner.
FIGURE 4.
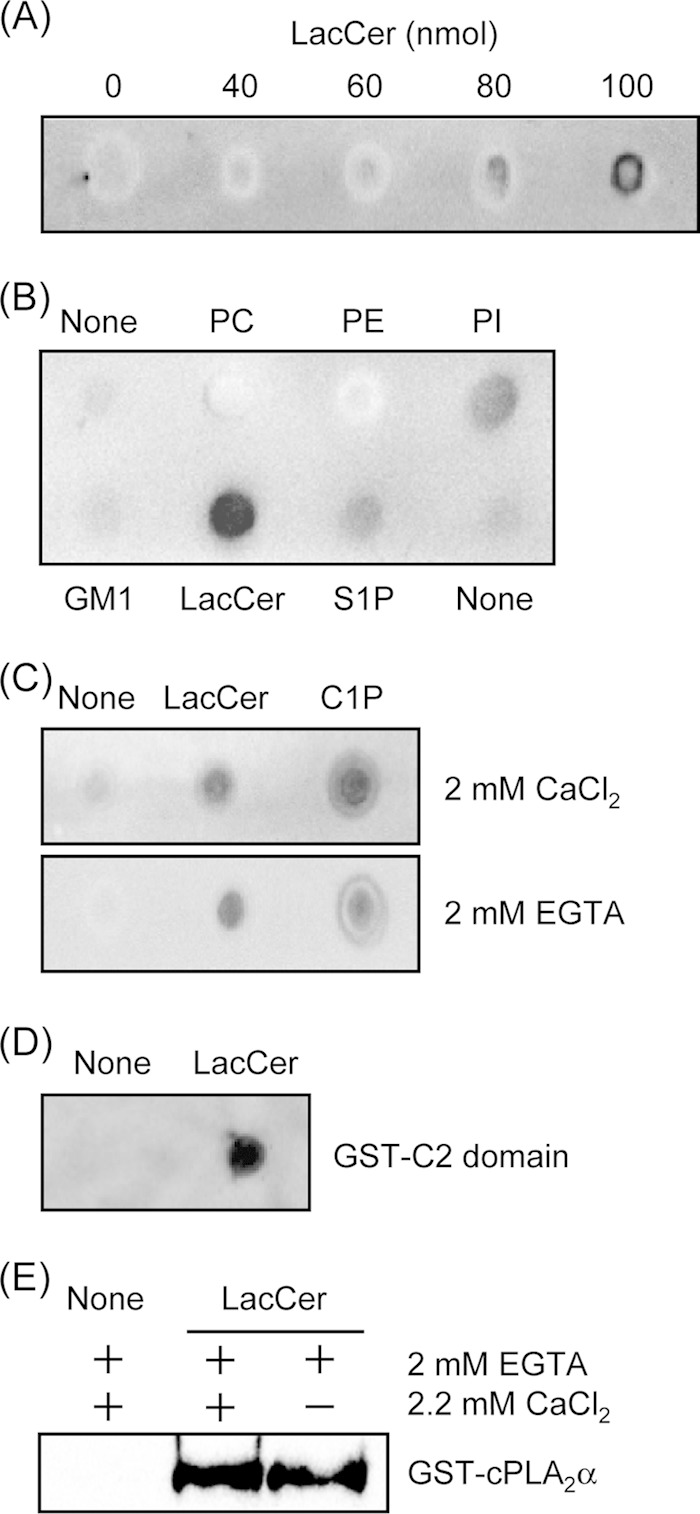
cPLA2α binds to LacCer in a calcium-independent manner. A, the binding of cPLA2α to LacCer was examined using the lipid-protein overlay assay as described under “Experimental Procedures.” The indicated amounts of LacCer were spotted onto a Hybond C membrane. The membrane was exposed to purified GST-cPLA2α overnight at 4 °C. B, the lipid-protein overlay assay was repeated with 100 nmol of lipids and GST-cPLA2α. PC, 1-palmitoyl-2-oleoylphosphatidylcholine; PE, 1-palmitoyl-2-oleoylphosphatidylethanolamine; PI, 1-palmitoyl-2-oleoylphosphatidylinositol; S1P, sphingosine 1-phosphate. C, the lipid-protein overlay assay was repeated with 100 nmol of LacCer, 20 nmol of C1P, and GST-cPLA2α with or without 2 mm EGTA. D, the membrane spotted with 100 nmol of LacCer was exposed to the GST-tagged C2 domain of cPLA2α overnight at 4 °C. E, the large multilamellar vesicle binding assay was performed as described under “Experimental Procedures.” In A–E, data are representative of three independent experiments.
To examine whether LacCer directly increases the enzymatic activity of cPLA2α, liposomes containing PAPC and LacCer were prepared and tested for activity in vitro. As shown in Fig. 5, the cPLA2α activity in the presence of PAPC liposomes alone was ∼1100 dpm. The cPLA2α activity in the liposomes with PAPC and LacCer combined at a molar ratio of 95:5 was increased by 1.5-fold compared with that with PAPC alone. LacCer could not activate cPLA2α in the presence of pyrrophenone or EGTA, indicating that the enzymatic activation of cPLA2α by LacCer requires Ca2+.
FIGURE 5.
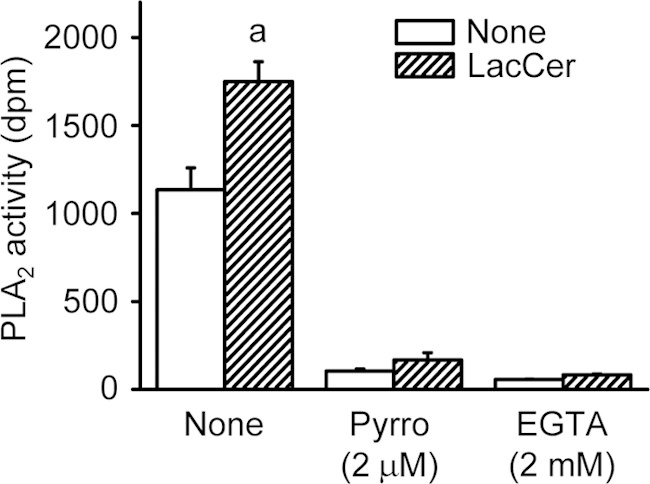
LacCer activates cPLA2α in vitro. PLA2 activity in the cytosolic fraction from HEK293T cells expressing human cPLA2α was measured as described under “Experimental Procedures.” PLA2 activities toward PAPC and 95:5 PAPC/LacCer-mixed vesicles were measured in reaction buffer with or without 2 μm pyrrophenone (Pyrro) or 2 mm EGTA. The data shown are the means ± S.E. for three experiments. a, p < 0.05, significantly different from the control without LacCer.
TNF-α Induces cPLA2α-dependent AA Release via LacCer Generation in L929 Cells
TNF-α is known to up-regulate the activity of cPLA2α and the generation of LacCer (8, 18, 19). We next examined the role of LacCer generation in TNF-α-induced activation of cPLA2α. As shown in Fig. 6A, treatment of L929 cells with 10 nm TNF-α significantly increased intracellular LacCer levels. Treatment of cells with TNF-α induced the release of AA in a time-dependent manner for up to 6 h, and the release at 6 h following stimulation was 2-fold of that in the control (Fig. 6B). In cPLA2α knockdown cells, TNF-α did not increase the release of AA (Fig. 6C). In addition, the TNF-α-induced release of AA from L929 cells was suppressed to near the control level by both treatment with PPMP (Fig. 6C) and silencing of GlcCer synthase (Fig. 6D). We next determined the effect of TNF-α on the translocation of cPLA2α in L929 cells. When the cells were treated with TNF-α for 2 h, the expression level of cPLA2α in membrane fractions was increased (Fig. 6E). In addition, TNF-α triggered the translocation of GFP-cPLA2α in L929 cells, which was inhibited by pretreatment with PPMP (Fig. 6F). These results indicate that TNF-α increases the cellular LacCer levels, thereby activating cPLA2α.
FIGURE 6.

TNF-α induces cPLA2α-dependent AA release via LacCer formation in L929 cells. A, L929 cells labeled with [14C]serine for 18 h were stimulated with or without 10 nm TNF-α for 2 h. After the cells were washed, lipids were extracted and separated by TLC as described under “Experimental Procedures.” B, [3H]AA-labeled L929 cells were stimulated with 10 nm TNF-α for the indicated time periods at 37 °C. ○, none; ●, TNF-α. C, L929 cells were pretreated with vehicle or 3 μm PPMP for 30 min, and then the cells and L929-cPLA2α-shRNA cells were stimulated with 10 nm TNF-α for 6 h at 37 °C. D, down-regulation of GlcCer synthase were performed as described under “Experimental Procedures.” L929 cells transfected with or without GlcCer synthase siRNA were stimulated with 10 nm TNF-α for 6 h at 37 °C. The expression levels of GlcCer synthase (GCS) in cells are shown. E, L929 cells were pretreated with vehicle or 3 μm PPMP for 30 min and then stimulated with 10 nm TNF-α for 2 h at 37 °C. The cytosolic and membrane fractions were prepared and subjected to immunoblot analysis. Upper panels, immunoblotting with antibodies against cPLA2α. Lower panel, the histogram represents the expression levels of cPLA2α in membrane fractions quantified using ImageJ. The data shown are the means ± S.E. for three experiments. a, p < 0.05, significantly different from the control without TNF-α; b, p < 0.05, significantly different from the control without PPMP or cPLA2α knockdown. F, L929 cells transiently transfected with expression vector for GFP-cPLA2α were treated with or without TNF-α for 2 h. PPMP were pretreated for 30 min before stimulation with TNF-α.
DISCUSSION
In this study, we found that LacCer is a direct activator of cPLA2α. Although LacCer has been reported to be a bioactive lipid in various cell physiological processes, such as smooth muscle proliferation (20), expression of adhesion molecules (8), and β1-integrin clustering and endocytosis (21), the direct targets of LacCer and cellular events mediated by LacCer are largely unknown. We found that LacCer interacted with and activated cPLA2α directly. In addition, we showed that depletion of glycosphingolipids by treatment with PPMP attenuated the induction of AA release in response to agonists. Although it is possible that the amounts of other sphingolipids were altered by treatment with PPMP, the rescue of glycosphingolipids by exogenous addition of GlcCer and LacCer could overcome the effect of PPMP on the release of AA (Fig. 1B). Furthermore, exogenous LacCer increased the release of AA, and that induction was decreased by treatment with an inhibitor of cPLA2α or silencing of the enzyme (Fig. 1, E and F). Thus, LacCer plays an important role in the activation of cPLA2α and AA release.
Translocation of cPLA2α from the cytosol to the membrane is necessary for its access to phospholipid substrate. In this study, depletion of glycosphingolipids by treatment with PPMP for 48 h did not inhibit the A23187-mediated translocation of cPLA2α (data not shown). Thus, LacCer is required for induction of the release of AA, but not for translocation of cPLA2α in response to A23187. LacCer may increase the retention time of cPLA2α in the membrane, resulting in induction of cPLA2α activity and AA release. cPLA2α is translocated in response to Ca2+ binding, and its binding to Asp43 is critical for mediating the translocation (17). In this study, the translocation of GFP-D43N-cPLA2α was induced by LacCer, but not by A23187 (Fig. 3B). In addition, treatment of CHO-W11A cells with LacCer, which did not mediate Ca2+ mobilization, induced the translocation of cPLA2α even in the presence of EGTA (Fig. 3, A and C). These results suggest that the translocation of cPLA2α induced by LacCer is Ca2+-independent. However, it is also possible that exogenous LacCer accumulated in the membrane affects the Ca2+ dependence of cPLA2α.
It has been reported that the C2 domain of cPLA2α binds to phosphatidylcholine and C1P in the presence of micromolar Ca2+ (22, 23). The domain also shows weak binding to phosphatidic acid vesicles in a Ca2+-independent manner (22). Anionic phosphatidylmethanol binds to full-length cPLA2α in the absence of Ca2+, but Ca2+ is required for the binding of the C2 domain to this lipid (24). Thus, cPLA2α displays both Ca2+-dependent and Ca2+-independent interfacial binding to vesicles. In this study, we showed that cPLA2α bound to LacCer in a Ca2+-independent manner (Fig. 4). However, Ca2+ was required for the activation of cPLA2α by LacCer in vitro (Fig. 5). Thus, it may be possible that, even if cPLA2α binds to LacCer, Ca2+ binding-mediated conformational changes of cPLA2α are essential for promoting the penetration of the enzyme into the PAPC vesicles that were used for measuring the enzymatic activity. Treatment of cells with LacCer caused the translocation of the GFP-D43N-cPLA2α mutant. Thus, this suggests that cPLA2α can bind to the LacCer-containing cell membrane in a Ca2+-independent manner, but Ca2+ is required for exerting its enzymatic activity.
C1P and PIP2 bind to cPLA2α and increase its activity. PIP2 activates the enzyme by increasing catalytic efficiency through increased penetration of the membrane (5). The binding site of PIP2 includes four lysine residues, which are located in the highly basic region of the catalytic domain on the side close to the membrane (25, 26). C1P enhances the activity of cPLA2α by increasing the resident time of the enzyme in the membrane through electrostatic interactions with cationic residues in the C2 domain (6). In this study, we showed that LacCer binds to the C2 domain. The C2 domain is known to bind to various lipids by hydrophobic or electrostatic interactions. Because LacCer is a neutral glycosphingolipid, hydrophobic forces might be responsible for this interaction.
The liberation of AA from membrane phospholipids by activation of cPLA2α is known to occur mainly in the perinuclear region, such as the Golgi and the endoplasmic reticulum. LacCer is located predominantly in the outer leaflet of the plasma membrane and the luminal leaflet of the Golgi, endosomes, and lysosomes. Several studies determined that the P-glycoprotein functions as a flippase in both the plasma membrane and the Golgi for phospholipids and simple glycosphingolipids, such as GlcCer, galactosylceramide, and LacCer (27, 28). Thus, the results from this study suggest that LacCer in the cytosolic leaflet of the Golgi promotes the binding of cPLA2α to the membrane.
TNF-α regulates various physiological functions, such as cell proliferation, differentiation, adhesion, and death. It has been reported that various cell functions induced by TNF-α are mediated through the activation of cPLA2α (18, 19). However, the cPLA2α-activating mechanisms of TNF-α have not been well elucidated. In this study, inhibition of synthesis of glycosphingolipids attenuated cPLA2α translocation and AA release in response to TNF-α (Fig. 6). Thus, TNF-α activates cPLA2α through the production of glycosphingolipids, especially LacCer. Previously, we reported that sphingomyelin disturbs the binding of cPLA2α to glycerophospholipids and inhibits the release of AA (11). Numerous reports have shown that sphingomyelin is hydrolyzed to ceramide by sphingomyelinase in response to TNF-α (9, 29, 30). In addition, TNF-α induces the activation of β1,4-galactosyltransferase and the formation of LacCer, which is generated via the galactosylation of GlcCer produced from ceramide (7). Thus, this suggests that, in response to TNF-α, the level of sphingomyelin as an inhibitor of cPLA2α is decreased and that of LacCer as an activator is increased in the membrane, thereby inducing the catalytic ability of the enzyme.
In summary, we have shown that LacCer is a direct activator of cPLA2α. LacCer binds to the C2 domain of cPLA2α and induces its translocation in a Ca2+-independent manner. Furthermore, we have demonstrated the potential role of LacCer in TNF-α-induced cPLA2α activation and AA release. These findings provide new insights into the regulatory mechanisms of cPLA2α and the physiological functions of LacCer as a signaling molecule.
Acknowledgments
We thank Dr. Kohji Hanasaki for providing pyrrophenone and Dr. Tetsuya Hirabayashi for plasmids.
This work was supported in part by Grant-in-aid for Young Scientists (B) 22790056 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
- AA
- arachidonic acid
- cPLA2α
- cytosolic phospholipase A2α
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- C1P
- ceramide 1-phosphate
- LacCer
- lactosylceramide
- PAPC
- 1-palmitoyl-2-[14C]arachidonylphosphatidylcholine
- PAF
- platelet-activating factor
- PPMP
- 1-phenyl-2-palmitoylamino-3-morpholino-1-propanol
- GlcCer
- glucosylceramide.
REFERENCES
- 1. Murakami M., Taketomi Y., Miki Y., Sato H., Hirabayashi T., Yamamoto K. (2011) Recent progress in phospholipase A2 research: from cells to animals to humans. Prog. Lipid Res. 50, 152–192 [DOI] [PubMed] [Google Scholar]
- 2. Hirabayashi T., Murayama T., Shimizu T. (2004) Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biol. Pharm. Bull. 27, 1168–1173 [DOI] [PubMed] [Google Scholar]
- 3. Evans J. H., Spencer D. M., Zweifach A., Leslie C. C. (2001) Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J. Biol. Chem. 276, 30150–30160 [DOI] [PubMed] [Google Scholar]
- 4. Tucker D. E., Ghosh M., Ghomashchi F., Loper R., Suram S., John B. S., Girotti M., Bollinger J. G., Gelb M. H., Leslie C. C. (2009) Role of phosphorylation and basic residues in the catalytic domain of cytosolic phospholipase A2α in regulating interfacial kinetics and binding and cellular function. J. Biol. Chem. 284, 9596–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Subramanian P., Vora M., Gentile L. B., Stahelin R. V., Chalfant C. E. (2007) Anionic lipids activate group IVA cytosolic phospholipase A2 via distinct and separate mechanisms. J. Lipid Res. 48, 2701–2708 [DOI] [PubMed] [Google Scholar]
- 6. Stahelin R. V., Subramanian P., Vora M., Cho W., Chalfant C. E. (2007) Ceramide 1-phosphate binds group IVA cytosolic phospholipase A2 via a novel site in the C2 domain. J. Biol. Chem. 282, 20467–20474 [DOI] [PubMed] [Google Scholar]
- 7. Pannu R., Singh A. K., Singh I. (2005) A novel role of lactosylceramide in the regulation of tumor necrosis factor α-mediated proliferation of rat primary astrocytes. J. Biol. Chem. 280, 13742–13751 [DOI] [PubMed] [Google Scholar]
- 8. Bhunia A. K., Arai T., Bulkley G., Chatterjee S. (1998) Lactosylceramide mediates tumor necrosis factor-α-induced intercellular adhesion molecule-1 (ICAM-1) expression and the adhesion of neutrophil in human umbilical vein endothelial cells. J. Biol. Chem. 273, 34349–34357 [DOI] [PubMed] [Google Scholar]
- 9. Martin S. F., Williams N., Chatterjee S. (2006) Lactosylceramide is required in apoptosis induced by N-Smase. Glycoconj. J. 23, 147–157 [DOI] [PubMed] [Google Scholar]
- 10. Gong N., Wei H., Chowdhury S. H., Chatterjee S. (2004) Lactosylceramide recruits PKCα/ϵ and phospholipase A2 to stimulate PECAM-1 expression in human monocytes and adhesion to endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 101, 6490–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakamura H., Wakita S., Suganami A., Tamura Y., Hanada K., Murayama T. (2010) Modulation of the activity of cytosolic phospholipase A2α (cPLA2α) by cellular sphingolipids and inhibition of cPLA2α by sphingomyelin. J. Lipid Res. 51, 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakamura H., Hirabayashi T., Shimizu M., Murayama T. (2006) Ceramide 1-phosphate activates cytosolic phospholipase A2α directly and by PKC pathway. Biochem. Pharmacol. 71, 850–857 [DOI] [PubMed] [Google Scholar]
- 13. Nakamura H., Hirabayashi T., Someya A., Shimizu M., Murayama T. (2004) Inhibition of arachidonic acid release and cytosolic phospholipase A2α activity by d-erythro-sphingosine. Eur. J. Pharmacol. 484, 9–17 [DOI] [PubMed] [Google Scholar]
- 14. Shimizu M., Matsumoto Y., Kurosawa T., Azuma C., Enomoto M., Nakamura H., Hirabayashi T., Kaneko M., Okuma Y., Murayama T. (2008) Release of arachidonic acid induced by tumor necrosis factor-α in the presence of caspase inhibition: evidence for a cytosolic phospholipase A2α-independent pathway. Biochem. Pharmacol. 75, 1358–1369 [DOI] [PubMed] [Google Scholar]
- 15. Bligh E. G., Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 16. Singh R. D., Liu Y., Wheatley C. L., Holicky E. L., Makino A., Marks D. L., Kobayashi T., Subramaniam G., Bittman R., Pagano R. E. (2006) Caveolar endocytosis and microdomain association of a glycosphingolipid analog is dependent on its sphingosine stereochemistry. J. Biol. Chem. 281, 30660–30668 [DOI] [PubMed] [Google Scholar]
- 17. Gijón M. A., Spencer D. M., Kaiser A. L., Leslie C. C. (1999) Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J. Cell Biol. 145, 1219–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayter H. L., Pettus B. J., Ito F., Obeid L. M., Hannun Y. A. (2001) TNFα-induced glutathione depletion lies downstream of cPLA2 in L929 cells. FEBS Lett. 507, 151–156 [DOI] [PubMed] [Google Scholar]
- 19. Jayadev S., Hayter H. L., Andrieu N., Gamard C. J., Liu B., Balu R., Hayakawa M., Ito F., Hannun Y. A. (1997) Phospholipase A2 is necessary for tumor necrosis factor α-induced ceramide generation in L929 cells. J. Biol. Chem. 272, 17196–17203 [DOI] [PubMed] [Google Scholar]
- 20. Chatterjee S. (1991) Lactosylceramide stimulates aortic smooth muscle cell proliferation. Biochem. Biophys. Res. Commun. 181, 554–561 [DOI] [PubMed] [Google Scholar]
- 21. Sharma D. K., Brown J. C., Cheng Z., Holicky E. L., Marks D. L., Pagano R. E. (2005) The glycosphingolipid, lactosylceramide, regulates β1-integrin clustering and endocytosis. Cancer Res. 65, 8233–8241 [DOI] [PubMed] [Google Scholar]
- 22. Nalefski E. A., McDonagh T., Somers W., Seehra J., Falke J. J., Clark J. D. (1998) Independent folding and ligand specificity of the C2 calcium-dependent lipid binding domain of cytosolic phospholipase A2. J. Biol. Chem. 273, 1365–1372 [DOI] [PubMed] [Google Scholar]
- 23. Pettus B. J., Bielawska A., Subramanian P., Wijesinghe D. S., Maceyka M., Leslie C. C., Evans J. H., Freiberg J., Roddy P., Hannun Y. A., Chalfant C. E. (2004) Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J. Biol. Chem. 279, 11320–11326 [DOI] [PubMed] [Google Scholar]
- 24. Hixon M. S., Ball A., Gelb M. H. (1998) Calcium-dependent and -independent interfacial binding and catalysis of cytosolic group IV phospholipase A2. Biochemistry 37, 8516–8526 [DOI] [PubMed] [Google Scholar]
- 25. Six D. A., Dennis E. A. (2003) Essential Ca2+-independent role of the group IVA cytosolic phospholipase A2 via distinct and separate mechanisms. J. Biol. Chem. 278, 23842–23850 [DOI] [PubMed] [Google Scholar]
- 26. Das S., Cho W. (2002) Roles of catalytic domain residues in interfacial binding and activation of group IV cytosolic phospholipase A2. J. Biol. Chem. 277, 23838–23846 [DOI] [PubMed] [Google Scholar]
- 27. Eckford P. D. W., Sharom F. J. (2005) The reconstituted P-glycoprotein multidrug transporter is a flippase for glucosylceramide and other simple glycosphingolipids. Biochem. J. 389, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buton X., Hervé P., Kubelt J., Tannert A., Burger K. N. J., Fellmann P., Müller P., Herrmann A., Seigneuret M., Devaux P. F. (2002) Transbilayer movement of monohexosylsphingolipids in endoplasmic reticulum and Golgi membranes. Biochemistry 41, 13106–13115 [DOI] [PubMed] [Google Scholar]
- 29. Jenkins R. W., Canals D., Idkowiak-Baldys J., Simbari F., Roddy P., Perry D. M., Kitatani K., Luberto C., Hannun Y. A. (2010) Regulated secretion of acid sphingomyelinase. Implications for selectivity of ceramide formation. J. Biol. Chem. 285, 35706–35718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clarke C. J., Truong T. G., Hannun Y. A. (2007) Role for neutral sphingomyelinase-2 in tumor necrosis factor α-stimulated expression of vascular cell adhesion molecule-1 (VCAM) and intercellular adhesion molecule-1 (ICAM) in lung epithelial cells. p38 MAPK is an upstream regulator of nSMase2. J. Biol. Chem. 282, 1384–1396 [DOI] [PubMed] [Google Scholar]