

Background: Che-1 is an RNA polymerase II-binding protein involved in gene transcription, cell proliferation, and DNA damage response.
Results: Che-1 localizes at interphase centrosomes. Che-1 inhibition abolishes Chk1 localization at centrosomes, advancing entry into mitosis.
Conclusion: Che-1 acts like an upstream regulator of Chk1 centrosomal functions.
Significance: Che-1 inhibition might potentiate tumor cell sensitivity to antimitotic drugs.
Keywords: Centrosome, Checkpoint Control, DNA Damage Response, Mitosis, Mitotic Spindle, Che-1, Chk1, PCNT
Abstract
To combat threats posed by DNA damage, cells have evolved mechanisms, collectively termed DNA damage response (DDR). These mechanisms detect DNA lesions, signal their presence, and promote their repair. Centrosomes integrate G2/M checkpoint control and repair signals in response to genotoxic stress, acting as an efficient control mechanism when G2/M checkpoint function fails and mitosis begins in the presence of damaged DNA. Che-1 is an RNA polymerase II-binding protein involved in the regulation of gene transcription, induction of cell proliferation, and DDR. Here we provide evidence that in addition to its nuclear localization, Che-1 localizes at interphase centrosomes, where it accumulates following DNA damage or spindle poisons. We show that Che-1 depletion generates supernumerary centrosomes, multinucleated cells, and multipolar spindle formation. Notably, Che-1 depletion abolishes the ability of Chk1 to bind pericentrin and to localize at centrosomes, which, in its turn, deregulates the activation of centrosomal cyclin B-Cdk1 and advances entry into mitosis. Our results reinforce the notion that Che-1 plays an important role in DDR and that its contribution seems to be relevant for the spindle assembly checkpoint.
Introduction
Proper genome maintenance, ensured by the cellular DNA damage response (DDR)3 machinery, is a prerequisite for the normal development and prevention of premature aging, diverse malfunctions, and numerous pathological states including cancer (1). Indeed, one reason for relative low cancer incidence appears to be the intrinsic ability of cells to detect and deal with the DNA damage caused by exogenous genotoxic agents such as radiation and chemicals, as well as endogenous events such as oncogene-related replication stress and telomere erosion during early stages of cancer development (1, 2). Actually, even if some DNA lesions occurring during tumorigenesis remain unrepaired, sustained signaling and effector pathways within the DDR “anticancer barrier” machinery usually eliminate the damaged cells by inducing apoptosis or senescence, both of which have potential antitumorigenic functions (2).
Centrosomes are cytoplasmic organelles that organize the interphase cytoskeleton and contribute to bipolar spindle formation during mitotic cell division. Hence, centrosomes are important for the correct segregation of chromosomes and positioning of the cleavage plane in mitosis. In normal mammalian cells, the single centrosome present at interphase, consisting of a pair of centrioles surrounded by pericentriolar material, is duplicated during the progression to S phase. This duplication occurs only once in every cell cycle, with the formation of a single daughter centriole adjacent to each parental centriole. Dysfunction of the centrosome/centriole regulatory controls can generate supernumerary centrosomes, abnormal mitotic spindles and, finally, chromosomal instability. For a long time now, supernumerary centrosomes have been associated with the formation of aneuploid cells, although it is still unclear whether these centrosome defects are a cause or a consequence of cancer (3, 4).
Surprisingly, recent findings have indicated that centrosomes are part of a network that integrates cell cycle arrest and repair signals. Crucial regulators of DDR, such as the checkpoint kinases ATM, ATR, Chk1, and Chk2 as well as the tumor suppressors p53 and BRCA1, have been found to localize to the centrosome (5, 6). Moreover, accumulating evidence suggests that centrosomes integrate G2/M checkpoint control and repair signals in response to genotoxic stress (7). These findings have led to a complete re-evaluation of centrosome function(s) in cellular physiology as well as in cancer.
Che-1 is a human RNA polymerase II-binding protein highly conserved from yeast to man and is involved in the regulation of gene transcription (8). Che-1 interacts with retinoblastoma protein and affects its growth suppression activity by interfering with the retinoblastoma protein-mediated recruitment of histone deacetylase I on the promoters of E2F1-responsive genes (9). Despite this pro-proliferative role of Che-1/Traube/Apoptosis antagonizing transcription factor (AATF), it has been described that this protein is involved in the regulation of apoptosis, exhibiting strong antiapoptotic activity in response to several kinds of cellular stress (8, 10). Notably, DNA damage by different genotoxic agents is associated with Che-1 phosphorylation and extended half-life (11). These post-translational modifications are induced by ATM/ATR and Chk2, which phosphorylate Che-1 on specific residues and are functionally linked to DNA damage-induced G2/M checkpoint. Moreover, in response to DNA damage, ATM-Chk2-phosphorylated Che-1 relocates to the TP53 promoter, activating its transcription and, consequently, that of several p53 target genes (11). It is noteworthy that Che-1 inhibition intensifies the cytotoxicity of DNA-damaging anticancer drugs, in such way reverting the chemoresistance of several tumor cell lines (11–13). Consistent with these findings, Che-1 depletion strongly decreases mutant p53 expression in human cancer cells, activates DNA damage checkpoint, and induces p73 transcription and apoptosis in these cells (14).
In the present study, we provide evidence beyond nuclear localization, showing that Che-1 localizes at interphase centrosomes, where it accumulates after DNA or spindle damage. Depletion of Che-1 prevents centrosomal recruitment of the checkpoint kinase Chk1, triggering a premature activation of centrosome-associated Cdk1 and early entry into mitosis. We also show that Che-1 interacts with pericentrin (PCNT), which, in its turn, is required for both Che-1 (this work) and Chk-1 centrosomal localization (15). Given that PCNT has an established role in targeting regulatory proteins to the centrosome (16), we suggest that PCNT mediates the localization of Che-1 to centrosomes. Our results show that depletion of Che-1 expression leads to abnormal centrosome amplification, accumulation of multinucleated cells, and abnormal spindle formation.
EXPERIMENTAL PROCEDURES
Cell Culture and Drug Treatments
HCT116 human colon carcinoma cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS); human foreskin fibroblasts and ATM−/− human fibroblasts (a kind gift from Dr. L. Wood) were grown in DMEM supplemented with 15% FBS; HCT15 cells (Chk2-deficient) were grown in RPMI 1640 plus 15% FBS; and control (AHH1) and PCNTS629fs lymphoblastoid cells (PCNT−/−) (CV1576; affected by Seckel, PCNT 1887delA) (17), were cultured in RPMI 1640 supplemented with 15% FBS. When required, Doxorubicin, nocodazole, or cytochalasin B was directly added to the growth medium. Double thymidine block was performed by treating cells with 2.5 mm thymidine for 16 h and then releasing them into fresh medium for 8 h, performing a second block with 2.5 mm thymidine for 17 h, and performing a second release into fresh medium.
Transfections and RNA Interference
Transfections were carried out by Lipofectamine 2000 (Life Technologies) following the manufacturer's instructions. Plasmids containing Myc-Che-1 wild type or the mutant Myc-S4A have already been described (11). The 22-nucleotide siRNA duplexes corresponding to nucleotides 1062–1083 (siChe-1-1) and 1473–1492 (siChe-1-2) of human Che-1 sequence and to nucleotides 122–143 of the negative control green fluorescent protein (GFP) sequence were in vitro synthesized by the Silencer siRNA construction kit (Ambion) following the manufacturer's instructions. To silence Chk1 expression, cells were transfected with a Chk1-shRNA vector (18).
Flow Cytometry
Cells were trypsinized and resuspended in ice-cold PBS at a density of 1 × 106/ml, fixed by adding 2 ml of ice-cold 70% ethanol in PBS with vortexing, and incubated at least 30 min on ice. Cells were collected by centrifugation at 1,000 × g and treated with RNase A at 37 °C for 30 min. Finally, the cells were stained with propidium iodide and incubated in the dark for 60 min or overnight before analysis. Mitotic cells were determined by incubation with polyclonal anti-phosphorylated histone H3 antibody (1:100) and then stained with Alexa Fluor 488 anti-rabbit IgG antibody. The samples were analyzed through flow cytometry using a Beckman-Coulter Epics XL. A total of 10,000 cells were counted for each sample. Gating of cell population and quantitation of the cells were carried out using WinMDI.
Western Blot Analysis and Immunoprecipitations
Whole cell extracts were prepared as described previously (11). Hypotonic lysis buffer (50 mm Tris-HCl, pH 7.5, 5 mm EDTA, 10 mm NaCl, 0.05% Nonidet P-40) was used to isolate nuclear and cytoplasmic fractions. Solubilized proteins (25 μg) were resolved on MOPS NuPAGE precast 4–12% gels or on NuPAGE 3–8% Tris-acetate gels (Invitrogen). Western blots were performed using the following rabbit polyclonal antibodies: anti-Che-1 (19), PARP p85 fragment (Promega), cyclin B1 (Calbiochem), anti-phospho-Ser-10-histone H3 (Millipore), histone H3 (Millipore), pericentrin (Abcam), cleaved caspase-7 (Cell Signaling), phospho-Tyr-15-Cdc2 (Santa Cruz Biotechnology), Cdc2 (Santa Cruz Biotechnology), and Chk2 (Cell Signaling). Mouse monoclonal antibodies β-actin (Sigma), γ-tubulin (Sigma), Plk1 (Abcam), and Chk1 (DCS-310, Thermo Scientific) were also used. Secondary antibodies used were goat anti-mouse and goat anti-rabbit, conjugated to horseradish peroxidase (Bio-Rad). Immunostained bands were detected by the chemiluminescent method (Amersham Biosciences).
Densitometric analyses of immunoblots were performed using UVitec Alliance software version 16.07 (serial number 12-630588). Ratios were calculated by the following formula.
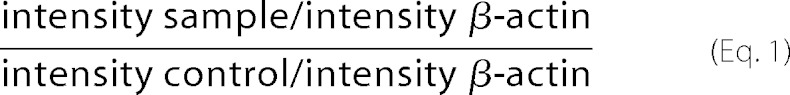 |
For immunoprecipitation experiments, cells were rinsed three times with ice-cold PBS, harvested, and centrifuged at 4 °C, and cell pellets were lysed by incubation at 0 °C for 30 min in lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 10% glycerol, 1% Nonidet P-40, 0.5 mm EDTA, 0.5 mm EGTA, 100 mm NaF, 3 mm Na3VO4, 10 nm okadaic acid, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin). After high speed centrifugation, the lysates were precleared with 20 μl of protein A/protein G beads (Santa Cruz Biotechnology) and immunoprecipitated by standard procedures. Western blots were prepared by standard procedures using antibodies described above. Immunoreactivity was detected by ECL chemiluminescence reaction (Amersham Biosciences).
Centrosome Isolation and Analysis
Centrosomes were isolated from all cell lines as described previously (20). In brief, exponentially growing cells were subjected to cytochalasin B and nocodazole treatment to disrupt microtubules and actin cytoskeleton and then lysed in a solution of 1 mm Tris-HCl (pH 8.0), 0.1% 2-mercaptoethanol, 0.5% Triton X-100. Centrosomes were harvested by centrifugation onto a 20% Ficoll cushion, and the centrosome fraction used for centrosome assays.
Microscopy
Adherent cells grown on glass coverslips or control and PCNT−/− lymphoblastoid cells cytocentrifuged on glass slides at 800 rpm for 4 min using a cytocentrifuge were fixed in 4% formaldehyde for 15 min and then permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 5 min. Indirect immunofluorescence staining was done using mouse monoclonal antibodies to γ-tubulin (GTU-88; Sigma), Chk1 (DCS-310), and α-tubulin (Calbiochem) and rabbit polyclonal antibodies to anti-Che-1 (19), pericentrin (Abcam), and cleaved caspase-7 (Cell Signaling). Antibody binding was revealed by incubation with the appropriate secondary antibodies: Alexa Fluor® 488 FITC-conjugated anti-mouse IgG, Alexa Fluor® 488 FITC-conjugated anti-rabbit IgG, Alexa Fluor® 594 TRITC-conjugated anti-mouse IgG, and Alexa Fluor® 594 TRITC-conjugated anti-rabbit IgG (Molecular Probes). Nuclei were visualized by staining with 1 μg/ml Hoechst dye 33258 (Sigma-Aldrich). Fluorescence images were captured using an Axioskop2 plus microscope (Zeiss) equipped with 40×, 63×, and 100× oil immersion objectives, a charge-coupled device camera (Zeiss, Oberkochen, Germany), and with AxioVision 4.7.1 software (Zeiss). Images were cropped and processed using Photoshop (CS5; Adobe).
Statistical Analysis
All analyses were performed in at least three independent experiments. Data were reported as mean ± S.D.
RESULTS
Che-1 Localizes at Interphase Centrosomes during Unperturbed Cell Cycle Progression
As reported above, Che-1 is a nuclear protein involved in the transcriptional regulation of the cell cycle and in the response to cellular stress (21). Nevertheless, it has been previously described that Che-1 interacts with centrosomal proteins (22, 23), and these observations prompted us to evaluate a possible Che-1 localization in this structure besides its well known nuclear localization. To do this, we examined the presence of Che-1 in centrosomes in HCT116 human colon carcinoma cells because these cells show a normal centrosome duplication and mitotic checkpoint. Isolated centrosomes analyzed by Western blotting revealed co-sedimentation of Che-1 with the centrosomal markers γ-tubulin and Plk1 (Fig. 1A), suggesting Che-1 as a centrosomal protein. These findings were confirmed by immunoprecipitation assays in which anti-Che-1 antibody was able to immunoprecipitate γ-tubulin (Fig. 1B), therefore indicating a specific interaction between these two proteins. To evaluate whether the centrosomal localization of Che-1 is regulated during the cell cycle, exponentially growing HCT116 cells in interphase or in M phase were analyzed by Western blot, and as shown in Fig. 1C, centrosomal accumulation of Che-1 was strongly reduced during mitosis. These results where confirmed from immunofluorescence analysis. Indeed, together with the expected strong nuclear signal, anti-Che-1 antibody decorated the centrosomes and colocalizes with γ-tubulin of the vast majority of growing HCT116 cells (78% out of 300 counted cells), and in contrast to centrosomes in interphase cells, mitotic spindle poles did not stain with anti-Che-1 antibody (Fig. 1D). Depletion of endogenous Che-1 by small interfering RNA resulted in an almost complete disappearance of both the nuclear and the centrosomal signal 24 h after transfection (Fig. 1E), thus confirming the specificity of the centrosomal Che-1 signal. Taken together, these data indicate that under physiological conditions, a fraction of human Che-1 associates with interphase centrosomes, from where it dissociates at mitosis.
FIGURE 1.

Che-1 localizes at interphase centrosomes during unperturbed cell cycle progression. A, cytoplasmic, nuclear, and centrosomal preparations from HCT116 cells were subjected to immunoblotting for the indicated proteins. The absence of β-actin expression was used as centrosome purification control. The purity of the fractions was documented with immunoblotting for histone 3 (nuclear) and γ-tubulin (cytoplasmic). B, total cellular lysates were immunoprecipitated with rabbit polyclonal Che-1 antibody, and resulting immunocomplexes were tested by immunoblotting using Che-1 and γ-tubulin antibodies. Immunoprecipitation (I.P.) negative control was performed by using normal rabbit IgG. C, Western blotting analysis of isolated centrosome preparations or whole cell lysates from HCT116 cells arrested in interphase or G2/M phase by double thymidine block. Ser-10 phosphorylated H3 histone (H3-S10-P) antibody was used as M phase control. D, HCT116 cells in interphase or metaphase were stained with Che-1 (red) and γ-tubulin (green) antibodies. Nuclei were visualized by staining with 1 μg/ml Hoechst dye 33258 (Sigma). Scale bar represents 5 μm. Insets show higher magnifications of the centrosomal Che-1 load. E, Che-1-depleted HCT116 cells were costained with Che-1 (red) and γ-tubulin (green) antibodies. Immunofluorescence microscopy was performed 24 h after transfection with siChe-1-1. Arrowheads point to centrosomes, which are shown enlarged in insets.
Che-1 Depletion Induces the Formation of Supernumerary Centrosomes and Leads to Aberrant Spindles
To determine a possible role of centrosomal Che-1, we transiently transfected HCT116 cells with GFP siRNA (siControl) or with two different Che-1 siRNAs (siChe-1-1 and siChe-1-2) (Fig. 2A, left panel) and examined the status of the centrosomes 24 h after depletion (Fig. 2A, middle panel). Che-1 depletion produced a strong increase of cells with centrosome amplification as compared with siControl cells (Fig. 2A, right panel). To determine whether the supernumerary centrosomes were able to mature and become functional, mitotic spindle structures were examined by using anti-pericentrin and anti-α-tubulin antibodies (Fig. 2B, left panel). In control cells, multipolar spindles were found in 1.2% of total cells, whereas in Che-1-depleted cells, the percentage dramatically increased to 5.3% (siChe-1-1) and to 4.9% (siChe-1-2) (Fig. 2B, right panel). Consistent with these results, Che-1 siRNA produced a significant increase of phospho-Ser-10-histone H3 levels (a mitotic marker) with respect to the control (Fig. 2C, left and middle panel), thus suggesting Che-1 involvement in the control of mitotic entry. To more carefully examine the effects of Che-1 depletion, we coupled siRNA transfection with cell synchronization by double thymidine block. As shown in Fig. 2D (right panel), control cells began to enter mitosis 8 h after release, reaching a peak of phospho-H3 signal at 10 h. In contrast, Che-1-depleted cells began to enter mitosis 4 h after release, indicating that Che-1 depletion led to premature mitotic entry. Altogether, these results support the involvement of Che-1 in maintaining numerical centrosome integrity and in controlling cell cycle progression during unperturbed mitosis.
FIGURE 2.

Che-1 depletion induces the formation of supernumerary centrosomes and leads to aberrant spindles. A, left, HCT116 cells were transiently transfected with GFP siRNA (siControl) or with two different Che-1 siRNAs (siChe-1-1 and siChe-1-2), and Western blotting was performed 24 h after transfection. Middle, supernumerary centrosomes analysis was performed by immunofluorescence after staining with pericentrin (green) antibody. Scale bar represents 10 μm. Insets show higher magnifications of centrosomes. Right, bar diagrams show the percentage of cells containing more than two centrosomes. B, left, multiple spindles analysis was performed by immunofluorescence after staining with pericentrin (green) and α-tubulin (red) antibodies. Scale bar represents 10 μm. Nuclei were visualized by staining with Hoechst dye 33258. Right, bar diagrams show the percentage of cells containing aberrant spindles. Error bars represent the S.D. after combining the results of three different experiments. C, left, HCT116 cells were transiently transfected as in A, and Western blotting was performed using the indicated antibodies 24 h after transfection. Right, bar diagrams show the percentage of mitotic cells assessed by immunofluorescence using an antibody Ser-10-histone H3 (H3-S10-P) as a mitotic marker. D, Che-1 was depleted with siRNA in well synchronized HCT116 cells by using the double thymidine block. After second release from the block, cells were harvested at different times and analyzed by Western blotting using the antibodies indicated.
Che-1 Accumulates at Centrosomes in Response to DNA Damage
Several findings demonstrated that Che-1 contributes to the maintenance of the G2/M checkpoint in response to genotoxic stress (8). At the same time, it is now generally accepted that the centrosome is involved in the DNA damage response (7). To directly address whether centrosomal Che-1 is responsive to DNA damage, HCT116 cells were treated with Doxorubicin (Dox) or ionizing radiation (I.R.) and immunostained with anti-γ-tubulin and anti-Che-1 antibodies. Both types of DNA-damaging treatments led to centrosomal accumulation of endogenous Che-1, as demonstrated by the increase of the intensity of centrosomal Che-1 signal (Fig. 3A). In agreement with this observation, centrosome extracts from HCT116 cells treated with genotoxic stresses contained increased levels of endogenous Che-1 (Fig. 3B).
FIGURE 3.

Che-1 accumulates at centrosomes in response to DNA damage. A, HCT116 cells were treated with 10 grays of I.R. or with 1 μm Dox. Top, cells were stained with antibodies to Che-1 (red) and to γ-tubulin (green) for identification of centrosomes. Bottom, quantification of pixel intensity profiles constructed from fluorescence images of HCT116 cells untreated or treated with 10 grays of I.R. or with 1 μm Dox, using ImageJ software (National Institutes of Health, Bethesda, MD). B, isolated centrosomes or whole cell lysates from HCT116 cells treated as in A were subjected to immunoblotting with the indicated antibodies. C and D, HCT116 cells were transiently transfected with GFP siRNA (siControl) or Che-1 siRNA (siChe-1-1) and treated with 10 grays of I.R. Cells were stained with the indicated antibodies. Nuclei were visualized by staining with Hoechst dye 33258. Bar diagrams show the percentage of cells containing multipolar spindles or more than two centrosomes. In diagrams, gray bars indicate siControl transfected cells, and black bars indicate siChe-1-1-depleted cells. Error bars represent the S.D. after combining the results of three different experiments. Scale bar represents 5 μm. Arrowheads point to centrosomes, which are shown enlarged in insets. E and F, isolated centrosomes or whole cell lysates from HCT116 and HCT15 (Chk2-deficient) cells (E) or from normal Human and ATM−/− human fibroblasts (F) were subjected to immunoblotting with the indicated antibodies (left). Cells were immunostained with the indicated antibodies (right). G, left, isolated centrosome preparations or whole cell lysates from HCT116 transfected with Myc-Che-1 or Myc-S4A were analyzed by Western blot using the indicated antibodies. Right, cells were immunostained with antibodies to γ-tubulin (green) and Myc (red) antibodies. Nuclei were visualized by staining with 1 μg/ml Hoechst dye 33258. The scale bar represents 5 μm. Arrowheads point to centrosomes, which are shown enlarged in insets.
Recent studies suggest that various elements of DDR can regulate the centrosome cycle (24). Thus, we sought to determine the effect of Che-1 depletion on centrosome duplication in response to DNA damage. To do this, we evaluated the number of centrosomes and the mitotic spindle structures in the siControl or siChe-1 cells in response to I.R. Immunofluorescence analysis revealed that irradiation further exacerbated the extent of centrosome amplification and of multipolar spindles in the Che-1 knockdown cells (Fig. 3, C and D).
It has been reported that in response to DNA damage, ATM/ATR and Chk2 kinases phosphorylate Che-1, increasing its half-life and relocalizing it onto the chromatin (11). Because ATM is responsible for p53 centrosomal localization (25), to verify whether Che-1 phosphorylation is required for its localization at centrosomes, we evaluated Che-1 centrosomal localization in cells lacking ATM or Chk2 expression. As shown in Fig. 3, E and F, Che-1 was found at the centrosome in ATM- or Chk2-deficient cells (HCT15 cells), indicating that in this case, these kinases do not regulate Che-1 localization. Consistent with these results, a Che-1 mutant lacking ATM and Chk2 phosphorylation sites (Myc-S4A, 11) localized at centrosomes as well as wild type protein (Fig. 3G). These data collectively indicated that Che-1 plays an important role in regulating centrosome duplication, both in physiological conditions and in response to DNA damage.
Che-1 Depletion Overrides Spindle Assembly Checkpoint and Induces Apoptosis
It has been previously reported that Che-1 contributes to the maintenance of the G2/M checkpoint in response to genotoxic stresses (11). Aside from this important function, the results shown above lead to hypothesize an involvement of Che-1 in the spindle assembly checkpoint (SAC) (26). To reveal the potential regulation of Che-1 in response to spindle damage, HCT116 cells were treated with the microtubule-depolymerizing agent nocodazole. As shown in Fig. 4A, nocodazole treatment significantly increased Che-1 cellular levels and its accumulation at the centrosome. To explore the role of Che-1 in the spindle checkpoint, we examined whether Che-1 was required for mitotic arrest in response to nocodazole. After drug treatment, siControl cells exhibited a clear G2/M accumulation, as shown by the increase of cyclin B1 levels (Fig. 4B, left panel) and by the augmentation of the percentage of cells expressing phospho-Ser-10-histone H3 (Fig. 4B, right panel). In contrast, Che-1-depleted cells exhibited a weakened SAC in response to nocodazole or I.R. treatments, resulting in those cells being unable to maintain high levels of cyclin B1 (Fig. 4B, left panel, and 4C) and phospho-Ser-10-histone H3 (Fig. 4B, right panel), with consequent failure to sustain mitotic arrest. Consistent with these results, the majority of cells transfected with Che-1 siRNA and treated with nocodazole underwent cell death, as emphasized by the increase in the apoptotic sub-G1 population (Fig. 4D) and by the expression of the cleaved forms of poly(ADP-ribose) polymerase and caspase-7, specific hallmarks of apoptosis (Fig. 4, B and C). Importantly, Che-1-depleted cells showed a high degree of nuclear fragmentation and micronuclei formation (Fig. 4E), a sign usually associated with mitotic catastrophe (27, 28), as confirmed by the presence of cleaved caspase-3 in such cells. Taken together, these results clearly indicate that Che-1 is involved in SAC and that its depletion induces mitotic arrest slippage and apoptosis.
FIGURE 4.

Che-1 depletion overrides spindle assembly checkpoint and induces apoptosis. A, isolated centrosomes or whole cell lysates from asynchronous HCT116 cells or treated with 0.2 μg/ml nocodazole (noc) were subjected to immunoblotting with the indicated antibodies. H3-S10-P, anti-Ser-10-histone H3. B, left, HCT116 cells transiently transfected with GFP siRNA (siControl) or Che-1 siRNA (siChe-1-1) were treated with 0.2 μg/ml nocodazole for the indicated times, and total cellular extracts were analyzed by Western blot with the indicated antibodies. Right, the percentage of mitotic cells was assessed by flow cytometry using an antibody anti-Ser-10-histone H3 as a mitotic marker. Parp, poly(ADP-ribose) polymerase. Cleav. Caspase7, cleaved caspase-7. C, HCT116 cells transiently transfected as in B were treated with 10 grays (10Gy) of I.R., and total cellular extracts were analyzed by Western blotting with the indicated antibodies at the indicated times. D, FACS profiles of HCT116 treated as in B and stained with propidium iodide at the indicated times. E, top, cells were fixed in methanol and stained with antibody to cleaved caspase-3 (red) and counterstained with Hoechst (blue). The scale bar represents 10 μm. Bottom, bar diagrams show the percentage of cells positive for cleaved caspase-3. At least 3 × 100 cells were counted per data bar. Results are given as mean ± S.D.
Che-1 Regulates Chk1 Centrosomal Localization
The data described above demonstrate that in unperturbed cells, Che-1 localizes at centrosomes in interphase, but dissociates from centrosomes in mitosis, whereas following mitotic spindle damage, Che-1 accumulates at mitotic centrosomes and is involved in SAC activation. As another checkpoint protein, the kinase Chk1, exhibits a similar behavior (29), we examined a possible relationship between centrosomal Che-1 and Chk1. To this aim, endogenous Chk1 was immunoprecipitated from HCT116 cells that were untreated or treated with Dox. Dox treatment substantially increased the levels of Che-1 that coprecipitated with Chk1 as compared with untreated cells (Fig. 5A), indicating that the two proteins belong to the same complex. Next, we analyzed whether Che-1 may impact the abundance of Chk1 at centrosomes. As shown in Fig. 5B, immunofluorescence analysis revealed a marked Chk1 centrosomal staining in control cells, but this localization was lost in Che-1-depleted cells, thus indicating that Che-1 is required for Chk1 centrosomal localization. These results were confirmed by Western blot analysis, in which Che-1-depleted cells exhibited a strong reduction of centrosomal Chk1 as compared with control cells (Fig. 5C). Conversely, Chk1 depletion did not impact the abundance of Che-1 at centrosomes (Fig. 5, D and E), thus indicating that no reciprocal regulation between Chk1 and Che-1 exists. It has been described that centrosome-associated Chk1 controls premature activation of cyclin B-Cdk1 complex at the centrosome, regulating in such a way mitotic entry (15, 30). Consistent with these observations, Che-1 depletion produced a marked reduction of centrosomal levels of phosphorylated Cdk1 and cyclin B1 (Fig. 5F), thus confirming that Che-1 regulates Chk1 centrosomal localization and its mitotic regulation.
FIGURE 5.

Che-1 regulates Chk1 centrosomal localization. A, total cellular extracts from HCT116 cells, untreated or treated with 1 μm Doxorubicin for 2 h, were immunoprecipitated (I.P.) with anti-Chk1 monoclonal antibody and analyzed by Western blot using the indicated antibodies. Neg. control, negative control. B, HCT116 cells transiently transfected with GFP siRNA (siControl) or Che-1 siRNA (siChe-1-1) were immunostained with the indicated antibodies. Bar diagrams show the percentages of cells with centrosomal colocalization of γ-tubulin and Chk1. C, isolated centrosomes or whole cell lysates from HCT116 cells transiently transfected as in B were analyzed by Western blot with the indicated antibodies. D, HCT116 cells transiently transfected with shRNA GFP (shControl) or shRNA Chk1 (shChk1) were immunostained with the indicated antibodies. Bar diagrams show the percentages of cells with centrosomal colocalization of γ-tubulin and Che-1. E, isolated centrosomes or whole cell lysates from HCT116 cells transiently transfected as in D were analyzed by Western blot using the indicated antibodies. F, isolated centrosomes or whole cell lysates from HCT116 cells transiently transfected as in B were analyzed by Western blot with the indicated antibodies. Nuclei were visualized by staining with 1 μg/ml Hoechst dye 33258.The scale bar represents 5 μm. At least 3 × 100 cells were counted per data bar. Results are given as mean ± S.D.
Che-1 Binds Pericentrin and Mediates Its Binding to Chk1
It has been described that another important protein involved in DNA damage response, PCNT, is required for Chk1 localization at centrosomes (15). As PCNT has an important role in targeting regulatory proteins to the centrosome (16, 31), we evaluated whether deficiency of PCNT impacts on the abundance of Che-1 at centrosomes. As shown in Fig. 6A, Che-1 centrosomal levels were reduced in lymphoblastoid cells lacking PCNT expression (17) as compared with control lymphoblastoid cells. Similar results were obtained by Western blots analyzing centrosome preparations from these cells (Fig. 6B), whereas whole cell lysates from control and PCNT−/− lymphoblastoid cells exhibited similar Che-1 levels. To examine the relationship between Che-1 and PCNT in more detail, endogenous Che-1 was immunoprecipitated from Dox-treated and untreated HCT116 cells. Endogenous PCNT was detectable in both immunoprecipitations, but it increased in response to DNA damage (Fig. 6C). Notably, Che-1 depletion reduced Chk1/PCNT interaction (Fig. 6D), whereas Che-1 was able to bind Chk1 in PCNT−/− cells (Fig. 6E) and to interact with PCNT in Chk1-depleted cells (Fig. 6F). Altogether, these findings indicate that Che-1 is part of a complex containing PCNT and Chk1 and that its presence is required for this interaction.
FIGURE 6.

Che-1 binds pericentrin and mediates its binding to Chk1. A, human lymphoblastoid AHH1 cells and CV1576 lymphoblastoid cells lacking PCNT expression were immunostained with the indicated antibodies. Bar diagrams show the percentages of cells with centrosomal colocalization of γ-tubulin and Che-1. B, Western blot analysis of isolated centrosome preparations or whole cell lysates from human lymphoblastoid AHH1 cells and PCNT-deficient cells. C, Western blot analysis of HCT116 cells untreated or treated with 1 μm Doxorubicin for 2 h. Total cellular extracts were immunoprecipitated (I.P.) with anti-Che-1 antibody and analyzed by Western blot using the indicated antibodies. D, total cellular extracts from HCT116 cells transiently transfected with GFP siRNA (siControl) or Che-1 siRNA (siChe-1) were immunoprecipitated with anti-Chk1 antibody and analyzed by Western blot using the indicated antibodies. The scale bar represents 5 μm. E, total cellular extracts from human lymphoblastoid AHH1 cells and CV1576 PCNT-deficient cells were immunoprecipitated (I.P.) with anti-Che-1 antibody and analyzed by Western blot using the indicated antibodies. F, total cellular extracts from HCT116 cells transiently transfected with GFP siRNA (siControl) or Chk1 siRNA (siChk1) were immunoprecipitated (I.P.) with anti-Che-1 antibody and analyzed by Western blot using the indicated antibodies. Immunoprecipitation negative control (Neg. control) was performed by using normal rabbit IgG. Scale bar represents 5 μm. Error bars represent S.D. after combining the results of three different experiments. Arrowheads point to centrosomes, which are shown enlarged in insets.
DISCUSSION
The DNA damage response and the spindle assembly checkpoint are two critical mechanisms by which mammalian cells maintain genome stability, and there is a growing body of evidence of a cross-talk between these two checkpoint, Indeed, a number of DNA damage responsive elements, including ATM, Chk1, Chk2, and Brca1, have been reported to play a role in spindle checkpoint activation (6, 17, 32, 33). In addition, several proteins involved in SAC such as Bub1 and Mad2 have been shown to play an important role in G2/M checkpoint (33–36). In particular, Chk1 kinase has been found to sustain activation of SAC in response to DNA damage (36). Therefore, altogether these findings show that the two checkpoints are not independent but that they cooperate to restrict mitotic progression in response to DNA damage.
In this study, we provide evidence that Che-1, another protein involved in G2/M checkpoint (11), contributes to integrate DNA damage response and spindle assembly checkpoint. Indeed, we found that human Che-1 localizes to interphase centrosomes, from where it is released at the onset of mitosis, whereas in response to DNA damage, it is specifically enriched at this structure. In addition, Che-1-depleted cells exhibit abnormal spindle structure and an advanced entry into mitosis both in unperturbed cells and following spindle poisons, suggesting Che-1 involvement in SAC regulation.
Consistently, we show here for the first time that Che-1 functions as an upstream regulator of Chk1 centrosomal functions. As a matter of fact, Chk1 centrosomal fraction appears to play a pivotal role in coupling S-phase and mitosis both in unperturbed cells and in damaged cells. Chk1 has been shown to prevent premature entry into mitosis by inhibiting Cdc25B at centrosomes (30). Chk1 strongly accumulates at centrosomes and mediates centrosome amplification following DNA damage (29). In unperturbed mitosis, Chk1 is phosphorylated at Ser-345 and detaches from centrosomes at prophase (37). Finally, when Chk1 is stably tagged to centrosomes, centrosome amplification occurs both in untreated cells and in DNA-damaged cells (29, 30).
Our results show that Che-1 exhibits a similar behavior, leading to the hypothesis that Che-1, besides its well known function in the G2/M checkpoint (11), is also involved in SAC, at least in part, by regulating Chk1 functions. Consistently, centrosome preparations from Che-1-depleted cells contained reduced levels of Chk1. Moreover, Che-1 binds PCNT, and this interaction is required for Che-1 centrosome localization because cells lacking PCNT expression do not exhibit centrosomal Che-1. At the same time, Che-1 depletion strongly affected PCNT/Chk1 interaction, thus suggesting a centrosomal complex PCNT-Che-1-Chk1, in which PCNT is crucial in directing the complex to centrosomes and Che-1 is crucial in connecting PCNT to Chk1.
Accumulating evidence reveals that in response to several cellular stresses, Che-1 is subjected to post-translational modifications that regulate its functions and localization (11, 13). Although we observed that ATM/ATR and Chk2 are not involved in Che-1 centrosomal localization, we cannot exclude that other modifications are responsible for this localization or for its removal from this structure. Further experiments are required to test this hypothesis.
In summary, our findings identify Che-1 as an important regulator of SAC response, underscoring integration between DDR and SAC at the centrosome and strengthening the notion that Che-1 can be considered an important target for enhancing the efficacy of antitumoral chemotherapeutic treatments.
This work was supported by the Italian Association for Cancer Research (AIRC) (to M. F.).
- DDR
- DNA damage response
- I.R.
- ionizing radiation
- Dox
- Doxorubicin
- PCNT
- pericentrin
- SAC
- spindle assembly checkpoint
- ATM
- ataxia telangiectasia-mutated
- ATR
- ATM and Rad3-related
- TRITC
- tetramethylrhodamine isothiocyanate
- FITC
- fluorescein isothiocyanate.
REFERENCES
- 1. Jackson S. P., Bartek J. (2009) The DNA-damage response in human biology and disease. Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Halazonetis T. D., Gorgoulis V. G., Bartek J. (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 [DOI] [PubMed] [Google Scholar]
- 3. Salisbury J. L., Whitehead C. M., Lingle W. L., Barrett S. L. (1999) Centrosomes and cancer. Biol. Cell 91, 451–460 [PubMed] [Google Scholar]
- 4. Marx J. (2001) Cell biology. Do centrosome abnormalities lead to cancer? Science 292, 426–429 [DOI] [PubMed] [Google Scholar]
- 5. Ciciarello M., Mangiacasale R., Casenghi M., Zaira Limongi M., D'Angelo M., Soddu S., Lavia P., Cundari E. (2001) p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 276, 19205–19213 [DOI] [PubMed] [Google Scholar]
- 6. Oricchio E., Saladino C., Iacovelli S., Soddu S., Cundari E. (2006) ATM is activated by default in mitosis, localizes at centrosomes, and monitors mitotic spindle integrity. Cell Cycle 5, 88–92 [DOI] [PubMed] [Google Scholar]
- 7. Zhang S., Hemmerich P., Grosse F. (2007) Centrosomal localization of DNA damage checkpoints proteins. J. Cell. Biochem. 101, 451–465 [DOI] [PubMed] [Google Scholar]
- 8. Floridi A., Fanciulli M. (2007) Che-1: a new effector of checkpoints signaling. Cell Cycle 6, 804–806 [DOI] [PubMed] [Google Scholar]
- 9. Bruno T., De Angelis R., De Nicola F., Barbato C., Di Padova M., Corbi N., Libri V., Benassi B., Mattei E., Chersi A., Soddu S., Floridi A., Passananti C., Fanciulli M., (2002) Che-1 affects cell growth by interfering with the recruitment of HDAC1 by Rb. Cancer Cell 2, 387–399 [DOI] [PubMed] [Google Scholar]
- 10. Ishigaki S., Fonseca S. G., Oslowski C. M., Jurczyk A., Shearstone J. R., Zhu L. J., Permutt M. A., Greiner D. L., Bortell R., Urano F. (2010) AATF mediates anti-apoptotic effect of the unfolded protein response through transcriptional regulation of AKT1. Cell. Death Differ. 17, 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bruno T., De Nicola F., Iezzi S., Lecis D., D'Angelo C., Di Padova M., Corbi N., Dimiziani L., Zannini L., Jekimovs C., Scarsella M., Porrello A., Chersi A., Crescenzi M., Leonetti C., Khanna K. K., Soddu S., Floridi A., Passananti C., Delia D., Fanciulli M. (2006) Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 10, 473–486 [DOI] [PubMed] [Google Scholar]
- 12. Bruno T., Iezzi S., De Nicola F., Di Padova M., Desantis A., Scarsella M., Di Certo M. G., Leonetti C., Floridi A., Passananti C., Fanciulli M. (2008) Che-1 activates XIAP expression in response to DNA damage. Cell. Death Differ. 15, 515–520 [DOI] [PubMed] [Google Scholar]
- 13. Höpker K., Hagmann H., Khurshid S., Chen S., Hasskamp P., Seeger-Nukpezah T., Schilberg K., Heukamp L., Lamkemeyer T., Sos M. L., Thomas R. K., Lowery D., Roels F., Fischer M., Liebau M. C., Resch U., Kisner T., Röther F., Bartram M. P., Müller R. U., Fabretti F., Kurschat P., Schumacher B., Gaestel M., Medema R. H., Yaffe M. B., Schermer B., Reinhardt H. C., Benzing T. (2012) AATF/Che-1 acts as a phosphorylation-dependent molecular modulator to repress p53-driven apoptosis. EMBO J. 31, 3961–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bruno T., Desantis A., Bossi G., Di Agostino S., Sorino C., De Nicola F., Iezzi S., Franchitto A., Benassi B., Galanti S., La Rosa F., Floridi A., Bellacosa A., Passananti C., Blandino G., Fanciulli M. (2010) Che-1 promotes tumor cell survival by sustaining mutant p53 transcription and inhibiting DNA damage response activation. Cancer Cell 18, 122–134 [DOI] [PubMed] [Google Scholar]
- 15. Tibelius A., Marhold J., Zentgraf H., Heilig C. E., Neitzel H., Ducommun B., Rauch A., Ho A. D., Bartek J., Krämer A. (2009) Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. J. Cell Biol. 185, 1149–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diviani D., Langeberg L. K., Doxsey S. J., Scott J. D. (2000) Pericentrin anchors protein kinase A at the centrosome through a newly identified RII-binding domain. Curr. Biol. 10, 417–420 [DOI] [PubMed] [Google Scholar]
- 17. Griffith E., Walker S., Martin C. A., Vagnarelli P., Stiff T., Vernay B., Al Sanna N., Saggar A., Hamel B., Earnshaw W. C., Jeggo P. A., Jackson A. P., O'Driscoll M. (2008) Mutations in Pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 40, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krause D. R., Jonnalagadda J. C., Gatei M. H., Sillje H. H., Zhou B. B., Nigg E. A., Khanna K. (2003) Suppression of Tousled-like kinase activity after DNA damage or replication block requires ATM, NBS1 and Chk1. Oncogene 22, 5927–5937 [DOI] [PubMed] [Google Scholar]
- 19. Fanciulli M., Bruno T., Di Padova M., De Angelis R., Iezzi S., Iacobini C., Floridi A., Passananti C. (2000) Identification of a novel partner of RNA polymerase II subunit 11, Che-1, which interacts with and affects the growth suppression function of Rb. FASEB J. 14, 904–912 [DOI] [PubMed] [Google Scholar]
- 20. Blomberg-Wirschell M., Doxsey S. J. (1998) Rapid isolation of centrosomes. Methods Enzymol. 298, 228–238 [DOI] [PubMed] [Google Scholar]
- 21. Passananti C., Fanciulli M. (2007) The anti-apoptotic factor Che-1/AATF links transcriptional regulation, cell cycle control, and DNA damage response. Cell Div. 2, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Felten A., Leister P., Burgdorf S., Uhlmann L., Scheidtmann K. H. (2007) Characterization of rat BLOS2/Ceap, a putative yeast She3 homolog, as interaction partner of apoptosis antagonizing transcription factor/Che-1. Biol. Chem. 388, 569–582 [DOI] [PubMed] [Google Scholar]
- 23. Robert A., Margall-Ducos G., Guidotti J. E., Brégerie O., Celati C., Bréchot C., Desdouets C. (2007) The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cell. J. Cell Sci. 120, 628–637 [DOI] [PubMed] [Google Scholar]
- 24. Wang Y., Ji P., Liu J., Broaddus R. R., Xue F., Zhang W. (2009) Centrosome-associated regulators of the G2/M checkpoint as targets for cancer therapy. Mol. Cancer 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tritarelli A., Oricchio E., Ciciarello M., Mangiacasale R., Palena A., Lavia P., Soddu S., Cundari E. (2004) p53 localization at centrosomes during mitosis and postmitotic checkpoint are ATM-dependent and require serine 15 phosphorylation. Mol. Biol. Cell 15, 3751–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Musacchio A., Salmon E. D. (2007) The spindle assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8, 379–393 [DOI] [PubMed] [Google Scholar]
- 27. Castedo M., Perfettini J. L., Roumier T., Andreau K., Medema R., Kroemer G. (2004) Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825–2837 [DOI] [PubMed] [Google Scholar]
- 28. Castedo M., Kroemer G. (2004) Mitotic catastrophe: a special case of apoptosis. J. Soc. Biol. 198, 97–103 [PubMed] [Google Scholar]
- 29. Löffler H., Bochtler T., Fritz B., Tews B., Ho A. D., Lukas J., Bartek J., Krämer A. (2007) DNA damage-induced accumulation of centrosomal Chk1 contributes to its checkpoint function. Cell Cycle 6, 2541–2548 [DOI] [PubMed] [Google Scholar]
- 30. Krämer A., Mailand N., Lukas C., Syljuåsen R. G., Wilkinson C. J., Nigg E. A., Bartek J., Lukas J. (2004) Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 6, 884–891 [DOI] [PubMed] [Google Scholar]
- 31. Chen D., Purohit A., Halilovic E., Doxsey S. J., Newton A. C. (2004) Centrosomal anchoring of protein kinase C βII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J. Biol. Chem. 279, 4829–4839 [DOI] [PubMed] [Google Scholar]
- 32. Kim E. M., Burke D. J. (2008) DNA damage activates the SAC in an ATM/ATR-dependent manner, independently of the kinetochore. PLoS Genet. 4, e1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang C., Wang H., Xu Y., Brinkman K. L., Ishiyama H., Wong S. T. C., Xu B. (2012) The kinetochore protein Bub1 partecipates in the DNA damage response. DNA repair 11, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dotiwala F., Harrison J. C., Jain S., Sugawara N., Haber J. E. (2010) Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr. Biol. 20, 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collura A., Blaisonneau J., Baldacci G., Francescani S. (2005) The fission yeast Crb2/Chk1 pathway coordinates the DNA damage and spindle checkpoint in response to replication stress induced by topoisomerase I inhibitor. Mol. Cell. Biol. 25, 7889–7899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chilà R., Celenza C., Lupi M., Damia G., Carrassa L. (2013) Chk1-Mad2 interaction: a crosslink between the DNA damage checkpoint and the mitotic spindle checkpoint. Cell Cycle 12, 1083–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilsker D., Petermann E., Helleday T., Bunz F. (2008) Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc. Natl. Acad. Sci. U.S.A. 105, 20752–20757 [DOI] [PMC free article] [PubMed] [Google Scholar]