

Background: RLIP76 homozygous knock-out mice (RLIP76−/−) display a partially anti-metabolic syndrome phenotype characterized by insulin sensitivity, hypoglycemia, and hypolipidemia.
Results: RLIP76−/− mice are highly resistant to obesity as well as these other features of metabolic syndrome (MetS) caused by a high-fat diet.
Conclusion: Our findings confirm a fundamental role of RLIP76 in regulating the function of obesity-promoting pro-inflammatory cytokines.
Significance: Present studies provide a novel mechanism for targeted therapy of obesity and MetS.
Keywords: Adiponectin, Adipose Tissue, Insulin, Kidney, Leptin, Obesity
Abstract
Feeding a Western high-fat diet (HFD) to C57BL/6 mice induces obesity, associated with a chronic inflammatory state, lipid transport, and metabolic derangements, and organ system effects that particularly prominent in the kidneys. Here, we report that RLIP76 homozygous knock-out (RLIP76−/−) mice are highly resistant to obesity as well as these other features of metabolic syndrome caused by HFD. The normal increase in pro-inflammatory and fibrotic markers associated with HFD induced obesity in wild-type C57B mice was broadly and nearly completely abrogated in RLIP76−/− mice. This is a particularly striking finding because chemical markers of oxidative stress including lipid hydroperoxides and alkenals were significantly higher in RLIP76−/− mice. Whereas HFD caused marked suppression of AMPK in wild-type C57B mice, RLIP76−/− mice had baseline activation of AMP-activated protein kinase, which was not further affected by HFD. The baseline renal function was reduced in RLIP76−/− mice as compared with wild-type, but was unaffected by HFD, in marked contrast to severe renal impairment and glomerulopathy in the wild-type mice given HFD. Our findings confirm a fundamental role of RLIP76 in regulating the function of obesity-promoting pro-inflammatory cytokines, and provide a novel mechanism for targeted therapy of obesity and metabolic syndrome.
Introduction
Obesity is a global epidemic disease affecting 300 million people worldwide (1, 2), and about 2/3 of the United States population are obese (body mass index >30 kg/m2) or overweight (body mass index 25–29.9 kg/m2) (3). Obesity is characterized by hypertrophy of adipose tissue (4, 5) due to dysregulation of chronic inflammatory pathways (6–9) and signaling pathways for hormones responsible for regulation of long-term nutrient metabolism, energy balance, appetite, and satiety. It predisposes other morbidities including atherosclerosis, hyperlipidemia, type 2 diabetes, coronary artery disease, metabolic syndrome (MetS),3 hypertension, stroke, degenerative arthritis, sleep apnea, preterm fetal death, as well as cancer (10). A relatively newly defined disorder to join this list is renal disease of obesity based on recent clinical and experimental studies demonstrating characteristic features of obesity-induced renal injury include glomerular hypertrophy, thickening of the glomerular basement membrane, messangial matrix expansion, and increased renal inflammation (11, 12). The pathophysiology of how obesity predisposes these conditions is still incompletely understood. A series of recent reports have convincingly linked chronic inflammation as a cause as well a consequence of obesity (13–15). The chronic inflammatory state propagated by cytokines from inflammatory leukocytes (16, 17) and adipokines (18, 19) released from hypertrophied adipose tissue in obesity (5, 20, 21) may also be playing a central role in promoting carcinogenesis (10, 17) as well as other oxidative stress-related degenerative disorders including degenerative arthritis (22), atherosclerosis (23–25), and MetS (8). The link between obesity and cancer is particularly evident from the commonality of kinase signaling pathways activated downstream of cytokines in both obesity and cancer (10). MetS characterized by hyperlipidemia, low high-density lipoprotein (HDL) cholesterol, high blood pressure, and high fasting glucose frequently coexists with obesity (26, 27). Insulin resistance is observed frequently in both obesity and MetS; despite the fact that not all MetS-related conditions are associated with insulin resistance (IR); the very frequent occurrence of IR in MetS warrants its inclusion as a hallmark of this disease (27).
The signaling components deranged in obesity include intracellular as well as secreted signaling molecules that are made in and/or have effects on adipose tissues including adipocytes and peri-adipocyte stromal and inflammatory cells, various brain nuclei, particularly the hypothalamus, liver, skeletal muscle, and the pancreatic islets. The secreted signaling molecules that play a key pathogenic role in obesity include neurotransmitters and peptide hormones from the brain (NPY, AgRP, POMC, αMSH, and CART) (28, 29), adipokines from adipocytes (leptin, adiponectin, resistin, visfatin, and retinol-binding protein-4) (10, 30–32), cytokines from adipocytes and adipose mesangial cells (IL1β, IL1Rα, IL-6, -8, -10, MCP-1, TNFα, TGFβ, and VEGF) (6, 24, 33), and powerful non-hormonal metabolites in extracellular fluid (glucose, free-fatty acids, polyunsaturated fatty acids and amino acids such as leucine) (34) that play a role in regulating nutrient-metabolism balance.
High calorie, high-fat, and highly pleasurable (sweet and fatty) foods can suppress the normal anorexigenic signals that are turned on after eating, and chronically high calorie diets can result in resistance to the normal anorexigenic control mechanism, with the resultant imbalance between hyperphagia and anorexia signals potentiating excessive eating behaviors (28, 34). This model of obesity is experimentally perhaps best demonstrated in mouse models of high-fat diet (HFD)-induced obesity (35). HFD causes excessive caloric consumption and is known to cause resistance to the normal anorexic effects of leptin, the chief anorexic adipokine (31) that functions to regulate food intake, energy expenditure, and neuroendocrine function.
The gene for leptin is defective in the congenitally obese ob/ob mice, and the leptin receptor is defective in db/db mice (36, 37). Administration of recombinant leptin has no effect in db, but exerts anorexigenic effects in leptin-deficient ob/ob mice (37). However, normal mice made obese by feeding a HFD have very high levels of leptin, but are resistant to the anorexigenic effects and normal energy homeostasis mediated by leptin (38). The cause of this leptin resistance is not clear, but multiple etiologies are proposed (39). The combined central and peripheral effects of leptin normally cause increases glucose turnover, glucose uptake, decreases hepatic glycogen content, improved immune function, improved thyroid hormone and corticosteroid hormone signaling, stimulated fatty acid oxidation in skeletal muscle through activation of AMPK, and inhibition of acetyl-CoA carboxylase. HFD-mediated leptin resistance, the central anorexigenic effects of leptin are curtailed, but peripheral effects that promote monocytic infiltration of fat and resulting chronic oxidative stress, are not inhibited.
Adiponectin is the major anti-obesity adipokine that functions to antagonize pro-inflammatory signals in adipose and other tissues (10, 40). Thus, as expected, hypo-adiponectinemia leads to type 2 diabetes, inflammation, atherogenesis, and obesity-associated malignancies. Adiponectin signals through the Adipo-R1 and Adio-R2 receptors and functions to antagonize the chronic oxidative inflammatory signals associated with JNK, NFκB, IL6, IFNγ, and TNFα to limit inflammation. Furthermore, it activates pPARγ2, a transcription factor that antagonizes the transcription of pro-inflammatory genes (41). Through activation of AMPK, the master regulator of cellular energy homeostasis, adiponectin can exert broad control of carbohydrate and lipid metabolism. The activity of AMPK is additionally regulated by protein kinase A (cAMP-activated kinase). In the setting of HFD-mediated obesity, resistance also develops to the beneficial energy homeostatic and anti-obesity effects of adiponectin, also for incompletely understood reasons.
We have recently reported that homozygous knock-out of the mercapturic acid pathway transporter RLIP76 (a 76-kDa membrane protein encoded by the human RALBP1 gene (18p11.22)) results in a condition that is the opposite of MetS in some respects, including hypolipidemia, hypoglycemia, and marked peripheral and central insulin sensitivity. Indeed, drugs that are used to treat MetS including rosiglitazone, metformin, atorvastatin, or gemfibrozil failed to lower already low lipid and glucose levels in RLIP76−/− mice (42, 43). Our previous studies also showed that AMPK, a key regulator of carbohydrate and lipid metabolism, as well as transcription factors PPARα and PPARγ, which are targets of hypolipidemic and insulin-sensitizing drugs, are already maximally activated in RLIP76−/− mice tissues (43). Taken together, their previous finding showed that RLIP76 is an important component of mechanisms that function to cause hyperlipidemia and insulin resistance that are characteristic of MetS. These findings further suggested that RLIP76−/− mice should also fail to develop obesity, MetS, or type 2 diabetes when placed on a HFD. Furthermore, obesity-associated renal disease should also be prevented in RLIP76−/− mice. The present studies were undertaken to test this postulate by comparing weight gain, inflammatory markers, and renal function between wild-type and RLIP76−/− C57B mice on a HFD. Using results of these studies presented in this article, we conclude that RLIP76 is a necessary component of mechanisms that translate oxidative stress into inflammatory cytokines and adipokines that mediate obesity, and that targeted inhibition of RLIP76 could be a novel anti-obesity therapy.
EXPERIMENTAL PROCEDURES
Reagents
An avidin/biotin complex (ABC) detection kit was purchased from Vector (Burlingame, CA). Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies were procured from Sigma. Angiotensin II type 1 receptor (AT1), renin, connective tissue growth factor (CTGF), β-actin, fibronectin, collagen IV, F4/80 (CD68), receptor for advanced glycation end products (RAGE), TGF-β, GAPDH, nitrotyrosine, and pAMPK (Thr172) antibodies were purchased from Santa Cruz Biotechnology (Columbus, OH), Novus Biologicals (Littleton, CO), and Abcam (Cambridge, MA). Phosphorylated acetyl-CoA carboxylase (pACC; Ser79), fatty acid synthase (FAS), pAKT (Ser473), and pP38-MAPK (Thr180/Tyr182) antibodies were procured from Cell Signaling Technologies (Danvers, MA). Insulin and glucagon antibodies were obtained from BioGenex (Fremont, CA). The source of anti-RLIP76 antibodies were the same as previously described (44–46).
Animals
All animal experiments were carried out in accordance with a protocol approved by the Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). RLIP76 heterozygous knock-out animals (RLIP76+/−) were generated by Lexicon Genetics (The Woodlands, TX). RLIP76−/− mice derived from RLIP76+/− × RLIP76+/− mating were genotyped by PCR strategy as described previously (45). Forty-eight (24 RLIP76+/+ and 24 RLIP76−/−) 12-week-old mice were fed a HFD (60% fat, 20% protein, 20% carbohydrate) or standard diet/normal chow diet (NC, 10% fat, 20% protein, 70% carbohydrate) for 12 weeks (12 RLIP76+/+ and 12 RLIP76−/− mice) and 24 weeks (12 RLIP76+/+ and 12 RLIP76−/− mice) (Research Diets, New Brunswick, NJ). Body weights and food consumptions were recorded twice a week. Twenty-four mice were sacrificed after 12 weeks, and the remaining 24 were sacrificed after 24 weeks by carbon dioxide inhalation followed by cervical dislocation. Blood pressure was measured by tail-cuff plethysmography (CODA2 Blood Pressure System, Kent Scientific, Torrington, CT).
Quantitative Real-time PCR
Real-time quantitative PCR of the cDNA using indicated gene primers (Table 1) was performed on an ABI-7500 fast real-time PCR system using SYBR Green master mix (47). Briefly, 1 μg of total RNA from kidney cortex was used to synthesize cDNA by reverse transcription using the RT kit (Applied Biosystems). The following primers: TGFβ, collagen type I and IV, TNFα, IL-6, MCP-1, AT1, renin, and β-actin as a housekeeping gene, were used for real-time PCR.
TABLE 1.
List of PCR primers
| Gene | Forward | Reverse |
|---|---|---|
| IL-6 | ACAAAGCCAGAGTCCTTCAGAG | ACCACAGTGAGGAATGTCCAC |
| MCP1 | AGGTCCCTGTCATGCTTCTGG | CAGCACTTCTTTGGGACACCTGCTG |
| TNFα | TGTTGCCTCCTCTTTTGCTT | TGGTCACCAAATCAGCGTTA |
| TGF-β1 | CGGGTCTACAACCAACACAACCCG | GCAGGAGCGCACAATCATGT |
| Col4a1 | GCCTTCCGGGCTCCTCAG | CGGATTATCACCAGTGGGTCCG |
| Col1a2 | CAGAACATCACCTACCACTGCAA | TTCAACATCGTTGGAACCCTG |
| AT1 | CTCTGTTTTATGGCTTTCTGGG | CATGTTATCTGAAGGGCGGTAG |
| Renin | ATGAAGGGGGTGTCTGTGGGGTC | ATGTCGGGGAGGGTGGGCACCTG |
| β-Actin | ACCTTCTACAATGAGCTGCG | CTGGATGGCTACGTACATGG |
Renal Function
Progression of renal dysfunction was assessed by measuring urinary albumin to creatinine ratio (UA/Cr), and serum creatinine. For urinary albumin and creatinine concentrations, mice were housed in metabolic cages (Nalgene, Rochester, NY) for 24 h and urine was collected in a collection beaker. Body weight-adjusted creatinine clearance (Ccr) was calculated.
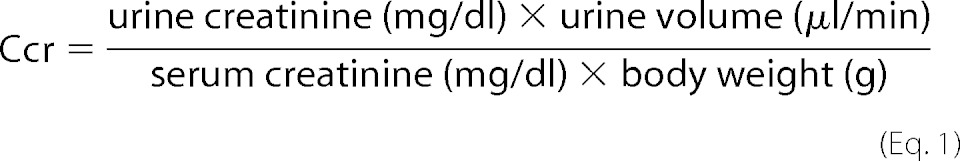 |
Urine and Plasma
The urine albumin and creatinine were measured using a mouse albumin ELISA kit and a creatinine companion kit (Exocell, Philadelphia, PA). As an index of oxidative stress, urine samples were also analyzed for H2O2 using Amplex red assay (Invitrogen). Plasma insulin levels were determined using the “Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem Inc., Downers Grove, IL). Plasma leptin and adiponectin levels were determined by ELISA kits (Invitrogen). The level of urine MCP-1 was measured by ELISA (Invitrogen). Blood was drawn by heart puncture and transferred into heparinized Microtainers on ice, centrifuged at 3000 × g for 10 min, and plasma was used for blood chemistries (cholesterol, triglycerides, glucose, creatinine, ALT, AST, LDH, alkaline phosphatase, and albumin) performed at the Pathology Core Lab, City of Hope, Duarte, CA.
Glucose Tolerance Test
Twenty-four (12 RLIP76+/+ and 12 RLIP76−/−) 12-week-old mice divided into four groups of 6 animals were used for this study. Twelve mice (6 RLIP76+/+ and 6 RLIP76−/−) were fed with NC and the other 12 mice (6 RLIP76+/+ and 6 RLIP76−/−) with a HFD for 12 weeks. Thereafter these mice were fasted for 16 h followed by intraperitoneal administration of 2 g/kg body weight of glucose (42).
Histopathological Examination for Angiogenic, Proliferative, and Differentiation Markers
NC and HFD-fed mice tissues that were formalin-fixed sections were used for histopathologic analyses. Immunohistochemistry analyses were performed by staining the sections with hematoxylin and eosin (H&E), periodic acid/methenamine silver (PAMS), F4/80 (CD68) (marker of macrophage), trichrome (collagen marker), fibronectin, nitrotyrosine, CTGF, TGFβ, collagen IV (fibrotic/cell cycle marker), AT1, renin (hypertension marker), RLIP76 (oxidative stress marker), RAGE (inflammatory marker), insulin, Oil Red O (matrix/lipid accumulation marker), and pAMPK (cellular regulator of lipid and glucose metabolism) from tissue from the NC- and HFD-fed groups of mice. Neutral lipid accumulation was evaluated by Oil Red O staining. Statistical significance of difference was determined by two-tailed Student's t test, with p < 0.001 considered a significant difference between parameters comparing HFD with NC. Photomicrographs at ×40 magnification were acquired using Olympus DP 72 microscope and processed with DP2-BSW software. The intensity of antigen staining was quantified by digital image analysis using Image Pro Plus-6.3 software (Media Cybernetics Inc., Bethesda, MD). Bars represent means with 95% confidence intervals (n = 6); *, p < 0.001 compared with control.
Statistical Analyses
Body weight, blood glucose (BG) levels, changes in tissue size and weight during the course of the experiments were shown using mean curves with 95% confidence intervals (error bars) over time. Insulin, serum lipids, renal function parameters, blood pressure, fold-expression of inflammatory markers were presented using bar plots with 95% confidence intervals. For body weight and blood glucose measurements over time, a repeated measures ANOVA model was used to examine possible time and group interactions. An effect was considered statistically significant when the corresponding two-sided p value was less than 0.05. All statistical analysis was carried out using both R software and Excel.
RESULTS
RLIP76−/− Mice Are Resistant to HFD-induced Weight Gain
The significantly greater weight gain in RLIP76+/+ as compared with RLIP76−/− mice was grossly evident (Fig. 1A), and plots of raw body weight showed a significantly lower total weight gain in the RLIP76−/− mice over the 24-week period (Fig. 1B) despite identical caloric intake between the wild-type and RLIP76−/− mice.
FIGURE 1.

Effect of HFD in RLIP76+/+ and RLIP76−/− mice. The experiment was carried out in wild-type (RLIP76+/+) and RLIP76 knock-out homozygous (RLIP76−/−) C57B male mice. In these experiments, 24 12-week-old RLIP76+/+ and 24 RLIP76−/− mice were used, and divided into 2 groups (normal diet, NC; and high-fat diet, HFD fed; 12 mice per group). Designated animals were fed NC or a HFD for 12 weeks, and animal pictures and body weight were monitored on a weekly basis before sacrificing for biochemical analyses. Animal pictures (panel A), animal body weight (panel B), and organ weights normalized to tibia length (panel C) are presented. Blood from mice of each group was collected and the serum was separated and used for adipokine (leptin and adiponectin) levels (panel D). Data are expressed as the means with 95% confidence intervals (p < 0.01 by two-sided Student's t test, compared with NC). A repeated measure of the ANOVA model applied to the body weight data in panel B showed that genotype, group, time, and their interactions are all very significant (p < 0.0001). This result indicates that the body weight of a mouse will depend on the genotype of the mouse, the diet of the mouse, and the time of measurement.
The daily food intake in the NC group was 2.8 ± 0.2 g/day, whereas for the HFD group the intake was 2.2 ± 0.2 g/day. RLIP76−/− mice had lower body weight compared with RLIP76+/+ mice; however, food intake was identical (NC, 2.6 ± 0.2 g/day; HFD, 2.0 ± 0.2 g/day) to RLIP76+/+ mice in the respective groups. Because of higher calories per gram in the high-fat diet, the caloric intake was almost the same in both NC as well as HFD groups (RLIP76+/+ NC, 10.8 ± 0.8 kcal; RLIP76+/+ HFD, 11.5 ± 1.1 kcal; RLIP76−/− NC, 10.1 ± 0.8 kcal; and RLIP76−/− HFD, 10.4 ± 1.1 kcal) (Table 2). Furthermore, the repeated measures of the ANOVA model showed very significant genotype effects, diet group effects, and genotype by diet interaction (all p < 0.0001). In addition, the within subject test demonstrated there are very significant time effects, its interactions with diet and genotype, and a three-way interaction of time, diet, and genotype (all p < 0.0001). This indicates that groups (in terms of diet and genotype) do change in weight over time and they change in different ways. Normalized to tibia length, organ weights were not affected differentially overall, but HFD caused a statistically significant increase in kidney size of the RLIP76+/+ mice (p < 0.02), whereas the kidneys of the RLIP76−/− were unchanged (Fig. 1C).
TABLE 2.
Mice food and calorie intake per day
Food was measured twice a week over the 24-week period of the dietary intervention.
|
RLIP76+/+ |
RLIP76−/− |
|||
|---|---|---|---|---|
| NCa | HFDb | NC | HFD | |
| Food intake per day (g) | 2.8 ± 0.2 | 2.2 ± 0.2 | 2.6 ± 0.2 | 2.0 ± 0.2 |
| Calorie intake per day (kcal) | 10.8 ± 0.8 | 11.5 ± 1.1 | 10.1 ± 0.8 | 10.4 ± 1.1 |
a NC, normal chow.
b HFD, high-fat diet.
RLIP76−/− Mice Are Resistant to HFD-induced Adipokine and Insulin Resistance
RLIP76+/+ mouse adipokines were increased upon HFD, with the effect on leptin being quite marked (p < 0.0001), in contrast, the adiponectin level was increased marginally but not significantly (Fig. 1D), whereas neither leptin nor adiponectin were affected significantly in the RLIP76−/− mouse (Fig. 1D). At baseline, the BG was significantly lower in RLIP76−/− mice as compared with the wild-type (91 ± 7 versus 156 ± 11 mg/dl, respectively; p < 0.001). BG increased from the pre-experiment baseline, when the mice were 12 weeks of age, consistently over the 24-week experiment (Fig. 2A). In the RLIP76+/+ mice fed a HFD and sacrificed at 12 weeks, BG was significantly higher than the RLIP76+/+ mice fed NC (212 ± 18 versus 156 ± 11 mg/dl, respectively; p < 0.001). This was accompanied by a nearly 10-fold higher insulin level (20.64 ± 0.51 versus 2.26 ± 0.16 ng/ml serum, respectively; p < 0.001). In comparison, RLIP76−/− mice fed a HFD versus NC had only a slight but significant increase in BG (110 ± 9 versus 91 ± 7 mg/dl, respectively; p < 0.05), accompanied by only a 1.6-fold increase in insulin (1.53 ± 0.08 versus 0.96 ± 0.06 ng/ml of serum, respectively; p < 0.02). The insulin/glucose ratios, in RLIP76+/+ mice increased dramatically with HFD as compared with NC (Fig. 2B). In contrast, the insulin/glucose ratio was lower and did not change significantly from baseline in the RLIP76−/− mice fed a HFD. These results confirm peripheral and hepatic insulin sensitivity of the knock-out mice in glucose-clamp studies (42), and indicate that genetic loss of RLIP76 nearly completely prevents HFD-induced obesity and insulin resistance.
FIGURE 2.

Glucose tolerance test and pancreata staining in HFD-fed RLIP76+/+ and RLIP76−/− mice. Twenty-four (12 RLIP76+/+ and 12 RLIP76−/−) 12-week-old mice divided into four groups of 6 animals were used for this study. Twelve mice (6 RLIP76+/+ and 6 RLIP76−/−) were fed NC and the other 12 mice (6 RLIP76+/+ and 6 RLIP76−/−) a HFD for 12 weeks. Groups of 6 mice in each group also left on NC as well as HFD for 24 weeks for blood sugar level measurements. Blood sugar (panel A) as well as insulin and glucose measurements in serum were performed and data are presented as insulin-to-glucose ratio (panel B). For the glucose tolerance test experiment (panel C), mice were fasted for 16 h followed by intraperitoneal administration of 2 g/kg body wt of glucose. We collected a drop of blood from tail nick at 0, 15, 30, 60, 90, 120, and 180 min after glucose administration for measurement of the glucose level. We used silver nitrate sticks to stop bleeding from the tail nick at the end of blood sampling. A repeated measures of ANOVA of such data suggested the time, diet, and genotype effects are all very significant (p < 0.0001), which indicates that the blood glucose of a mouse will depend on the genotype, diet type, and the time it is measured. NC and HFD mice pancreas sections used for histopathologic analyses, represents H&E staining (panel D), and insulin (red) and glucagon (green) staining (panel E). Results shown are representative images from pancreas samples from each group. Percent staining was determined by measuring positive immunoreactivity per unit area. The intensity of antigen staining was quantified by digital image analysis using Image Pro plus 6.3 software (Media Cybernetics Inc., Bethesda, MD). Bars represent means with 95% confidence intervals (n = 6); *, p < 0.002, insulin staining, HFD compared with NC.
Glucose tolerance was significantly impaired in RLIP76+/+ mice given a HFD as compared with RLIP76−/− mice (Fig. 2C). A repeated measures ANOVA of these data showed that time, diet type, and genotype effects are all very significant (all p < 0.0001). Specifically, there are very significant genotype effects, diet group effects, and genotype by diet interaction (all p < 0.0001). The within subject test demonstrated there are very significant time effects, its interactions with diet and genotype, and a three-way interaction of time, diet, and genotype (all p < 0.0001). This indicates that the blood glucose level will depend on the diet type, genotype, and the time it was measured. The peak glucose concentration after the glucose tolerance test were elevated in HFD-fed RLIP76+/+ mice sufficient to define the presence of type 2 diabetes; by 24 weeks, RLIP76+/+ baseline BG reached 250 ± 19 mg/dl consistent with development of adult onset diabetes; in contrast, RLIP76−/− mice remained non-diabetic (130 ± 14 mg/dl) (Fig. 2A). Islet hypertrophy in RLIP76+/+ mouse pancreas was evident by histological exam and immunohistochemistry but no significant change was seen in the pancreas of RLIP76−/− mice (Fig. 2, D and E).
RLIP76−/− Mice Are Resistant to HFD-induced Hyperlipidemia
Hyperlipidemia is a key pathological component of MetS as well as obesity. We have previously shown that, in addition to BG, both serum cholesterol and triglycerides are also considerably lower in RLIP76−/− mice (42, 43). Results of the present studies (Fig. 3A) confirmed that both cholesterol and triglycerides are indeed significantly lower in RLIP76−/− than RLIP76+/+ mice (p < 0.001). The HFD caused a further rise in these serum lipids in the RLIP76+/+ mice and in contrast, lipid levels were minimally affected in RLIP76−/− mice on HFD. These results indicate that HFD alone is insufficient to cause hyperlipidemia in the absence of RLIP76.
FIGURE 3.

Effect of HFD on lipids, signaling proteins, and adipose tissue staining. Blood from each group of mice was collected and the serum was separated and used for cholesterol and triglycerides levels (panel A). NC and HFD mice adipose sections were used for histopathologic analyses. H&E staining, PAMS, Oil Red O, Masson's trichrome, Collagen IV, fibronectin, F4/80 (CD68), and pAMPK, showing increased positive staining in HFD RLIP76+/+ mice and its marked reduction in RLIP76−/− mice, except pAMPK. pAMPK expression was significantly reduced in HFD RLIP76+/+ mice and its marked induction in RLIP76−/− mice. Immunoreactivity is evident as a dark brown stain, whereas non-reactive areas display only the background color. Sections were counterstained with hematoxylin (blue). Percentage positive staining was reduced significantly in RLIP76−/− mice. Results shown are representative images from adipose samples from each group. Photomicrographs at ×40 magnification were acquired using an Olympus DP 72 microscope and processed with DP2-BSW software. Percent staining was determined by measuring positive immunoreactivity per unit area. The intensity of antigen staining was quantified by digital image analysis using Image Pro plus 6.3 software. Bars represent means with 95% confidence intervals (n = 6); asterisks denote statistically significant differences (p < 0.003) compared with NC by two-sided Student's t test (panel B). Western blot analyses of signaling proteins (RLIP76, pAMPK (Thr172), pACC (Ser79), FAS, pP38-MAPK (Thr180/Tyr182), and pAKT (Ser473)) in adipose tissue lysates (used 40 μg of protein in each lane) in NC and HFD fed groups from RLIP76+/+ and RLIP76−/− mice are indicated. GAPDH was used as an internal control. Numbers below the blots represent the fold-change in the levels of proteins as compared with wild-type NC fed mice tissue (i.e. lanes 1–3, in each blot) as determined by densitometry using a Alpha Imager HP. The blots are from three different mice tissues from each group as indicated (panel C).
RLIP76−/− Mouse Adipose Resists HFD-induced Inflammation and Fibrosis
Adipose tissue hypertrophy was evident by H&E-stained histological sections (Fig. 3B) prepared from fat taken from HFD-fed RLIP76+/+ mice. PAMS stain showed increased mesangial staining, and Oil Red O stains showed increased fat globule deposition in HFD-fed RLIP76+/+ mice. The fibrotic markers trichrome stain, collagen IV, fibronectin, and the macrophage marker (CD68) were increased significantly as well in RLIP76+/+ mice, but not in RLIP76−/− mice fed HFD. Interestingly, levels of pAMPK were reduced in adipose tissues of HFD-fed RLIP76+/+ mice; in contrast, RLIP76−/− mice on NC had significantly increased pAMPK at baseline, and HFD feeding of these mice caused only a slight insignificant decrease in this elevated AMPK.
The suppression of activated pAMPK (phospho-Thr172) by HFD-induced obesity was further confirmed by Western blot analyses of adipose tissue (Fig. 3C). Phospho-acetyl-CoA carboxylase (pACC-Ser79), which is downstream of AMPK, was suppressed, but FAS, which is upstream of AMPK, was markedly up-regulated in the adipose tissue of RLIP76+/+ mice fed a HFD. Remarkably, pACC-Ser79, FAS, or AMPK were unaffected by HFD in the adipose of RLIP76−/− mice. Additional signaling mechanisms involved in obesity including activation of p38-MAPK (Thr180/Tyr182) and pAKT (Ser473) were also activated in HFD-fed RLIP76+/+ mice; these markers were already activated at baseline in RLIP76−/− mice and were not further affected by HFD. RLIP76 expression was also significantly higher upon HFD (Fig. 3C). Our findings confirm that inflammatory, fibrotic, and kinase signaling pathways that promote obesity are increased in adipose tissue during obesity caused by HFD in RLIP76+/+ mice, whereas RLIP76−/− mice are quite resistant to these.
RLIP76−/− Mice Are Resistant to HFD-induced Liver Abnormalities
HFD induced obesity in the RLIP76+/+ mice resulted in abnormal elevations of all four liver enzymes examined (Fig. 4, A and B). Albumin was also increased slightly only in the RLIP76+/+ mice consistent with an acute phase reaction (Fig. 4C); in contrast, liver enzymes and albumin remained unchanged in RLIP76−/− mice on HFD. The elevated liver enzymes were reflected in changes of steatosis by H&E examination (Fig. 4D) and accumulation for fat droplets by Oil Red O examination of liver tissue (Fig. 4E). Changes in pAMPK (Thr172), pACC (Ser79), p38-MAPK (Thr180/Tyr182), FAS, RLIP76, and pAKT (Ser473) were the same as discussed above for adipose tissue (Fig. 4F). These results show that the usual hepatic abnormalities of MetS and obesity were reproduced by HFD-mediated obesity in wild-type mice, but the RLIP76−/− mice appeared immune to these hepatotoxic effects of obesity. Furthermore, inflammatory and fibrotic pathways that are activated as a vicious cycle during obesity are mitigated by the absence of RLIP76, indicating that targeted depletion of RLIP76 could play an intrinsic anti-obesity role in a pharmacological setting.
FIGURE 4.

Effect of HFD on liver enzymes and albumin levels, signaling proteins, and liver tissue staining. Blood from mice of each group was collected and the serum was separated and used for SGOT, SGPT, alkaline phosphatase (panel A), lactate dehydrogenase (panel B), and albumin levels (panel C). NC and HFD mice liver sections were used for histopathologic analyses, and H&E & Oil Red O staining are presented. Sections were counterstained with hematoxylin (blue). Results shown are representative images from liver samples from each group (panels D and E). Statistical significance of difference was determined by two-tailed Student's t test. Western blot analyses of signaling proteins (RLIP76, pAMPK (Thr172), pACC (Ser79), FAS, pP38-MAPK (Thr180/Tyr182), and pAKT (Ser473)) in liver tissues lysates (used 40 μg of protein in each lane) in NC and HFD fed groups from RLIP76+/+ and RLIP76−/− mice was used. GAPDH was used as internal control. Numbers below the blots represent the fold-change in the levels of proteins as compared with wild-type NC-fed mice tissue (i.e. lanes 1–3, in each blot) as determined by densitometry using Alpha Imager HP. The blots are from three different mice tissues from each group as indicated (panel F).
RLIP76−/− Mice Are Hypertensive Without or With HFD
Despite a lower baseline weight and a lower degree of weight gain on the HFD mice, RLIP76−/− mice had a higher baseline blood pressure (BP) as compared with RLIP76+/+ mice (Fig. 5A); furthermore, HFD caused both systolic and diastolic BP to increase significantly (p < 0.03) in RLIP76−/− mice as compared with RLIP76+/+ mice in which no significant change in BP was noted with HFD. This highly significant finding implies that weight gain alone caused by HFD in non-stressed mice is not alone and is sufficient to induce hypertension, and that perhaps the presence of RLIP76 plays a role in preventing hypertension caused by weight gain caused by HFD. Unaffected pulse in all groups argues against basal adrenergic tone as a cause of hypertension. Higher BP at baseline, associated with elevated levels of chemical oxidative stress may also be responsible for a shorter lifespan of RLIP76−/− mice as compared with their wild-type counterparts (29 ± 1.4 versus 36 ± 1.6 months, respectively, p < 0.001) observed in a group of sentinel animals fed normal chow for their lifetime. Significantly increased baseline renin and angiotensin type 1 receptor (AT1) levels in renal tissues of RLIP76−/− mice without or with HFD as compared with the RLIP76+/+ mice in Western blots indicated that hypertension in the RLIP76−/− mice was due to an overactive renin-angiotensin mechanism (Fig. 5B). These results were further confirmed by immunohistochemical studies (Fig. 5C) and real-time quantitative PCR (Fig. 5D), showing that AT1 and renin were maximally induced in RLIP76−/− mice, and no further effect of HFD on AT1 and renin expression was found. Although not definitive because of the possible role of other hypertension mediating factors that may be affected, our findings suggest that hypertension seen in RLIP76−/− is due to abnormally increased signaling in the rennin-angiotensin mechanism.
FIGURE 5.


Effect of HFD on blood pressure, urine, and hypertension markers, signaling proteins, and kidney cortex tissue staining. Blood pressure and pulse rate were measured by tail-cuff plethysmography (CODA2 Blood Pressure System; Kent Scientific, Torrington, CT) (panel A). Western blot analyses of AT1 and renin in kidney tissue lysates (used 40 μg of protein in each lane) in NC and HFD fed groups from RLIP76+/+ and RLIP76−/− mice. GAPDH was used as internal control. Numbers below the blots represent the fold-change in the levels of proteins as compared with wild-type NC-fed mice tissue (i.e. lanes 1–3, in each blot) as determined by densitometry. The blots are from three different mice tissues from each group as indicated (panel B). NC and HFD mice kidney sections were used for histopathologic analyses of AT1 and renin, showing significantly increased positive staining in RLIP76−/− mice without or with HFD as compared with RLIP76+/+ mice. Immunoreactivity is evident as a dark brown stain, whereas non-reactive areas display only the background color. Results shown are representative images from kidney samples from each group. Bars represent means with 95% confidence intervals (n = 6); asterisks denote statistically significant differences (p < 0.001) compared with RLIP76+/+ NC by two-sided Student's t test (panel C). Quantitative real-time PCR was performed with kidney cortex from all four groups, and each normalized against β-actin. Values are means with 95% confidence intervals (n = 4 in each group). All values were compared with RLIP76+/+ NC (*, p < 0.003) (panel D). Blood from the mice of each group was collected and the serum was separated and used for creatinine level. Urine from the mice of each group was collected and used for creatinine albumin (panel E), MCP-1, and H2O2 levels (panel F). NC and HFD mice kidney sections were used for histopathologic analyses. H&E staining, PAMS, Oil Red O, Masson's trichrome, collagen IV, nitrotyrosine, fibronectin, F4/80 (CD68), RAGE, CTGF, TGFβ, and pAMPK, showing increased positive staining in HFD RLIP76+/+ mice and its marked reduction in RLIP76−/− mice, except pAMPK. pAMPK expression was significantly reduced in HFD RLIP76+/+ mice and its marked induction in RLIP76−/− mice. Immunoreactivity is evident as a dark brown stain, whereas non-reactive areas display only the background color. Sections were counterstained with hematoxylin (blue). Percentage positive staining was reduced significantly in RLIP76−/− mice. Results shown are representative images from kidney samples from each group. Photomicrographs at ×40 magnification were acquired using Olympus DP 72 microscope and processed with DP2-BSW software. Percent staining was determined by measuring positive immunoreactivity per unit area. The intensity of antigen staining was quantified by digital image analysis using Image Pro plus 6.3 software. Bars represent means with 95% confidence intervals (n = 6); asterisks denote statistically significant differences (p < 0.001) compared with NC by two-sided Student's t test (panel G). Western blot analyses of signaling proteins (RLIP76, pAMPK (Thr172), pACC (Ser79), FAS, pP38-MAPK (Thr180/Tyr182), and pAKT (Ser473)) in kidney tissues lysates (used 40 μg of protein in each lane) in NC- and HFD-fed groups from RLIP76+/+ and RLIP76−/− mice. GAPDH was used as internal control. Numbers below the blots represent the fold-change in the levels of proteins as compared with wild-type NC-fed mice tissue (i.e. lanes 1–3, in each blot) as determined by densitometry using Alpha Imager HP. The blots are from three different mice tissues from each group as indicated (panel H).
RLIP76−/− Mice Are Resistant to HFD-mediated Renal Disease of Obesity
The RLIP76−/− mice had significantly poorer renal filtration as reflected by decreased creatinine clearance compared with RLIP76+/+ mice fed the NC (Fig. 5E). Albuminuria was not increased in the RLIP76−/− mice (Fig. 5E). In the RLIP76+/+ mice, HFD caused a significant rise in serum creatinine and concomitant drop in weight-adjusted creatinine clearance accompanied by increased albuminuria (Fig. 5E) as well as increased markers of oxidative stress including hydrogen peroxide excretion and urinary MCP-1 levels (Fig. 5F); in contrast, renal function, albuminuria, hydrogen peroxide, and MCP-1 were unaffected by HFD in RLIP76−/− mice.
Histological analyses of kidneys revealed glomerular loss and smaller than normal glomeruli in RLIP76−/− mice as compared with RLIP76+/+ mice on NC diet. HFD-fed RLIP76+/+ mice had an increased glomerular area and matrix accumulation, large glomeruli, and vacuolated cytoplasm (Fig. 5G). Oil Red O staining on kidney sections on a HFD shows the accumulated neutral lipid. Mesangial hypertrophy, glomerular loss, and fibrotic changes were analyzed by staining for PAMS and analyzing PAMS-positive area in glomerulus. Masson's trichrome staining was used to analyze collagen deposition and cortical tubule degeneration. These studies confirmed histological impressions, showing a robust increase in glomerular matrix by stains for fibronectin collagen type IV deposition by HFD in RLIP76+/+ mice, whereas these effects were significantly blunted in HFD-fed RLIP76−/− mice.
TGFβ and CTGF proteins were significantly increased in kidneys of HFD compared with NC fed RLIP76+/+ mice. Immunostaining for TGFβ was increased in the glomerular, epithelial, mesangial, and tubulointerstitial tissues of HFD mice. CTGF immunostaining was also increased in HFD-fed RLIP76+/+ mice, being more prominent in the tubulointerstitial areas. In contrast, RLIP76 knockdown significantly attenuated expression and protein production of TGFβ and CTGF in the kidneys of HFD mice. Immunohistochemical staining of both extracellular matrix proteins, fibronectin and collagen IV, indicated that both could be observed in the kidneys of NC-fed mice; fibronectin staining was mostly in the intra-glomerular mesangium, with collagen IV weakly stained in the mesangium and tubulointerstitium. Both the intensity and area of fibronectin and collagen IV staining were dramatically increased in RLIP76+/+ HFD mice compared with NC-fed mice, this increase being strongly associated with increased PAMS-positive materials in the glomeruli. In contrast, these extracellular matrix proteins were lower in RLIP76−/− mice, and less significantly affected by HFD.
The RAGE (receptor of AGE), a marker for inflammation, aging and diabetic complications (12, 48–52) was increased significantly in the cortical tubulointerstitium of NC-fed RLIP76+/+ as compared with RLIP76−/− mice. HFD caused a further increase in RAGE staining in RLIP76+/+ mice, as compared with RLIP76−/− mouse renal tissues. Nitrotyrosine, a marker for protein oxidation, was predominantly detected in the renal tubules, and was increased in HFD-fed as compared with NC-fed RLIP76+/+ mice; again, the effect of HFD was significantly blunted in RLIP76−/− mouse renal tissues (Fig. 5G). AMPK was shut off by HFD in the RLIP76+/+ mouse; in contrast, AMPK was elevated at baseline in the NC-fed RLIP76−/− mouse, and it was not affected significantly by HFD in these mice.
The pattern of effects of HFD on pAMPK (Thr172), pACC (Ser79), p38-MAPK (Thr180/Tyr182), FAS, RLIP76, and pAKT (Ser473) (Fig. 5H) were similar to those seen in adipose and liver. Unlike in adipose and liver, pACC (Ser79) was lower at baseline in renal tissues of NC-fed RLIP76+/+ mice, consistent with known lower expression of this enzyme in the kidneys; the NC-fed RLIP76−/− mice had baseline elevations of pACC (Ser79), but as in other tissues, this was not changed by the HFD diet. In both liver and kidney, pAKT (Ser473) was lower than in adipose tissues, but was significantly induced in these tissues from HFD-fed RLIP76+/+ animals. In contrast, pAKT (Ser473) was activated in NC-fed RLIP76−/− renal tissues and was not further affected by HFD.
The Western blot findings of renal tissue effects of HFD on inflammatory and fibrotic signaling were confirmed at the mRNA expression level using real-time quantitative PCR. Results of these studies (Fig. 6, A and B) showed in a more dramatic fashion that HFD-mediated activation of expression of MCP1, TNFα, TGFβ, collagen-1α2, and collagen-4α1 are lost in the absence of RLIP76. Interestingly, significant activation of IL6 was preserved, a likely cause of the residual obesity mediating effect of HFD in RLIP76−/− mice.
FIGURE 6.

Effect of HFD on renal gene expression. Quantitative real-time PCR was performed with kidney cortex from all four groups, and each normalized against β-actin. Values are means with 95% confidence intervals (n = 4 in each group). In panel A, all values were compared with RLIP76+/+ NC (*, p < 0.01 compared with NC). In panel B, comparison was between NC and HFD in RLIP76−/− kidney. Values were compared with RLIP76−/− NC (*, p < 0.05 compared with NC). Data were analyzed by 2−∧∧Ct method.
HFD-induced RLIP76
NC and HFD fed mice kidney cortex, adipose, liver, and pancreas sections were used for RLIP76 expression, represents increased positive staining in HFD RLIP76+/+ mice for RLIP76 and its marked reduction or nearly absence in RLIP76−/− mice (Fig. 7A). A model representing the schematic diagram of the role of RLIP76 in signaling of obesity is shown in Fig. 7B.
FIGURE 7.

Representative photomicrographs of mice tissue sections for RLIP76 expression. NC- and HFD-fed mice kidney cortex, adipose, liver, and pancreas sections were used for RLIP76 expression using ABC staining kit, representing increased positive staining in HFD RLIP76+/+ mice for RLIP76 and its marked reduction or nearly absent in RLIP76−/− mice. The intensity of antigen staining was quantified by digital image analysis using Image Pro plus 6.3 software. Bars represent means with 95% confidence intervals (n = 6); asterisks denote statistically significant differences (p < 0.004) compared with NC by two-sided Student's t test (panel A). A schematic diagram of the role of RLIP76 in signaling of obesity (panel B).
DISCUSSION
The present studies demonstrate that RLIP76−/− mice, known to have greater insulin sensitivity and lower baseline weight (42), are resistant to HFD-mediated obesity because the inflammation-mediated effects of HFD on obesity are impaired in these mice. This is remarkable because RLIP76−/− mice are known to have very high levels of tissue oxidative stress as compared with RLIP76+/+ mice (45, 46), and imply that oxidative stress alone is insufficient to mediate obesity caused by HFD. Instead, the direct logical conclusion from our studies is that RLIP76 activity is necessary to translate the effects of the HFD-mediated increased oxidative stress into obesity.
Increased oxidative stress was manifested by increased lipid hydroperoxides and their alkenal breakdown products, both of which are converted into glutathione conjugates by glutathione S-transferases (GST) for them to be excreted as mercapturic acids. Because RLIP76 is the dominant transporter of lipid peroxidation-derived glutathione conjugates (i.e. leukotrienes and alkenal-SG conjugates) and this ATPase-mediated transport activity is directly necessary for clathrin-dependent endocytosis (CDE), RLIP76 functions as a rate-limiting step of both the mercapturic acid pathway and CDE (53). Thus, RLIP76 activity correlates directly with the rate of internalization of peptide hormone-receptor complexes from the plasma membrane through CDE, and indirectly with the accumulation of oxidative stress promoting mercapturic acid precursor glutathione conjugates of lipid peroxidation-derived reactive oxygen species. CDE is a process critically involved in modulating (activating or inhibiting) intracellular signaling downstream of a variety of peptide hormones (i.e. insulin, EGF, and TGFβ). CDE is essential for activating signaling downstream of EGF and TGFβ because the signaling complex for these hormones is assembled on the surface of the CDE endosome after endocytosis. In contrast, signaling via insulin is initiated by receptor on the cell surface, and CDE-mediated endocytosis of insulin-IR is the first step in shutting off signaling (54). Thus, lack of endocytosis causes a greater dwell-time of the insulin-IR complex on the plasma membrane surface, translating into greater insulin sensitivity of the RLIP76−/− mice (42), and signaling pathways downstream of insulin including AKT and AMPK are activated at baseline in RLIP76−/− mice. In contrast, lack of endocytosis impairs the function of TGFβ and other inflammatory cytokines such that RLIP76−/− mice are protected from the obesity causing inflammation-mediated effects of HFD.
Adiponectin and leptin levels were nearly unaffected in RLIP76−/− mice. Because these hormones are produced in response to hyper-caloric or inflammatory stress, our findings imply that RLIP76−/− mice lack the ability to translate signaling that normally causes hyper-leptinemia in HFD. Because CDE is an integral process in inflammatory signaling, and nutrient sensing mechanisms such as mammalian target of rapamycin are regulated by multiple external signals that are modulated by CDE, lack of CDE in RLIP76−/− mice should play a key role in the observed lack of adipokine-resistance. RLIP76−/− mice even with HFD also lacked insulin resistance as compared with RLIP76+/+, as evident from insulin levels and the glucose tolerance test. Resistance to HFD-induced hyperlipidemia or hepatic function abnormalities in RLIP76−/− mice, despite increased oxidative stress, taken in context of the lack of activation of cytokines by HFD strengthen our assertion that RLIP76−/− mice lack a critical mechanism for translating oxidative stress into the manifestations of MetS. Key features of MetS are obesity and systemic fibrosis; whereas wild-type mice were quite susceptible to this effect of the HFD, the RLIP76−/− mice were resistant. AMPK inactivation is another key normal signaling event caused by caloric stress that functions to integrate carbohydrate and lipid metabolism with growth and inflammation promoting activities. Because RLIP76−/− mice are resistant to the inhibitory effect of HFD on AMPK, and because AMPK is controlled through a CDE-regulated process downstream of insulin, adrenergic receptors and adiponectin receptors, it is not unreasonable to propose that resistance to HFD-induced AMPK suppression can also be due to the loss of CDE in RLIP76−/− mice.
RLIP76−/− mice are not completely immune from obesity mediated by HFD. Because MCP1, TNFα, TGFβ, collagen-1α2, and collagen-4α1 cannot be activated by HFD in RLIP76−/− mice, it is likely that the residual obesity-mediating effects of HFD are mediated through IL-6. These findings are consistent with the known greater role of caveolin-dependent endocytosis (a slower form of endocytosis, which is preserved in RLIP76−/− mice) in signaling downstream of IL-6 (48, 55–58). However, the stimulus toward obesity mediated by IL-6 is insufficient to mediate the entire syndrome of obesity and end-organ damage in the absence of RLIP76. Dramatic end-organ damage to the kidneys was observed due to a HFD in RLIP76+/+ mice, whereas the RLIP76−/− mice were spared from the HFD-mediated effects of oxidative stress. Our results suggest that targeted depletion or inhibition of RLIP76 could protect from obesity as well obesity-induced renal disease. However, this assertion must be tempered, and benefits of RLIP76 depletion may be overcome by hypertension, a condition that RLIP76−/− mice appear to be afflicted with. This is a novel observation, and may lie at the heart of controversies regarding the very definition of MetS, and suggest that even if a single RLIP76-targeted therapy were to be developed treatment of MetS (in which hypertension is included) would still require additional anti-hypertensive agents.
Acknowledgments
We thank Linda Lanting for help in performing animal experiments especially in blood pressure and pulse rate measurements, and Mei Wang, Department of Diabetes and Metabolic Diseases Research, City of Hope, CA, for providing quantitative PCR primers as well as help in performing real-time PCR. We also thank Prof. Ismail Al-Abdullah and Sofia Loera for help in immunohistochemical analyses.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 CA77495 (to S. A.) from the NCI, and funds from the Perricone Family Foundation, Los Angeles, CA, and the Beckman Research Institute of the City of Hope (to S. A.).
- MetS
- metabolic syndrome
- ACC
- acetyl-CoA carboxylase
- AGE
- advanced glycation end-product
- AMPK
- AMP-activated protein kinase
- AT1
- angiotensin II type 1 receptor
- CTGF
- connective tissue growth factor
- FAS
- fatty acid synthase
- HFD
- high-fat diet
- MCP-1
- monocyte chemoattractant protein-1
- PAMS
- periodic acid-Schiff-methenamine silver
- RAGE
- receptor of advanced glycation end-products
- RLIP76
- ral-interacting protein
- CDE
- clathrin-dependent endocytosis
- NC
- normal chow
- BP
- blood pressure
- IR
- insulin resistance
- ANOVA
- analysis of variance.
REFERENCES
- 1. Ogden C. L, Yanovski S. Z., Carroll M. D., Flegal K. M. (2007) The epidemiology of obesity. Gastroenterology 132, 2087–2102 [DOI] [PubMed] [Google Scholar]
- 2. Nordsiek F. W. (1964) An epidemiological approach to obesity. Am. J. Public Health Nations Health 54, 1689–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pender J. R., Pories W. J. (2005) Epidemiology of obesity in the United States. Gastroenterol. Clin. North Am. 34, 1–7 [DOI] [PubMed] [Google Scholar]
- 4. Ito A., Suganami T., Miyamoto Y., Yoshimasa Y., Takeya M., Kamei Y., Ogawa Y. (2007) Role of MAPK phosphatase-1 in the induction of monocyte chemoattractant protein-1 during the course of adipocyte hypertrophy. J. Biol. Chem. 282, 25445–25452 [DOI] [PubMed] [Google Scholar]
- 5. Kim D. H., Puri N., Sodhi K., Falck J. R., Abraham N. G., Shapiro J., Schwartzman M. L. (2013) Cyclooxygenase-2 dependent metabolism of 20-HETE increases adiposity and adipocyte enlargement in mesenchymal stem cell derived adipocytes. J. Lipid Res. 54, 786–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gil A., María Aguilera C., Gil-Campos M., Cañete R. (2007) Altered signalling and gene expression associated with the immune system and the inflammatory response in obesity. Br. J. Nutr. 98, S121-S126 [DOI] [PubMed] [Google Scholar]
- 7. Hotamisligil G. S. (2008) Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int. J. Obes. 32, S52–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harvey A. E., Lashinger L. M., Hursting S. D. (2011) The growing challenge of obesity and cancer. An inflammatory issue. Ann. N.Y. Acad. Sci. 1229, 45–52 [DOI] [PubMed] [Google Scholar]
- 10. Dalamaga M., Diakopoulos K. N., Mantzoros C. S. (2012) The role of adiponectin in cancer. A review of current evidence. Endocr. Rev. 33, 547–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bayliss G., Weinrauch L. A., D'Elia J. A. (2012) Pathophysiology of obesity-related renal dysfunction contributes to diabetic nephropathy. Curr. Diab. Rep. 12, 440–446 [DOI] [PubMed] [Google Scholar]
- 12. Tanner R. M., Brown T. M., Muntner P. (2012) Epidemiology of obesity, the metabolic syndrome, and chronic kidney disease. Curr. Hypertens. Rep. 14, 152–159 [DOI] [PubMed] [Google Scholar]
- 13. Singman H. S., Berman S. N., Cowell C., Maslansky E., Archer M. (1980) The anti-coronary club, 1957 to 1972. Am. J. Clin. Nutr. 33, 1183–1191 [DOI] [PubMed] [Google Scholar]
- 14. Galassetti P. (2012) Inflammation and oxidative stress in obesity, metabolic syndrome, and diabetes. Exp. Diabetes Res. 10.1155/2012/943706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bray G. A., Bellanger T. (2006) Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine 29, 109–117 [DOI] [PubMed] [Google Scholar]
- 16. Lee J. (2013) Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Arch. Pharm. Res. 36, 208–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Okwan-Duodu D., Umpierrez G. E., Brawley O. W., Diaz R. (2013) Obesity-driven inflammation and cancer risk. Role of myeloid derived suppressor cells and alternately activated macrophages. Am. J. Cancer Res. 3, 21–33 [PMC free article] [PubMed] [Google Scholar]
- 18. Blüher M. (2012) Clinical relevance of adipokines. Diabetes Metab. J. 36, 317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sahin-Efe A., Katsikeris F., Mantzoros C. S. (2012) Advances in adipokines. Metabolism 61, 1659–1665 [DOI] [PubMed] [Google Scholar]
- 20. Gornicka A., Fettig J., Eguchi A., Berk M. P., Thapaliya S., Dixon L. J., Feldstein A. E. (2012) Adipocyte hypertrophy is associated with lysosomal permeability both in vivo and in vitro. Role in adipose tissue inflammation. Am. J. Physiol. Endocrinol. Metab. 303, E597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Drolet R., Richard C., Sniderman A. D., Mailloux J., Fortier M., Huot C., Rhéaume C., Tchernof A. (2008) Hypertrophy and hyperplasia of abdominal adipose tissues in women. Int. J. Obes. 32, 283–291 [DOI] [PubMed] [Google Scholar]
- 22. El-Bikai R., Welman M., Margaron Y., Côté J-F., Macqueen L., Buschmann M. D., Fahmi H., Shi Q., Maghni K., Fernandes J. C., Benderdour M. (2010) Perturbation of adhesion molecule-mediated chondrocyte-matrix interactions by 4-hydroxynonenal binding. Implication in osteoarthritis pathogenesis. Arthritis Res. Ther. 12, R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Landsberg L., Aronne L. J., Beilin L. J. (2013) Obesity-related hypertension, pathogenesis, cardiovascular risk, and treatment. A position paper of the obesity society and the american society of hypertension. J. Clin. Hypertens. 15, 14–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malavazos A. E., Corsi M. M., Ermetici F., Coman C., Sardanelli F., Rossi A., Morricone L., Ambrosi B. (2007) Proinflammatory cytokines and cardiac abnormalities in uncomplicated obesity. Relationship with abdominal fat deposition. Nutr. Metab. Cardiovasc. Dis. 17, 294–302 [DOI] [PubMed] [Google Scholar]
- 25. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 16, 396–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Libby P. (2012) Inflammation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 2045–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alberti K. G., Eckel R. H., Grundy S. M., Zimmet P. Z., Cleeman J. I., Donato K. A., Fruchart J. C., James W.P., Loria C. M., Smith S. C. (2009) Harmonizing the metabolic syndrome. A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120, 1640–1645 [DOI] [PubMed] [Google Scholar]
- 28. Lubis A. R., Widia F., Soegondo S., Setiawati A. (2008) The role of SOCS-3 protein in leptin resistance and obesity. Acta Med. Indones. 40, 89–95 [PubMed] [Google Scholar]
- 29. Myers M. G., Cowley M. A., Münzberg H. (2008) Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 70, 537–556 [DOI] [PubMed] [Google Scholar]
- 30. Nogueiras R., Williams L. M., Dieguez C. (2010) Ghrelin. New molecular pathways modulating appetite and adiposity. Obes. Facts 3, 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Myers M. G., Jr., Heymsfield S. B., Haft C., Kahn B. B., Laughlin M., Leibel R. L., Tschöp M. H., Yanovski J. A. (2012) Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 15, 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bełtowski J. (2006) Apelin and visfatin. Unique “beneficial” adipokines upregulated in obesity? Med. Sci. Monit. 12, RA112-RA119 [PubMed] [Google Scholar]
- 33. O'Rourke R. W., Kay T., Lyle E. A., Traxler S. A., Deveney C. W., Jobe B. A. (2006) Alterations in peripheral blood lymphocyte cytokine expression in obesity. Clin. Exp. Immunol. 146, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woods S. C., Seeley R. J., Cota D. (2008) Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu. Rev. Nutr. 28, 295–311 [DOI] [PubMed] [Google Scholar]
- 35. Hariri N., Thibault L. (2010) High-fat diet-induced obesity in animal models. Nutr. Res. Rev. 23, 270–299 [DOI] [PubMed] [Google Scholar]
- 36. Tartaglia L. A., Dembski M., Weng X., Deng N., Culpepper J., Devos R. (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83, 1263–1271 [DOI] [PubMed] [Google Scholar]
- 37. Weigle D. S., Bukowski T. R., Foster D. C., Holderman S., Kramer J. M., Lasser G. (1995) Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J. Clin. Invest. 96, 2065–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frederich R. C., Hamann A., Anderson S., Löllmann B., Lowell B. B., Flier J. S. (1995) Leptin levels reflect body lipid content in mice. Evidence for diet-induced resistance to leptin action. Nat. Med. 1, 1311–1314 [DOI] [PubMed] [Google Scholar]
- 39. Könner A. C., Brüning J. C. (2012) Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 16, 144–152 [DOI] [PubMed] [Google Scholar]
- 40. Cheng X., Folco E. J., Shimizu K., Libby P. (2012) Adiponectin induces pro-inflammatory programs in human macrophages and CD4+ T cells. J. Biol. Chem. 287, 36896–36904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kadowaki T., Hara K., Kubota N., Tobe K., Terauchi Y., Yamauchi T. (2002) The role of pPARγ in high-fat diet-induced obesity and insulin resistance. J. Diabetes Complications 16, 41–45 [DOI] [PubMed] [Google Scholar]
- 42. Awasthi S., Singhal S. S., Yadav S., Singhal J., Vatsyayan R., Zajac E., Luchowski R., Borvak J., Gryczynski K., Awasthi Y. C. (2010) A central role of RLIP76 in regulation of glycemic control. Diabetes 59, 714–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singhal J., Nagaprashantha L., Vatsyayan R., Awasthi S., Singhal S. S. (2011) RLIP76, a glutathione-conjugate transporter, plays a major role in the pathogenesis of metabolic syndrome. PLoS ONE 6, e24688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Awasthi S., Cheng J., Singhal S.S., Saini M. K., Pandya U., Bandorowicz-Pikula J., Singh S. V., Zimniak P., Awasthi Y. C. (2000) Novel function of human RLIP76. ATP-dependent transport of glutathione-conjugates and doxorubicin. Biochemistry 39, 9327–9334 [DOI] [PubMed] [Google Scholar]
- 45. Awasthi S., Singhal S. S., Yadav S., Singhal J., Drake K., Nadkar A., Zajac E., Wickramarachchi D., Rowe N., Yacoub A., Boor P., Dwivedi S., Dent P., Jarman W. E., John B., Awasthi Y. C. (2005) RLIP76 is a major determinant of radiation sensitivity. Cancer Res. 65, 6022–6028 [DOI] [PubMed] [Google Scholar]
- 46. Singhal J., Singhal S. S., Yadav S., Suzuki S., Warnke M. M., Yacoub A., Dent P., Bae S., Sharma R., Awasthi Y. C., Armstrong D. W., Awasthi S. (2008) RLIP76 in defense of radiation poisoning. Int. J. Radiat. Oncol. Biol. Phys. 72, 553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun G., Reddy M. A., Yuan H., Lanting L., Kato M., Natarajan R. (2010) Epigenetic histone methylation modulates fibrotic gene expression. J. Am. Soc. Nephrol. 21, 2069–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strandberg L., Mellström D., Ljunggren O., Grundberg E., Karlsson M. K., Holmberg A. H., Orwoll E. S., Eriksson A. L., Svedberg J., Bengtsson M., Ohlsson C., Jansson J. O. (2008) IL6 and IL1B polymorphisms are associated with fat mass in older men. The MrOS Study Sweden. Obesity 16, 710–713 [DOI] [PubMed] [Google Scholar]
- 49. Declèves A.E., Mathew A.V., Cunard R., Sharma K. (2011) AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 22, 1846–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Putta S., Lanting L., Sun G., Lawson G., Kato M., Natarajan R. (2012) Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 23, 458–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamagishi S., Maeda S., Matsui T., Ueda S., Fukami K., Okuda S. (2012) Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta 1820, 663–671 [DOI] [PubMed] [Google Scholar]
- 52. Ramasamy R., Yan S. F., Schmidt A. M. (2011) Receptor for AGE (RAGE). Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N.Y. Acad. Sci. 1243, 88–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Singhal S. S., Wickramarachchi D., Yadav S., Singhal J., Leake K., Vatsyayan R., Chaudhary P., Lelsani P., Suzuki S., Yang S., Awasthi Y. C., Awasthi S. (2011) Glutathione-conjugate transport by RLIP76 is required for clathrin-dependent endocytosis and chemical carcinogenesis. Mol. Cancer Ther. 10, 16–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang J., Holman G. D. (2005) Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J. Biol. Chem. 280, 4070–4078 [DOI] [PubMed] [Google Scholar]
- 55. Vozarova B., Weyer C., Hanson K., Tataranni P. A., Bogardus C., Pratley R. E. (2001) Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes. Res. 9, 414–417 [DOI] [PubMed] [Google Scholar]
- 56. Kern P. A., Ranganathan S., Li C., Wood L., Ranganathan G. (2001) Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 280, E745–751 [DOI] [PubMed] [Google Scholar]
- 57. Skurk T., van Harmelen V., Lee Y. M., Wirth A., Hauner H. (2002) Relationship between IL-6, leptin and adiponectin and variables of fibrinolysis in overweight and obese hypertensive patients. Horm. Metab. Res. 34, 659–663 [DOI] [PubMed] [Google Scholar]
- 58. Bastard J. P., Maachi M., Van Nhieu J. T., Jardel C., Bruckert E., Grimaldi A., Robert J. J., Capeau J., Hainque B. (2002) Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 87, 2084–2089 [DOI] [PubMed] [Google Scholar]