

Background: The mechanisms for angiotensin (Ang) II-induced podocytopathy are not clear.
Results: Ang II induced calmodulin-dependent protein kinase (CaMK) II/cAMP response element-binding protein (CREB) and Wnt/β-catenin signaling activation in podocytes. Blocking CaMK II/CREB inhibited Ang II-induced Wnt/β-catenin signaling and blocking CaMK II/CREB/Wnt/β-catenin signaling attenuated Ang II-induced podocyte injury.
Conclusion: CaMK II/CREB/Wnt/β-catenin signaling cascade regulates Ang II-induced podocytopathy.
Significance: Targeting this pathway may offer protection against podocyte injury.
Keywords: Angiotensin II, β-Catenin, Calcium-calmodulin-dependent Protein Kinase (CaMK), Podocytes, Wnt Signaling
Abstract
Angiotensin II (Ang II) plays a pivotal role in promoting podocyte dysfunction and albuminuria, however, the underlying mechanisms have not been fully delineated. In this study, we found that Ang II induced Wnt1 expression and β-catenin nuclear translocation in cultured mouse podocytes. Blocking Wnt signaling with Dickkopf-1 (Dkk1) or β-catenin siRNA attenuated Ang II-induced podocyte injury. Ang II could also induce the phosphorylation of calmodulin-dependent protein kinase (CaMK) II and cAMP response element-binding protein (CREB) in cultured podocytes. Blockade of this pathway with CK59 or CREB siRNA could significantly inhibit Ang II-induced Wnt/β-catenin signaling and podocyte injury. In in vivo studies, administration of Ang II promoted Wnt/β-catenin signaling, aggregated podocyte damage, and albuminuria in mice. CK59 could remarkably ameliorate Ang II-induced podocyte injury and albuminuria. Furthermore, ectopic expression of exogenous Dkk1 also attenuated Ang II-induced podocytopathy in mice. Taken together, this study demonstrates that the CaMK II/CREB/Wnt/β-catenin signaling cascade plays an important role in regulating Ang II-induced podocytopathy. Targeting this signaling pathway may offer renal protection against the development of proteinuric kidney diseases.
Introduction
Podocytes are highly differentiated cells that reside on glomerular base membrane. They form the outer layer of the glomerular filtration barrier and prevent certain blood components such as albumin from leaking into urine (1). The maintenance of their highly specialized differentiated state is indispensable for keeping normal structure and function of glomerular filtration barrier. Under diseased conditions, podocytes may undergo several adaptive changes, such as hypertrophy and dedifferention, such adaptive changes will affect the podocyte differentiated state, leading to podocyte dysfunction and proteinuria. Various metabolic or hemodynamic factors can damage podocytes (2). Among them, angiotensin II (Ang II),4 a major component of renin-angiotensin system, plays a critical role in podocyte function and inducing albuminuria, such as in diabetic nephropathy (3, 4).
Ang II can induce changes in podocyte phenotype from a dynamically stable state to adaptively migratory state, which eventually exhausts podocytes with a high turnover of actin cytoskeletal and causes podocyte depletion and focal segmental glomerulosclerosis (5, 6). Podocytes express both AT1 and AT2 receptors that provide high-affinity binding sites for Ang II (7–13). Administration of exogenous Ang II or targeted overexpression of AT1 to glomerular podocytes resulted in albuminuria and focal segmental glomerular sclerosis in mice (4, 8, 14, 15). On the other hand, blocking the renin-angiotensin system with ACE-I or ARB could benefit patients with chronic kidney disease (13, 16–21). Previous studies have also demonstrated that Ang II could stimulate podocytes to undergo de-differentiation in an Ang II type 1 receptor-mediated fashion (7, 22, 23). Those studies have established an intimate linkage of Ang II to the pathogenesis of podocyte injury and proteinuria. However, the underlying mechanisms for Ang II inducing podocyte injury need more investigation.
The Wnt gene family, which encodes secreted growth and differentiation factors, has been implicated in controlling many biological processes (24, 25). Canonical Wnt/β-catenin signaling is an evolutionarily conserved signaling pathway. Emerging evidence suggests a pathogenic involvement for Wnt/β-catenin signaling in various kidney diseases, including kidney cancer, renal fibrosis, cystic kidney diseases, and diabetic nephropathy (26–30). Our previous studies have shown that activation of Wnt/β-catenin signaling via adriamycin or TGFβ1 led to podocyte injury and albuminuria (27, 31). Other studies reported that Ang II could induce Ca2+ accumulation in podocytes through both influx of Ca2+ and the release of Ca2+ from intracellular stores (32, 33). Calmodulin-dependent protein kinase (CaMK) II can be activated after intracellular calcium accumulation in many cell types (34). CaMK II activates the downstream pathway that is mediated by the transcription factor cAMP response element (CRE)-binding protein (CREB). Recent studies also found that CaMKs/CREB activation is accompanied by up-regulation of Wnt, suggesting a potential involvement of CaMK II/CREB/Wnt signaling in the pathogenesis of podocyte injury (35, 36). Considering Ang II is able to activate CaMK signaling in many cell types, it is highly possible that Wnt/β-catenin may be activated downstream of CaMK signaling by Ang II in podocytes, which likely mediates Ang II-induced podocyte injury and proteinuria.
Here we demonstrate that Ang II can promote canonical Wnt signaling activation and podocyte damage. Ang II induced Wnts expression is mediated by CaMK II/CREB signaling activation. Blocking this signaling cascade ameliorates Ang II-triggered podocyte injury in vitro and in vivo. This study provides a mechanistic insight about CaMK II/CREB/Wnt/β-catenin signaling in Ang II-induced podocyte damage and albuminuria.
MATERIALS AND METHODS
Cell Culture and Treatment
The conditionally immortalized mouse podocyte cell line was kindly provided by Dr. Peter Mundel. To propagate podocytes, cells were cultured at 33 °C in RPMI 1640 medium supplemented with 10% fetal bovine serum and recombinant mouse interferon-γ (R&D Systems, Minneapolis, MN). To induce differentiation, podocytes were grown under nonpermissive conditions at 37 °C in the absence of interferon-γ and in the presence of retinoic acid (10−6 m) and 1,25-dihydroxyvitamin D (10−7 m) (37). Podocytes were treated with recombinant Ang II (Sigma) at 1 μm, unless otherwise indicated. For some experiments, podocytes were transiently transfected with a FLAG-tagged Dkk1 expression plasmid (pDkk1, provided by Dr. Xi He, Harvard Medical School, Boston, MA), β-catenin siRNA, or CREB siRNA by using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. For the CK59 experiment, podocytes were pretreated with CK59 (208922, Merck KgcA, Germany) at 20 or 50 μm for 30 min followed by Ang II incubation.
DNA Constructs
Mouse Ang II full-length cDNA was generated from mouse kidney tissue by RT-PCR using the following primers: sense, 5′-AGCTTATGCACAGATCGGAGATGACTC-3′, antisense, 5′-GAATTCGAAGGGGTGGATGTATACGCGG-3′. The amplified gene was confirmed by sequencing and cloned into pcDNA3 vector for expression in mammalian cells. Plasmids encoding Dkk1 were used as described previously (31, 38).
Transfection and Luciferase Assay
The effect of Ang II on β-catenin-mediated gene transcription was detected using the TOP-flash TCF reporter plasmid containing two sets of three copies of the TCF binding site upstream of the thymidine kinase (TK) minimal promoter and luciferase open reading frame (Millipore, Billerica, MA). Podocytes were cotransfected with TOP-flash or FOP-flash plasmid (1 μg) and an internal control reporter plasmid (0.1 μg) Renilla reniformis luciferase driven under the TK promoter (pRL-TK; Promega). The transfected cells were incubated in serum-free medium without or with Ang II (10−6 m) as indicated. Luciferase assay was performed using a dual luciferase assay system kit according to the manufacturer's protocols (Promega). Relative luciferase activity (arbitrary units) was reported as fold-induction over the controls after normalizing for transfection efficiency.
Animal Models
Male CD1 or female Balb/c mice that weighed ∼20–22 g were acquired from the Specific Pathogen-Free Laboratory Animal Center of Nanjing Medical University and maintained according to the guidelines of the Institutional Animal Care and Use Committee at Nanjing Medical University. Ang II was infused at the rate of 1 μg/kg/min by a mini-osmotic pump (Alzet) for 4 weeks in male CD1 mice. The Ang II expression plasmid (pAng II) was injected intravenously at 1 mg/kg of body weight using a hydrodynamics-based in vivo gene transfer approach in female Balb/c mice. Naked Dkk1 expression plasmid (pDkk1) was preinjected intravenously at 1 mg/kg of body weight for 12 h followed by pAng II injection. In the CK59 treatment experiment, CK59 was administered intraperitoneally at 10 μmol/kg/day.
Urinary Albumin and Creatinine Assay
Urinary albumin was measured using a mouse albumin ELISA Quantification kit according to the manufacturer's protocol (Bethyl Laboratories, Montgomery, TX). Urine creatinine was determined by a creatinine assay kit, according to the protocols by the manufacturer.
Histologic and Immunohistochemical Staining
Renal sections were fixed in 4% paraformaldehyde, embedded in paraffin, and deparaffinized in xylene. The sections were then stained with hematoxylin/eosin for general histology. Paraffin-embedded kidney tissue sections were deparaffinized and hydrated and antigen-retrieved, and endogenous peroxidase activity was quenched by 3% H2O2. Tissue sections were then blocked with 10% normal donkey serum, followed by incubating with anti-Wnt1 (R&D, Minneapolis, MN), anti-β-catenin (Abcam, Cambridge, UK), anti-WT1 (Santa Cruz), or anti-p-CREB (Ser-133) overnight at 4 °C. After incubation with secondary antibody for 1 h, sections were then incubated with ABC reagents for 1 h at room temperature before being subjected to DAB incubation (Vector Laboratories, Burlingame, CA). Slides were viewed with a Nikon Eclipse 80i Epimicroscope equipped with a digital camera (DS-Ri1, Nikon).
Immunofluorescent Staining
Cells cultured on coverslips and kidney cryosections were fixed with cold methanol/acetone (1:1) for 10 min at −20 °C. Following three extensive washings with PBS, the cells were blocked with 2% normal donkey serum in PBS buffer for 40 min at room temperature and then incubated with the anti-β-catenin (Abcam, Cambridge, MA), anti-nephrin (Fitzgerald Industries International, Concord, MA), anti-podocin (Sigma), or anti-WT1 followed by staining with FITC or TRITC-conjugated secondary antibody. Cells were double stained with 4′,6-diamidino-2-phenylindole to visualize the nuclei. Slides were viewed with a Nikon Eclipse 80i epifluorescence microscope equipped with a digital camera (DS-Ri1, Nikon).
Western Blot Analysis
Cultured podocytes were lysed in 1× SDS sample buffer. The kidneys were lysed with radioimmunoprecipitation assay buffer containing 1% Nonidet P-40, 0.1% SDS, 100 μg/ml of PMSF, 1% protease inhibitor mixture, and 1% phosphatase I and II inhibitor mixture (Sigma) on ice. The supernatants were collected after centrifugation at 13,000 × g at 4 °C for 30 min. The protein concentration was determined by the bicinchoninic acid protein assay (BCA Protein Assay Kit, Pierce) according to the manufacturer's guide. An equal amount of protein was loaded onto the SDS-PAGE gel and separated, then transferred onto polyvinylidene difluoride membrane. The primary antibodies used were: anti-p-catenin (Ser-33/37) (Cell Signaling Technology), anti-p-catenin (Ser-675) (Cell Signaling Technology), anti-β-catenin (Abcam), anti-podocin (Sigma), anti-Wnt1 (R&D Systems), anti-WT1 (Sigma), anti-p-CaMK II α/β (Thr-286/287) (Novus Biologicals, Littleton, CO), anti-p-CREB (Ser-133), anti-CREB and anti-p-LRP6 (Ser-1490) (Cell Signaling Technology), and anti-β-actin (Santa Cruz Biotechnology). Quantification was performed by measuring the intensity of the signals with the aid of National Institutes of Health Image software package.
RT-PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. cDNAs were synthesized using 1 μg of total RNA, ReverTra Ace (Toyobo, Osaka, Japan), and oligo(dT)12–18 primers according to the manufacturer's instructions. Gene expression was measured by real-time PCR using cDNA, real-time PCR Master Mix Reagents (Roche Applied Science), and a set of gene primers with 7300 real-time PCR system (Applied Biosystems). The sequences of primer pairs were as follows: mouse podocin: sense, 5′-TCTGGTCAAGAGATCAGCCA-3′ and antisense, 5′-TATGGGCCCAAACATCTACA-3′; mouse Nephrin: sense, 5′-CCCAGGTACACAGAGCACAA-3′ and antisense, 5′-CTCACGCTCACAACCTTCAG-3′; mouse Wnt1: sense, 5′-AAATCGCCCAACTTCTGCA-3′ and antisense, 5′-AATACCCAAAGAGGTCACAGC-3′; mouse Wnt3: sense, 5′-TTCTTAGATGGGCTTGTCACAGC-3′ and antisense, 5′-TGGCTTCAGCATCTGTTACCTTC-3′; mouse Snail: sense, 5′-TCCTGCTTGGCTCTCTTGGT-3′ and antisense, 5′-GGGTACAAAGGCACTCCATCA-3′; mouse Axin2: sense, 5′-TGACTCTCCTTCCAGATCCCA-3′ and antisense 5′-TGCCCACACTAGGCTGACA-3′; mouse MMP7: sense, 5′-CACTCACTGGGTCCTCCATT-3′ and antisense, 5′-CATTCCGTCAAGATGACCCT-3′; mouse GAPDH: sense, 5′-CCTCGTCCCGTAGACAAAATG-3′, and antisense, 5′-TCTCCACTTTGCCACTGCAA-3′. The relative amount of miRNA or gene to internal control was calculated using the equation 2ΔCT, in which ΔCT = CT,gene − CT,control.
Electronic Microscopy
Mouse kidneys were fixed with 2.5% gluteraldehyde, 0.1 m sodium cacodylate, and 5 mm calcium chloride (pH 7.2) at room temperature. Postfixation was completed with 1% OsO4 for 1 h. Specimens were dehydrated in graded ethanols and infiltrated with a 1:1 mixture of propylene oxide-Polybed 812 epoxy resin for 3 h at room temperature, followed by a re-infiltration with pure epoxy resin overnight at 4 °C. Samples were embedded and polymerized at 60 °C for 48 to 72 h. Ultrathin sections (60 nm) were stained with 2% uranyl acetate, followed by 1% lead citrate. Electron micrographs were taken with a Zeiss 910 Advanced Transmission Electron Microscope (Zeiss).
Statistical Analysis
All data examined are presented as mean ± S.E. Statistical analysis of the data were performed using Sigma Stat software (Jandel Scientific Software, San Rafael, CA). Comparison between groups was made using one-way analysis of variance, followed by the Student-Newman-Keuls test. p < 0.05 was considered statistically significant.
RESULTS
Ang II Activates Wnt/β-Catenin Signaling and Induces Podocyte Injury in Cultured Podocytes
In cultured immortalized mouse podocytes, Ang II could inhibit nephrin and podocin expression in a time- and dose-dependent manner (data not show). To investigate if Ang II-induced podocyte damage is mediated by Wnt/β-catenin signaling, we treated mouse podocytes with Ang II. Ang II significantly induced Wnt1 and Wnt3 mRNA expression as early as 6 h and maintained a high level 24 h after treatment (Fig. 1A). Wnt1 protein abundance was also increased at 24 h after Ang II treatment (Fig. 1B). It has been reported that phosphorylation of β-catenin at Ser-33/37 promotes its degradation, whereas phosphorylation at Ser-675 induces β-catenin accumulation in the nucleus and increases its transcriptional activity (39, 40). To investigate whether β-catenin, the central component of the canonical Wnt signaling pathway, is also activated after Ang II treatment, p-β-catenin (Ser-33/37) and p-β-catenin (Ser-675) were detected in the podocytes after Ang II treatment, respectively. Fig. 1, C and D, show that Ang II could time dependently decrease p-β-catenin (Ser-33/37) abundance, accompanied by a increase of p-β-catenin (Ser-675), indicating the inhibition of β-catenin degradation in podocyte cytoplasm after Ang II treatment. To further investigate whether the cytoplasm-accumulated β-catenin undergoes nuclear translocation, podocyte cytoplasm and nuclei protein were extracted and β-catenin abundance was detected by Western blot, respectively. As shown in Fig. 1E, β-catenin abundance was reduced in the cytoplasm after Ang II treatment, which was in parallel with increased β-catenin in the nuclei, suggesting β-catenin underwent nuclei translocation after Ang II stimulation. Immunofluorescent staining for β-catenin further confirmed this phenomenon (Fig. 1F). We also assessed the functional consequence of β-catenin signaling activation by examining β-catenin-mediated gene transcription in a luciferase reporter system. As shown in Fig. 1G, Ang II could significantly induce β-catenin-mediated gene transcription in cultured podocytes. In addition, LRP6 phosphorylation was also induced in podocytes treated with Ang II in a time-dependent manner (Fig. 1H).
FIGURE 1.

Ang II activates Wnt/β-catenin signaling in cultured podocytes. A, real-time PCR analysis showing the induction of Wnt1 and Wnt3 mRNA at different time points after Ang II treatment as indicated, the asterisk indicates p < 0.05 compared with controls (n = 3). B, Western blot showing Wnt1 protein abundance in podocytes after Ang II treatment, β-actin was probed as normalization. C and D, Western blot showing p-β-catenin (Ser-33/37) and p-β-catenin (Ser-675) abundance after Ang II treatment, β-actin was probed as normalization (C); semi-quantitative analysis for p-β-catenin (Ser-33/37) and p-β-catenin (Ser-675) abundance (D). E, Western blot showing β-catenin abundance in podocyte cytoplasm or nuclei after Ang II treatment. F, representative immunofluorescent staining images showing β-catenin underwent nuclear translocation after Ang II treatment. Arrows indicate β-catenin positive nuclei. Cells were counterstained with DAPI for nuclear visualization. G, the graph showing the result for β-catenin-mediated gene transcriptional activity assay. Podocytes were transfected with TOP-flash reporter or FOP-flash plasmid. Relative luciferase activity was reported as the mean ± S.E., the asterisk indicates p < 0.05 versus control (n = 3). H, Western blotting assay showing the abundance of the phosphorylated LRP6 and quantitative analysis for p-LRP6 in mouse podocytes after Ang II treatment.
Wnt/β-Catenin Signaling Activation Is Involved in Ang II-induced Podocyte Injury
The data above showed that Ang II could promote Wnt/β-catenin signaling in podocytes. We then tested if Wnt/β-catenin signaling mediated Ang II-induced podocyte damage. We overexpressed Dickkopf-1 (Dkk1), a canonical Wnt signaling blocker through binding with LRP5/6, in cultured podocytes. Dkk1 overexpression inhibited the Ang II-induced p-β-catenin decrease (Ser-33/37) (Fig. 2A). The expression levels of podocin and nephrin were also largely restored in Dkk1 over-expressing podocytes (Fig. 2, B–D). To further investigate the role of canonical Wnt signaling in Ang II-induced podocyte injury, we transfected podocytes with β-catenin siRNA to reduce β-catenin expression. Compared with scramble siRNA, β-catenin siRNA transfection could remarkably reduce β-catenin protein abundance in podocytes (Fig. 2E). Similar to the results in Dkk1 treatment, knockdown of β-catenin could remarkably reverse Ang II-induced nephrin and podocin reduction, suggesting a pivotal role of Wnt/β-catenin signaling in Ang II-promoted podocyte damage.
FIGURE 2.

Wnt/β-catenin signaling mediates Ang II-induced podocyte injury. A, Western blot showing p-β-catenin (Ser-33/37) abundance in podocytes. B, Western blot showing podocin protein abundance in podocytes. C and D, real-time PCR analysis showing nephrin and podocin mRNA abundance, the asterisk indicates p < 0.05 compared with controls (n = 3); # indicates p < 0.05 compared with Ang II plus pcDNA3-treated cells (n = 3). E, Western blot showing β-catenin abundance after β-catenin siRNA transfection. F, Western blot showing podocin abundance. G and H, real-time PCR analysis showing nephrin and podocin mRNA abundance, the asterisk indicates p < 0.05 compared with controls (n = 3), # indicates p < 0.05 compared with Ang II plus scramble siRNA-treated cells (n = 3).
CaMK II/CREB Is Involved in Ang II-induced Wnt/β-Catenin Signaling Activation and Podocyte Injury
The data thus far suggest that Wnt/β-catenin signaling plays a critical role in mediating Ang II-induced podocyte injury. We further asked if the CaMK II/CREB pathway is involved in Ang II-induced Wnt/β-catenin activation and podocyte injury. We analyzed CaMK II and CREB phosphorylation in podocytes treated with Ang II (10−6 m) at different time points. CaMK II was phosphorylated at as early as 1 min after Ang II treatment and maintained at a high level for at least 3 h (Fig. 3, A and B). CREB phosphorylation was also increased at as early as 5 min and reached a peak at 10 to 15 min (Fig. 3, C and D). In addition, we treated podocytes with CK59, a CaMK II blocker. CK59 treatment inhibited Ang II-induced Wnt1 and Wnt3 mRNA expression, CREB phosphorylation, and β-catenin phosphorylation, indicating that CaMK II plays a major role in Ang II-induced CREB phosphorylation and canonical Wnt signaling activation (Fig. 4, A and B).
FIGURE 3.

Ang II activates CaMK II/CREB signaling in podocytes. A, Western blot showing the induction of p-CaMK II (Thr-286/287) after Ang II treatment, CaMK II was probed as normalization. B, semi-quantitative analysis for p-CaMK II abundance. C, Western blot showing the levels of p-CREB (Ser-133) after Ang II treatment. D, semi-quantitative analysis for p-CREB abundance, the asterisk indicates p < 0.05 compared with control (n = 3).
FIGURE 4.

Blockade of CaMK II with CK59 attenuates Ang II-induced Wnt/β-catenin signaling activation and podocyte damage. A, real-time PCR analysis showing the abundance of Wnt1 and Wnt3 mRNA in podocytes. Asterisk indicates p < 0.05 compared with controls (n = 3); # indicates p < 0.05 compared with Ang II-treated cells (n = 3). B, Western blot showing p-CREB and p-β-catenin (Ser-33/37) abundance in podocytes. C, real-time PCR analysis showing the abundance of nephrin and podocin mRNA in podocytes. The asterisk indicates p < 0.05 compared with controls (n = 3), # indicates p < 0.05 compared with Ang II-treated cells (n = 3). D, Western blot showing podocin protein abundance.
We then evaluated if blocking CaMK II could attenuate Ang II-induced podocyte damage. The mouse podocytes were pretreated with CK59, and then treated with Ang II (10−6 m) for 24 h. Ang II could significantly inhibit nephrin and podocin mRNA expression, whereas CK59 restored nephrin and podocin expression (Fig. 4C). Western blotting results also showed that CK 59 could restore podocin protein expression in the podocytes (Fig. 4D).
CREB functions as the downstream of CaMK II. We found that Ang II could induce CREB phosphorylation in cultured podocytes. To test the role of CREB in Ang II-induced podocyte injury, CREB siRNA was used to knockdown CREB expression. Similar to the observations derived from CK59 treatment, reduction of CREB expression could inhibit Ang II-induced Wnt1 and Wnt3 mRNA expression (Fig. 5A). Wnt1 protein expression and phosphorylated β-catenin were also inhibited (Fig. 5, B and C). Podocyte diffentiation markers including nephrin and podocin were also restored with CREB siRNA transfection (Fig. 5, D–F). These data indicate that CaMK II/CREB is involved in Ang II-induced Wnt signaling and podocyte damage.
FIGURE 5.

Knockdown of CREB expression inhibits Ang II-induced Wnt/β-catenin signaling activation and podocyte damage. A, real-time PCR analysis showing the abundance of Wnt1 and Wnt3 mRNA in podocytes, the asterisk indicates p < 0.05 compared with controls (n = 3); # indicates p < 0.05 compared with Ang II-treated cells (n = 3). B, Western blot showing CREB, Wnt1, and p-β-catenin (Ser-675) abundance in podocytes. C, semi-quantitative analysis for Wnt1 and p-β-catenin (Ser-675) abundance, the asterisk indicates p < 0.05 compared with control (n = 3). D, real-time PCR analysis showing the abundance of nephrin and podocin mRNA in podocytes, the asterisk indicates p < 0.05 compared with controls (n = 3), the asterisk indicates p < 0.05 compared with Ang II-treated cells (n = 3). E, Western blot for podocin protein. F, semi-quantitative analysis for podocin protein abundance, the asterisk indicates p < 0.05 compared with control (n = 3).
Wnt/β-Catenin Signaling Is Activated in Ang II-induced Podocytopathy in Mice
To decipher the effects of Ang II on podocytes in mice, we created two mouse models for Ang II-induced podocyte damage. Mini pumps filled with purified Ang II protein were embedded subcutaneously in mice. Two weeks later, mice embedded with Ang II-infused mini pumps developed heavy albuminuria (Fig. 6A). Alternatively, we injected Ang II-expressing plasmid by a hydrodynamic method through the tail vein to induce kidney disease. Ang II mRNA abundance was increased about 30-fold in liver at 16 h after Ang II plasmid injection compared with those injected with empty vector (data not show), suggesting a successful delivery of exogenous Ang II gene. Ang II plasmid injection also induced albuminuria as early as 1 week after injection, suggesting that Ang II plasmid injection could also cause podocyte damage (Fig. 6B). In the Ang II group, there was no obvious glomerular changes as evaluated under light microscopy at week 4 after injection (Fig. 6C), but foot process effacement could be detected via transmissive electronic microscopic analysis (Fig. 6D). Compared with those of sham control, the levels of nephrin, podocin, WT1, as well as CD2AP mRNAs were significantly reduced in kidney tissues of the Ang II group (Fig. 6E). Podocin and WT1 protein levels were also significantly decreased (Fig. 6, F and G). Immunofluorescent staining indicated that nephrin and podocin signals were attenuated in the Ang II group compared with those in controls. The number of WT1 positive cells per glomerular was also sharply decreased in Ang II-treated mice (Fig. 6, H–M). Taken together, these in vivo models suggest that exogenously delivered Ang II could induce podocyte damage and albuminuria in mice.
FIGURE 6.

Ang II induces podocyte injury and albuminuria in mice. A, male CD1 mice were subcutaneously embedded with a mini pump infused with purified Ang II protein. ELISA was used for urinary albumin excretion. The asterisk indicates p < 0.05 compared with sham controls (n = 5); B, female Balb/c mice were injected with Ang II expression plasmid through the tail vein. ELISA was used for urinary albumin excretion. The asterisk indicates p < 0.05 compared with control mice injected with pcDNA3 (n = 5). C, representative images for HE staining. D, representative images for transmissive electronic microscopic examination. E, real-time PCR analysis for the nephrin, podocin, CD2AP, and WT1 mRNA in kidney tissues, the asterisk indicates p < 0.05 compared with sham controls (n = 5). F, Western blot showing podocin and WT1 protein abundance. G, semi-quantitative analysis for podocin and WT1 protein abundance, the asterisk indicates p < 0.05 compared with Sham controls (n = 3). H–M, representative immunofluorescent staining images for nephrin, podocin, and WT1 as indicated from sham control (H, J, and L) and Ang II-treated mice (I, K, and M).
In the Ang II protein-infused mice, both Wnt1 and Wnt3 mRNA were increased about 4-fold compared with the vehicle controls, whereas the levels of Wnt5a and Wnt5b were not changed (Fig. 7A). The Wnt1 protein level was increased about 2-fold after Ang II treatment (Fig. 7, B and C). Phosphorylated β-catenin at Ser-33/37 was decreased about 60% in kidney tissues of the Ang II group compared with that in the control group (Fig. 7, D and E), suggesting the inhibition of β-catenin degradation in Ang II-treated kidneys. Immunohistochemical staining revealed that the Wnt1 protein was elevated in podocytes (Fig. 7F), and that β-catenin could be detected in podocyte nuclei, indicating β-catenin undergoing nuclear translocation in the podocytes of the Ang II group (Fig. 7G). We also detected the mRNA expression level of three β-catenin target genes including Snail, Axin2, and MMP7 in Ang II-treated kidneys. As shown in Fig. 7H, all of them were significantly up-regulated compared with those in control kidneys. Together, the data above demonstrates that Wnt/β-catenin signaling is activated in Ang-II treated kidneys.
FIGURE 7.

Ang II activates Wnt/β-catenin signaling in podocytes from mice. A, real-time PCR analysis for Wnt1, Wnt3, Wnt5a, and Wnt5b mRNA abundance in kidney tissues, the asterisk indicates p < 0.05 compared with sham controls (n = 5). B and C, Western blot and semi-quantitative analysis for Wnt1 protein abundance in kidney tissues from sham control and Ang II-infused mice. Asterisk indicates p < 0.05 compared with sham controls (n = 3). D and E, Western blot and semi-quantitative analysis for p-β-catenin (Ser-33/37) protein abundance in kidney tissues from sham control and Ang II-infused mice, the asterisk indicates p < 0.05 compared with sham controls (n = 3) (E). F and G, representative immunohistochemical staining images for Wnt1 and β-catenin protein in glomeruli, arrows indicate staining positive podocytes. H, real-time PCR analysis for the Snail, Axin2, and MMP7 mRNA expression in kidney tissues, the asterisk indicates p < 0.05 compared with sham control (n = 3–4).
CK59 Attenuates Ang II-induced Podocyte Injury and Albuminuria in Mice
After demonstrating the important roles of CaMK II/CREB in Ang II-induced Wnt/β-catenin signaling and podocyte injury in cultured cells, we performed additional experiments to delineate if blocking this pathway with CK59 could reverse Ang II-induced podocytopathy in mice. CK59 was injected intraperitoneally at 10 μmol/kg/day. Two weeks after Ang II-plasmid injection, mice developed albuminuria and CK59 treatment could largely abolish it (Fig. 8A). In isolated glomeruli, the extent of β-catenin phosphorylation (p-β-catenin at Ser-675) was decreased, suggesting a reduction of β-catenin signaling. Nephrin expression was markedly restored in the CK59-treated group (Fig. 8B). Immunohistochemical staining revealed that Ang II plasmid injection induced CREB phosphorylation and Wnt1 protein expression in podocytes, whereas CK59 could inhibit such effects. The immunofluorescent staining result of nephrin further confirmed the immunoblot observations (Fig. 8C).
FIGURE 8.
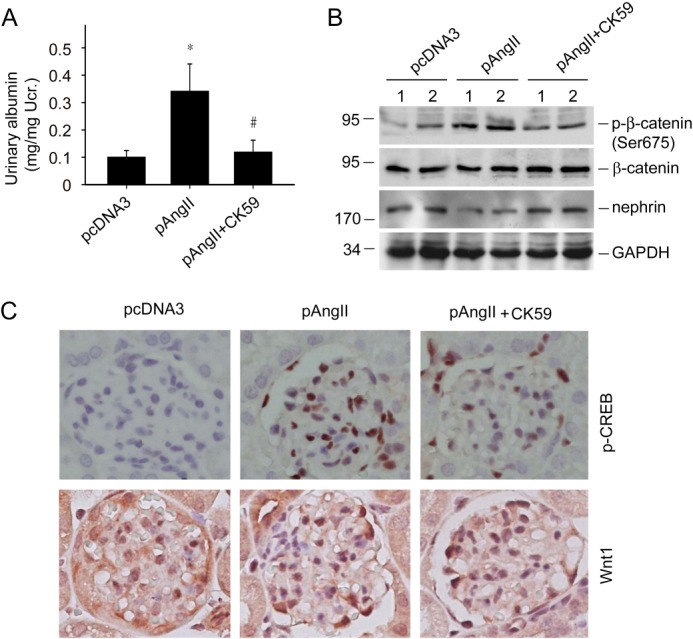
CK59 ameliorates the Ang II-induced podocyte injury and albuminuria in mice. A, urinary albumin excretion in mice, the asterisk indicates p < 0.05 compared with controls (n = 4–5), # indicates p < 0.05 compared with Ang II plus vehicle-treated mice (n = 5). B, Western blot for p-β-catenin (Ser-675) and nephrin protein abundance in isolated glomeruli. C, representative immunohistochemical staining images for p-CREB (Ser-133), Wnt1, and nephrin protein in glomeruli.
Dkk1 Ameliorates Ang II-induced Podocyte Injury and Albuminuria in Mice
To explore the role of Wnt/β-catenin signaling in Ang II-induced podocyte dysfunction and albuminuria, we injected mice with the Dkk1 expressing plasmid to induce exogenous Dkk1 overexpression. The control mice were injected with pcDNA3 plasmid. Sixteen hours after injection, mice received Ang II expressing plasmid or control vector injection, respectively. One week after Ang II plasmid injection, mice developed albuminuria compared with the pcDNA3 control. In contrast, the mice injected with Dkk1 plasmid showed reduced albuminuria (Fig. 9A). To further identify the podocyte damage in these mice, we isolated glomeruli and used real-time PCR to detect nephrin and podocin mRNA abundance. Ang II plasmid injection down-regulated glomerular nephrin and podocin mRNA expression (Fig. 9, B and C), whereas ectopic expression of Dkk1 remarkably restored their expression, indicating a protective role of blocking Wnt/β-catenin signaling in Ang II-promoted podocyte damage and albuminuria.
FIGURE 9.

Blockade of Wnt signaling with ectopic expression of Dkk1 ameliorates Ang II-induced podocyte injury and albuminuria in mice. A, urinary albumin excretion in mice, the asterisk indicates p < 0.05 compared with controls (n = 5), # indicates p < 0.05 compared with Ang II plus pcDNA3-treated mice (n = 5). B and C, real-time PCR analysis for nephrin and podocin mRNA, the asterisk indicates p < 0.05 compared with controls (n = 5), # indicates p < 0.05 compared with Ang II plus pcDNA3-treated mice (n = 5). D, schematic model illustrates that Ang II can stimulate a cascade of signaling pathway in podocytes, including activation of CaMK II, CREB, and Wnt/β-catenin signaling, which leads to podocyte injury and albuminuria. Inhibition of this signaling cascade with Dkk1, β-catenin siRNA, CREB siRNA, and CK59 ameliorates Ang II-induced podocyte injury.
DISCUSSION
We report here that Ang II can promote canonical Wnt signaling activation and podocyte damage. Ang II-induced Wnts up-regulation is mediated by the CaMK II/CREB signaling activation. Inhibition of this signaling cascade ameliorates the Ang II-triggered podocyte injury in vitro and in vivo. This study provides a mechanistic insight about CaMK II/CREB/Wnt/β-catenin signaling in Ang II-induced podocyte damage and albuminuria.
Although podocytes are specialized, terminally differentiated visceral epithelial cells that reside on the glomerular basement membrane outside the glomerular capillaries, they can undergo a range of adaptive changes, including hypertrophy, dedifferentiation, detachment, and apoptosis. Such adaptive changes can affect the podocyte differentiated state and will lead to podocyte dysfunction and proteinuria. Accumulated evidence suggests that alteration in the podocyte phenotype plays an important role in podocyte dysfunction and albuminuria. Clinically, in diabetic patients, podocytes that undergo phenotypic alterations can be detected in excreted urine samples. Sustained activation of the renin-angiotensin-aldosterone system is crucial to the pathogenesis of podocyte injury, but the mechanism by which Ang II modulates podocyte injury remains unclear (1, 41–43). Podocytes possess a local renin-angiotensin system that includes Ang II type 1 receptors (3, 8). Within the glomerulus, Ang II decreases the ultrafiltration coefficient and modulates glomerular capillary permselectivity, leading to proteinuria. It has been reported that Ang II directly influences the functions of podocytes dependent on the Ang II type 1 receptor (44, 45). A large body of evidence supports that inhibition of the renin-angiotensin system mitigates many deleterious processes in podocytes, thereby reducing the extent of injury. In this study, we found that Ang II could decrease nephrin and podocin expression in podocytes. Further experiments demonstrated that the exogenous Ang II could induce podocyte damage and albuminuria in mice. These findings suggest that Ang II can promote podocyte phenotypic alteration and podocyte dysfunction (17, 46).
It has been reported that Ang II induces Ca2+ accumulation in podocytes through both influx of Ca2+ and release of Ca2+ from intracellular stores (32, 33). CaMK II can be activated after intracellular calcium accumulation in many cell types (34). In podocytes, we found that Ang II could promote CaMK II phosphorylation, which was also found in other cell types such as myocytes (47). Inhibition of CaMK II with CK59 could largely restore Ang II-induced podocyte damage, suggesting a critical role of CaMK II in regulating Ang II-induced podocyte injury. CaMKs and the phosphatase calcineurin activate distinct downstream pathways that are mediated by the transcription factor, CRE-binding protein (CREB), and the nuclear factor of activated T cells, respectively. The CREB is common downstream of CaMKs, which functions in a broad array of biological and pathophysiological processes (48). In our study, Ang II stimulated CREB phosphorylation in podocytes, and reduction of its expression with CREB-specific siRNA could also inhibit the effects of Ang II on podocyte injury, suggesting that the CaMK II/CREB pathway is involved in Ang II-induced podocyte damage. Our results are confirmed by Kang et al. (49) studies that VEGF induction by Ang II treatment in podocyte was mediated by CREB activation, which played a pathogenic role in diabetic nephropathy. However, in HIV-associated nephropathy, CREB phosphorylation has been associated with podocyte differentiation (50). The reason behind such a discrepancy is still not clear.
CaMK II/CREB signaling activation can regulate many downstream target genes expression, among which Wnt has gained our attention (51–53). Previous studies including ours show that Wnt/β-catenin signaling activation could promote podocyte dedifferentiation and proteinuria (26, 54, 55). We have also reported that factors such as adriamycin and TGFβ1 could induce Wnt/β-catenin signaling activation in podocytes. Podocyte-specific deletion of the β-catenin gene protects against the development of albuminuria after injury. We found that Wnt/β-catenin activation induced the expression of the transcription factor Snail (27, 56). Targeting the Wnt signaling pathway with Dickkopf-1, a Wnt antagonist, blocks TGF-β1-induced podocyte injury and albuminuria (26, 29, 31). Here, we demonstrated that Ang II could activate canonical Wnt signaling, and blocking this signaling could ameliorate Ang II-induced alterations of podocyte phenotype and albuminuria. Concomitant expression of Dickkopf-1 gene abolished Ang II-triggered Wnt target gene expression, and mitigated albuminuria. Furthermore, we also demonstrated that blocking CaMK II/CREB signaling with CK59 or CREB siRNA could inhibit Ang II-induced Wnt/β-catenin signaling activation. Thus, it is likely that CaMK II/CREB as well as canonical Wnt/β-catenin signaling is involved in Ang II-driven podocyte injury and proteinuria.
In summary, this study demonstrates that Ang II can trigger CaMK II/CREB signaling, and this signaling mechanism can lead to subsequent activation of the Wnt/β-catenin signaling pathway that may underlie podocyte injury (Fig. 9D). Targeting these signaling cascades may protect against Ang II-induced podocytopathy and albuminuria.
This work was supported by National Basic Research Program of China 973 Program Grant 2012CB517601, National Science Foundation of China Grant 81070550/H0503 (to C. D.), National Basic Research Program of China 973 Program Grant 2011CB504005, and National Science Foundation of China Grant 3171093/C1102 (to J. W. Y.), National Natural Science Foundation of China, General Program Grant 81170780, and Ph.D. Programs Foundation of Ministry of Education of China Grant 20113234110005 (to A. Z. Z.).
- Ang II
- antiotensin II
- CaMK
- calmodulin-dependent protein kinase
- CREB
- cAMP response element-binding protein
- CRE
- cAMP-response element
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Lemley K. V. (2012) Protecting podocytes. How good do we need to be? Kidney Int. 81, 9–11 [DOI] [PubMed] [Google Scholar]
- 2. Attia D. M., Feron O., Goldschmeding R., Radermakers L. H., Vaziri N. D., Boer P., Balligand J. L., Koomans H. A., Joles J. A. (2004) Hypercholesterolemia in rats induces podocyte stress and decreases renal cortical nitric oxide synthesis via an angiotensin II type 1 receptor-sensitive mechanism. J. Am. Soc. Nephrol. 15, 949–957 [DOI] [PubMed] [Google Scholar]
- 3. Durvasula R. V., Shankland S. J. (2006) The renin-angiotensin system in glomerular podocytes. Mediator of glomerulosclerosis and link to hypertensive nephropathy. Curr. Hypertens. Rep. 8, 132–138 [DOI] [PubMed] [Google Scholar]
- 4. Izuhara Y., Nangaku M., Inagi R., Tominaga N., Aizawa T., Kurokawa K., van Ypersele de Strihou C., Miyata T. (2005) Renoprotective properties of angiotensin receptor blockers beyond blood pressure lowering. J. Am. Soc. Nephrol 16, 3631–3641 [DOI] [PubMed] [Google Scholar]
- 5. Hsu H. H., Hoffmann S., Endlich N., Velic A., Schwab A., Weide T., Schlatter E., Pavenstädt H. (2008) Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J. Mol. Med. 86, 1379–1394 [DOI] [PubMed] [Google Scholar]
- 6. Faul C., Donnelly M., Merscher-Gomez S., Chang Y. H., Franz S., Delfgaauw J., Chang J. M., Choi H. Y., Campbell K. N., Kim K., Reiser J., Mundel P. (2008) The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat. Med. 14, 931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crowley S. D., Vasievich M. P., Ruiz P., Gould S. K., Parsons K. K., Pazmino A. K., Facemire C., Chen B. J., Kim H. S., Tran T. T., Pisetsky D. S., Barisoni L., Prieto-Carrasquero M. C., Jeansson M., Foster M. H., Coffman T. M. (2009) Glomerular type 1 angiotensin receptors augment kidney injury and inflammation in murine autoimmune nephritis. J. Clin. Invest. 119, 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoffmann S., Podlich D., Hähnel B., Kriz W., Gretz N. (2004) Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J. Am. Soc. Nephrol. 15, 1475–1487 [DOI] [PubMed] [Google Scholar]
- 9. Wang L., Flannery P. J., Spurney R. F. (2003) Characterization of angiotensin II-receptor subtypes in podocytes. J. Lab. Clin. Med. 142, 313–321 [DOI] [PubMed] [Google Scholar]
- 10. Bianchi C., Gutkowska J., Thibault G., Garcia R., Genest J., Cantin M. (1986) Distinct localization of atrial natriuretic factor and angiotensin II binding sites in the glomerulus. Am. J. Physiol. 251, F594–F602 [DOI] [PubMed] [Google Scholar]
- 11. Durvasula R. V., Shankland S. J. (2008) Activation of a local renin angiotensin system in podocytes by glucose. Am. J. Physiol. Renal Physiol. 294, F830–F839 [DOI] [PubMed] [Google Scholar]
- 12. Herman-Edelstein M., Thomas M. C., Thallas-Bonke V., Saleem M., Cooper M. E., Kantharidis P. (2011) Dedifferentiation of immortalized human podocytes in response to transforming growth factor-β. A model for diabetic podocytopathy. Diabetes 60, 1779–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Campbell K. N., Raij L., Mundel P. (2011) Role of angiotensin II in the development of nephropathy and podocytopathy of diabetes. Curr. Diabetes Rev. 7, 3–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reiser J., Mundel P. (2007) Dual effects of RAS blockade on blood pressure and podocyte function. Curr. Hypertens. Rep. 9, 403–408 [DOI] [PubMed] [Google Scholar]
- 15. Velez J. C., Bland A. M., Arthur J. M., Raymond J. R., Janech M. G. (2007) Characterization of renin-angiotensin system enzyme activities in cultured mouse podocytes. Am. J. Physiol. Renal Physiol. 293, F398–F407 [DOI] [PubMed] [Google Scholar]
- 16. Benigni A., Morigi M., Rizzo P., Gagliardini E., Rota C., Abbate M., Ghezzi S., Remuzzi A., Remuzzi G. (2011) Inhibiting angiotensin-converting enzyme promotes renal repair by limiting progenitor cell proliferation and restoring the glomerular architecture. Am. J. Pathol. 179, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benigni A., Tomasoni S., Gagliardini E., Zoja C., Grunkemeyer J. A., Kalluri R., Remuzzi G. (2001) Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J. Am. Soc. Nephrol 12, 941–948 [DOI] [PubMed] [Google Scholar]
- 18. Blanco S., Bonet J., López D., Casas I., Romero R. (2005) ACE inhibitors improve nephrin expression in Zucker rats with glomerulosclerosis. Kidney Int. 67, S10–S14 [DOI] [PubMed] [Google Scholar]
- 19. Fukuda A., Wickman L. T., Venkatareddy M. P., Sato Y., Chowdhury M. A., Wang S. Q., Shedden K. A., Dysko R. C., Wiggins J. E., Wiggins R. C. (2012) Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int. 81, 40–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hiramatsu N., Hiromura K., Shigehara T., Kuroiwa T., Ideura H., Sakurai N., Takeuchi S., Tomioka M., Ikeuchi H., Kaneko Y., Ueki K., Kopp J. B., Nojima Y. (2007) Angiotensin II type 1 receptor blockade inhibits the development and progression of HIV-associated nephropathy in a mouse model. J. Am. Soc. Nephrol. 18, 515–527 [DOI] [PubMed] [Google Scholar]
- 21. Moeller M. J. (2010) Glomerular scarring. Can we delay or even reverse glomerulosclerosis by RAAS inhibition? Nephrol. Dial. Transplant. 25, 2101–2103 [DOI] [PubMed] [Google Scholar]
- 22. Durvasula R. V., Petermann A. T., Hiromura K., Blonski M., Pippin J., Mundel P., Pichler R., Griffin S., Couser W. G., Shankland S. J. (2004) Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 65, 30–39 [DOI] [PubMed] [Google Scholar]
- 23. Benigni A., Gagliardini E., Remuzzi G. (2004) Changes in glomerular perm-selectivity induced by angiotensin II imply podocyte dysfunction and slit diaphragm protein rearrangement. Semin. Nephrol. 24, 131–140 [DOI] [PubMed] [Google Scholar]
- 24. Moon R. T., Kohn A. D., De Ferrari G. V., Kaykas A. (2004) WNT and β-catenin signalling. Diseases and therapies. Nat. Rev. Genet. 5, 691–701 [DOI] [PubMed] [Google Scholar]
- 25. Saneyoshi T., Kume S., Amasaki Y., Mikoshiba K. (2002) The Wnt/calcium pathway activates NF-AT and promotes ventral cell fate in Xenopus embryos. Nature 417, 295–299 [DOI] [PubMed] [Google Scholar]
- 26. Shkreli M., Sarin K. Y., Pech M. F., Papeta N., Chang W., Brockman S. A., Cheung P., Lee E., Kuhnert F., Olson J. L., Kuo C. J., Gharavi A. G., D'Agati V. D., Artandi S. E. (2012) Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat. Med. 18, 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dai C., Stolz D. B., Kiss L. P., Monga S. P., Holzman L. B., Liu Y. (2009) Wnt/β-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol 20, 1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Y. (2010) New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 21, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kato H., Gruenwald A., Suh J. H., Miner J. H., Barisoni-Thomas L., Taketo M. M., Faul C., Millar S. E., Holzman L. B., Susztak K. (2011) Wnt/β-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J. Biol. Chem. 286, 26003–26015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heikkilä E., Juhila J., Lassila M., Messing M., Perälä N., Lehtonen E., Lehtonen S., Sjef Verbeek J., Holthofer H. (2010) β-Catenin mediates adriamycin-induced albuminuria and podocyte injury in adult mouse kidneys. Nephrol. Dial. Transplant. 25, 2437–2446 [DOI] [PubMed] [Google Scholar]
- 31. Wang D., Dai C., Li Y., Liu Y. (2011) Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1-driven podocyte injury and proteinuria. Kidney Int. 80, 1159–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nitschke R., Henger A., Ricken S., Gloy J., Müller V., Greger R., Pavenstädt H. (2000) Angiotensin II increases the intracellular calcium activity in podocytes of the intact glomerulus. Kidney Int. 57, 41–49 [DOI] [PubMed] [Google Scholar]
- 33. Henger A., Huber T., Fischer K. G., Nitschke R., Mundel P., Schollmeyer P., Greger R., Pavenstädt H. (1997) Angiotensin II increases the cytosolic calcium activity in rat podocytes in culture. Kidney Int. 52, 687–693 [DOI] [PubMed] [Google Scholar]
- 34. Saito T., Fukuzawa J., Osaki J., Sakuragi H., Yao N., Haneda T., Fujino T., Wakamiya N., Kikuchi K., Hasebe N. (2003) Roles of calcineurin and calcium/calmodulin-dependent protein kinase II in pressure overload-induced cardiac hypertrophy. J. Mol. Cell Cardiol. 35, 1153–1160 [DOI] [PubMed] [Google Scholar]
- 35. Wayman G. A., Impey S., Marks D., Saneyoshi T., Grant W. F., Derkach V., Soderling T. R. (2006) Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 50, 897–909 [DOI] [PubMed] [Google Scholar]
- 36. Fox K. E., Colton L. A., Erickson P. F., Friedman J. E., Cha H. C., Keller P., MacDougald O. A., Klemm D. J. (2008) Regulation of cyclin D1 and Wnt10b gene expression by cAMP-responsive element-binding protein during early adipogenesis involves differential promoter methylation. J. Biol. Chem. 283, 35096–35105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takano Y., Yamauchi K., Hiramatsu N., Kasai A., Hayakawa K., Yokouchi M., Yao J., Kitamura M. (2007) Recovery and maintenance of nephrin expression in cultured podocytes and identification of HGF as a repressor of nephrin. Am. J. Physiol. Renal Physiol. 292, F1573–F1582 [DOI] [PubMed] [Google Scholar]
- 38. Semënov M. V., Zhang X., He X. (2008) DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J. Biol. Chem. 283, 21427–21432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yost C., Torres M., Miller J. R., Huang E., Kimelman D., Moon R. T. (1996) The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 10, 1443–1454 [DOI] [PubMed] [Google Scholar]
- 40. Hino S., Tanji C., Nakayama K. I., Kikuchi A. (2005) Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase stabilizes β-catenin through inhibition of its ubiquitination. Mol. Cell Biol. 25, 9063–9072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hsu H. H., Hoffmann S., Di Marco G. S., Endlich N., Peter-Katalinić J., Weide T., Pavenstädt H. (2011) Down-regulation of the antioxidant protein peroxiredoxin 2 contributes to angiotensin II-mediated podocyte apoptosis. Kidney Int. 80, 959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lai K. N., Leung J. C., Tang S. C. (2011) The renin-angiotensin system. Contrib. Nephrol. 170, 135–144 [DOI] [PubMed] [Google Scholar]
- 43. Ye M., Wysocki J., William J., Soler M. J., Cokic I., Batlle D. (2006) Glomerular localization and expression of Angiotensin-converting enzyme 2 and angiotensin-converting enzyme. Implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 17, 3067–3075 [DOI] [PubMed] [Google Scholar]
- 44. Naito T., Ma L. J., Yang H., Zuo Y., Tang Y., Han J. Y., Kon V., Fogo A. B. (2010) Angiotensin type 2 receptor actions contribute to angiotensin type 1 receptor blocker effects on kidney fibrosis. Am. J. Physiol. Renal Physiol. 298, F683–F691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rüster C., Franke S., Wenzel U., Schmidthaupt R., Fraune C., Krebs C., Wolf G. (2011) Podocytes of AT2 receptor knockout mice are protected from angiotensin II-mediated RAGE induction. Am. J. Nephrol 34, 309–317 [DOI] [PubMed] [Google Scholar]
- 46. Sofue T., Kiyomoto H., Kobori H., Urushihara M., Nishijima Y., Kaifu K., Hara T., Matsumoto S., Ichimura A., Ohsaki H., Hitomi H., Kawachi H., Hayden M. R., Whaley-Connell A., Sowers J. R., Ito S., Kohno M., Nishiyama A. (2012) Early treatment with olmesartan prevents juxtamedullary glomerular podocyte injury and the onset of microalbuminuria in type 2 diabetic rats. Am. J. Hypertens. 25, 604–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palomeque J., Rueda O. V., Sapia L., Valverde C. A., Salas M., Petroff M. V., Mattiazzi A. (2009) Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ. Res. 105, 1204–1212 [DOI] [PubMed] [Google Scholar]
- 48. Finkbeiner S., Tavazoie S. F., Maloratsky A., Jacobs K. M., Harris K. M., Greenberg M. E. (1997) CREB. A major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047 [DOI] [PubMed] [Google Scholar]
- 49. Kang Y. S., Park Y. G., Kim B. K., Han S. Y., Jee Y. H., Han K. H., Lee M. H., Song H. K., Cha D. R., Kang S. W., Han D. S. (2006) Angiotensin II stimulates the synthesis of vascular endothelial growth factor through the p38 mitogen activated protein kinase pathway in cultured mouse podocytes. J. Mol. Endocrinol. 36, 377–388 [DOI] [PubMed] [Google Scholar]
- 50. Lu T. C., Wang Z., Feng X., Chuang P., Fang W., Chen Y., Neves S., Maayan A., Xiong H., Liu Y., Iyengar R., Klotman P. E., He J. C. (2008) Retinoic acid utilizes CREB and USF1 in a transcriptional feed-forward loop in order to stimulate MKP1 expression in human immunodeficiency virus-infected podocytes. Mol. Cell Biol. 28, 5785–5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bensman A., Niaudet P. (2010) Non-immunologic mechanisms of calcineurin inhibitors explain its antiproteinuric effects in genetic glomerulopathies. Pediatr. Nephrol. 25, 1197–1199 [DOI] [PubMed] [Google Scholar]
- 52. Kim J., Kim D. W., Chang W., Choe J., Kim J., Park C. S., Song K., Lee I. (2012) Wnt5a is secreted by follicular dendritic cells to protect germinal center B cells via Wnt/Ca2+/NFAT/NF-κB-B cell lymphoma 6 signaling. J. Immunol. 188, 182–189 [DOI] [PubMed] [Google Scholar]
- 53. Wang Y., Jarad G., Tripathi P., Pan M., Cunningham J., Martin D. R., Liapis H., Miner J. H., Chen F. (2010) Activation of NFAT signaling in podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol 21, 1657–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matsui I., Ito T., Kurihara H., Imai E., Ogihara T., Hori M. (2007) Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab. Invest. 87, 273–283 [DOI] [PubMed] [Google Scholar]
- 55. Naves M. A., Requião-Moura L. R., Soares M. F., Silva-Júnior J. A., Mastroianni-Kirsztajn G., Teixeira V. P. (2012) Podocyte Wnt/β-catenin pathway is activated by integrin-linked kinase in clinical and experimental focal segmental glomerulosclerosis. J. Nephrol. 25, 401–409 [DOI] [PubMed] [Google Scholar]
- 56. He W., Kang Y. S., Dai C., Liu Y. (2011) Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J. Am. Soc. Nephrol. 22, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]