

Abstract
Global cycling of environmental manganese requires catalysis by bacteria and fungi for MnO2 formation, since abiotic Mn(II) oxidation is slow under ambient conditions. Genetic evidence from several bacteria implicates multicopper oxidases (MCOs) as being required for MnO2 formation. However, MCOs catalyze one-electron oxidations, whereas conversion of Mn(II) to MnO2 is a two-electron process. Trapping experiments with pyrophosphate (PP), a Mn(III) chelator, have demonstrated that Mn(III) is an intermediate in Mn(II) oxidation when mediated by exosporium from the Mn-oxidizing bacterium Bacillus SG-1. The reaction of Mn(II) depends on O2 and is inhibited by azide, consistent with MCO catalysis. We show that the subsequent conversion of Mn(III) to MnO2 also depends on O2 and is inhibited by azide. Thus, both oxidation steps appear to be MCO-mediated, likely by the same enzyme, indicated by genetic evidence to be the MnxG gene product. We propose a model of how the manganese oxidase active site may be organized to couple successive electron transfers to the formation of polynuclear Mn(IV) complexes as precursors to MnO2 formation.
Keywords: Manganese oxidation, biogenic Mn oxides, multicopper oxidase, Bacillus sp. SG-1
Introduction
Redox cycling of manganese is a key chemical process in the natural environment. MnO2 is widespread in soils and sediments, and serves as an electron sink for microbial metabolism when O2 and nitrate are absent. In addition, MnO2 adsorbs other metal ions avidly [1,2], including pollutants like Pb2+, and thereby plays a role in controlling their mobility in the environment.
MnO2 and Mn(II) are the thermodynamically stable forms of manganese under aerobic and anaerobic conditions, respectively, but the uncatalyzed oxidation of Mn(II) is very slow [3,4]. The stability of its half-filled (d5) electronic shell makes extraction of an electron from Mn(II) highly endothermic (E0Mn3+/Mn2+ ∼ 1.5 V), unless strong chelating agents are available to stabilize the Mn(III). The lattice energy of MnO2 drives the two-electron oxidation, but the high initial barrier to electron transfer greatly slows the process.
However, a variety of bacteria and fungi can catalyze the conversion of Mn(II) to MnO2, by 3-5 orders of magnitude [2,5]. Thus, most of the MnO2 present in the environment has been processed by these organisms. Why they do so is uncertain. A number of functions have been suggested [2,6], including protection from toxic metals, or from UV light, or from predation or viruses. MnO2 can also break down natural organic matter into metabolizable substrates, thereby enhancing the bacterial food source [7]. An intriguing possibility is that the MnO2 deposits with which bacteria coat themselves (Fig. 1) may serve as a reservoir of Mn(II), which can protect them from oxidative damage [2,6]. A combination of benefits may have driven the evolution of Mn-oxidizing ability in these organisms.
Fig. 1. TEM of MnO2-coated spores from Bacillus sp. SG-1 [9].

Of considerable bioinorganic interest is the finding that the enzymes responsible for bacterial MnO2 formation are likely to be multicopper oxidases (MCOs). In three well-studied bacteria, Mn-oxidizing ability is lost upon disruption of a gene whose sequence identifies it as a MCO [8-11]. Cell homogenates of these and related organisms display Mn-oxidizing protein bands on acrylamide gels [12-14]; in one case, the sequence analysis of peptides derived from the band confirmed that the protein is a MCO gene product [15]. In another bacterium, antibodies raised to the protein in a Mn-oxidizing band were found to cross-react with a MCO gene product from an expression library [16].
However, the finding that bacterial Mn oxidases are likely to be MCOs raises a mechanistic conundrum. MCOs are one-electron catalysts. How can they catalyze a two-electron oxidation? In MCOs, single electrons are transferred from substrates to the type 1 Cu, from which they are relayed to O2 at the type 3 Cu site [17]. Thus, Mn(III), not MnO2, should be the product of MCO-mediated Mn(II) oxidation. Indeed, fungal laccase has been shown to convert Mn(II) to Mn(III) in the presence of the strong Mn(III) chelator, pyrophosphate (PP) [18,19].
Mn oxidases have yet to be purified in amounts sufficient to permit mechanistic studies. However, Webb et al. [20] utilized a Mn-oxidizing preparation of exosporium (the loose outer part of the spore coat) from Bacillus sp. SG-1 [9,13], to show, via PP trapping, that Mn(III) is an intermediate in the production of MnO2. In this bacterium, disruption of the MCO gene MnxG abolishes Mn oxidizing activity [9]. Following addition of Mn(II) and PP to the exosporium, the concentration of Mn(III)PP rose to a peak and then fell again, while MnO2 particles were observed to form. In addition, exogenous Mn(III)PP was shown to be a substrate for the exosporium, the Mn(III)PP concentration falling at about the same rate when Mn(III) instead of Mn(II) was added to the exosporium in the presence of PP. Thus, MnO2 formation occurred in two-steps, both of which required exosporium. Mn(II) oxidation required O2, as expected, and was abolished by azide, a known inhibitor of MCOs [21,22]. However, since the protein organization of exosporium is unknown, it was unclear whether both oxidation steps were MCO-mediated or only the initial oxidation of Mn(II). Conceivably, Mn(III) oxidation to MnO2 took place at a different site, either through further reaction with O2 or via a disproportionation mechanism. The present study was undertaken to answer this question, and to consider how the second step of the two-step oxidation might proceed.
Methods
UV-vis absorption spectra were monitored with an Agilent 8453 UV-vis spectrophotometer, using a thermostattable multicell transport configuration, with automated kinetic scan capability. Spectra were recorded at room temperature every 15 min during the 24-36 h, using a 10 mm pathlength cuvette with a magnetic stirrer. The samples were stirred continuously during the UV-vis time courses using a “Spinette” cell stirrer (Starna Cell). Typically, several reaction mixtures and a control were monitored in a parallel configuration.
Bacillus sp. SG-1 spores were harvested [23] and exosporium was prepared as previously described [13]. All experiments were carried out at pH = 7.5 in a 10 mM HEPES/50 mM NaCl buffer. For the UV-vis absorption time courses, 1 mL samples contained 50 μL aliquots of exosporium suspension, 0.1 mM Mn(II) chloride or Mn(III) acetate and 500 μM NaPP, added as a trapping agent for the Mn(III) ions. For experiments under anaerobic conditions, O2 was eliminated from the reaction mixture by continuous blowing of Ar over the solution for 1 h; after that the cuvette was sealed and transferred to the spectrophotometer. For inhibition experiments, sodium azide was added to the reaction mixture at a concentration of 10 mM.
Time-dependent absorption spectra were modeled with pure component spectra using a linear least squares fit. Three basis spectra were used (Fig. 2): (1) exosporium (50 μL in 1 mL of HEPES), (2) Mn(III)-PP (0.1 mM in HEPES) and (3) MnO2 in water (obtained as the supernatant, after centrifuging a “Mn(II)+PP+exosporium” mixture, that had reacted for 26 h). The 231-650 nm spectral region was used for fitting. The exosporium component was kept fixed during the fitting.
Fig. 2.

UV-vis absorption spectra of exosporium (50 μL in 1 mL of HEPES buffer) (black), Mn(III)-pyrophosphate complex (100 μM in HEPES buffer) (red), and MnO2 suspension in water (blue) used as basis spectra for a linear squares fit analysis of the UV-vis absorption spectra taken over 30 h experiments of Mn oxidation catalyzed by exosporium
Results
The experiments were much the same as those reported by Webb et al. [20], with the crucial addition of experiments to test the effects of oxygen exclusion and azide inhibition on Mn(III) oxidation. Also, instead of simply monitoring the Mn(III)PP absorption intensity at 258 nm, we performed a full analysis of the absorption spectra by fitting them to three-component spectra (Fig. 2): (1) exosporium in HEPES buffer plus PP, (2) Mn(III)PP, and (3) MnO2. A fourth component, Mn(II)PP, was present in the reaction mixtures, but contributed negligibly to the spectra at the concentrations in use. Since exosporium plus PP is unaltered throughout the course of the reaction, the first component was held constant in the fitting of Mn(III)PP and MnO2 contributions.
Exosporium and Mn(III)PP both have absorption bands at 220 nm and 255-260 nm, but their shapes and relative intensities differ, allowing for discrimination between them. The spectrum of MnO2 is variable, depending on particle size. We took the product spectrum to be that of the MnO2 colloid in the reaction mixture supernatant after 26 h of incubation. Beyond this time, the absorption spectrum of the reaction mixture did not change significantly, except for alterations due to aging of the MnO2, which formed larger particles. The MnO2 colloid displays broad UV-vis absorption, with a weak maximum at ∼420 nm (Fig. 2).
Exosporium converts Mn(II) to Mn(III) and then MnO2
When Mn(II) was added to exosporium in the presence of excess PP, the absorption spectra evolved with time (Fig. 3a) showing increased absorption in both UV and visible regions. Component fitting revealed a rise and fall in the Mn(III)PP fraction, with a maximum around 10 h (Fig. 3b). In addition, we see that MnO2 production commenced only after 10 h, when Mn(III)PP had reached its peak. The MnO2 fraction then rose steadily as the Mn(III)PP fraction fell. Thus, there is a clear two-stage reaction sequence, Mn(II)PP being first converted to Mn(III)PP and then to MnO2.
Fig. 3.
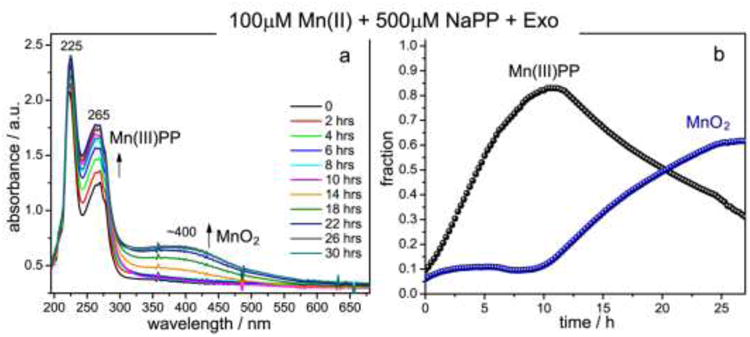
Time-resolved UV-vis absorption spectral measurements of Mn(II) oxidation by exosporium at room temperature. (a) Selected UV-vis absorption spectra of the reaction mixture taken at the indicated time points during the course of Mn(II) oxidation. The growth of 265 nm band indicates formation of the Mn(III) intermediate trapped by pyrophosphate; the 400 nm band is due to formation of particulate Mn oxides. (b) Time courses followed by Mn(III)-pyrophosphate and MnO2 species during the 30 h UV-vis absorption experiment and obtained from the fit of the absorption spectrum at each time point to a linear combination of the component spectra depicted in Fig.2
When Mn(III) was added to exosporium instead of Mn(II), the absorption spectra showed little change in the UV region and a rise in the visible region (Fig. 4a). Component fitting (Fig. 4b) revealed a steady decline of the Mn(III)PP fraction, while the MnO2 fraction rose from the beginning. Thus, the second stage in the reaction sequence proceeds independently of the first.
Fig. 4.

Time-resolved UV-vis absorption spectral measurements of Mn(III) oxidation by exosporium at room temperature. (a) Selected UV-vis absorption spectra of the reaction mixture taken at the indicated time points during Mn(III) oxidation. The growth of the 400 nm band indicates formation of the particulate Mn oxides. (b) Time courses followed by Mn(III)-pyrophosphate and MnO2 species during the 30 h UV-vis absorption experiment and obtained from the fit of the absorption spectrum at each time point to a linear combination of the component spectra depicted in Fig. 2
Eliminating O2 or adding azide blocks oxidation of both Mn(II) and Mn(III)
When O2 was eliminated from the reaction mixture (see experimental section), no reaction with Mn(II) was observed. Likewise, there was no reaction when 10 mM sodium azide was added in the presence of O2 (data not shown). In both cases, the Mn(II) plus exosporium UV-vis absorption spectrum remained essentially unchanged. Crucially, there was likewise no reaction of Mn(III) if the reaction mixture was de-aerated (Fig. 5a) or if 10 mM sodium azide was added (Fig. 5b). Again, the initial UV-vis absorption spectrum remained essentially unchanged (Fig. 5) over the time course of reaction with O2 (Fig. 4).
Fig. 5.

UV-vis absorption spectra of the reaction mixture taken at the indicated time points during Mn(III) oxidation by exosporium (a) under anaerobic condition; (b) with 10 mM azide present. Both conditions, exclusion of oxygen and addition of azide, resulted in inhibition of Mn(III) oxidation
Discussion
Our results corroborate those of Webb et al. [20], showing that oxidation of Mn(II) by SG-1 exosporium produces chelatable Mn(III), and that this reaction requires O2 and is blocked by azide. These results are consistent with Mn(II) oxidation by a MCO, since MCOs couple O2 reduction to one-electron oxidation of substrates, and are inhibited by azide binding. These findings corroborate the genetic and biochemical evidence that the manganese oxidase in SG-1 exosporium, MnxG, is an MCO.
We also confirm the observation [20] that in the presence of exosporium, chelated Mn(III) is further converted to MnO2. However, it was uncertain whether this step was also catalyzed by an MCO, since purified enzyme is unavailable. The exosporium might have harbored separate enzymes for Mn(II) oxidation and MnO2 production. Moreover, it was uncertain whether MnO2 production involved a one-electron oxidation of Mn(III). It might instead have resulted from enzyme-catalyzed disproportionation of Mn(III) followed by re-oxidation of the resulting Mn(II). The possible pathways are illustrated in Fig. 6.
Fig. 6.
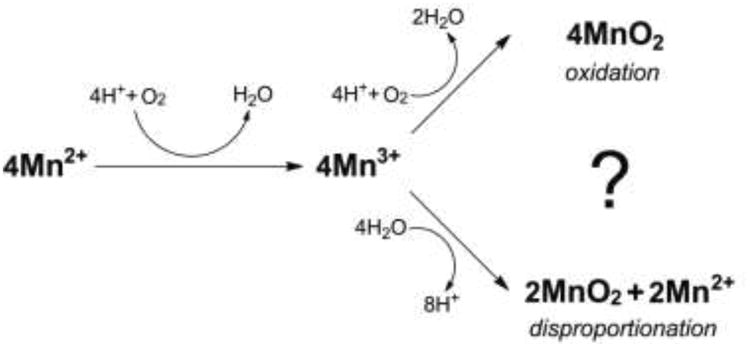
Possible pathways for bacterial MnO2 formation by MCO-catalyzed Mn(II) to Mn(III) oxidation followed by further oxidation (top) or by disproportionation (bottom) of complexed Mn(III)
The present experiments were designed to address these issues. If MnO2 production resulted from disproportionation, it should have proceeded in the absence of O2, although its production would have been cut in half because the Mn(II), produced in equal amount by disproportionation, would not have been reoxidized. However, de-aeration completely blocked MnO2 production, from Mn(III) as well as Mn(II). Thus, Mn(III) conversion to MnO2 requires O2.
There remained the question whether the oxidation of Mn(II) and of Mn(III) is catalyzed by the same or different enzymes. The observed inhibition by azide is consistent with an MCO being involved with Mn(III) as well as Mn(II) oxidation. It is possible that azide acts at other enzyme sites than at the MCO trinuclear Cu site (known to be the site of MCO inhibition [22,24-26]). Azide is an inhibitor of manganese catalase [27], whose active site is a dinuclear Mn(III) complex; such a complex is a likely intermediate in Mn(III) oxidation (see below). Azide could bind this intermediate and thereby inhibit oxidation. However, this intermediate is likely formed within the same protein as the MCO Cu centers. The genetic evidence points to MnxG as a single protein associated with MnO2 formation, and a single native gel protein band converts Mn(II) to MnO2 [9,15].
If a single MCO is responsible for both steps of the reaction, how might its active site be organized? The initial reaction, Mn(II) conversion to Mn(III) is straightforward enough. MCO-catalyzed Mn(II) oxidation has previously been observed in Mn(III) trapping experiments with a laccase enzyme [18,19], and the mechanism is likely similar to MCO-catalyzed Fe(II) oxidation by human ceruloplasmin [28,29,30] or yeast Fet3p [21,30-32], or Cu(I) oxidation by CueO from E. coli[33,34]. Crystallographic studies [28] of the ferroxidase MCO, ceruloplasmin, indicate that Fe(II) binds to a site adjacent to the type 1 Cu, and that, after electron transfer, the product Fe(III) moves several angstroms toward the solvent interface, to a ‘holding site’ (Fig. 7). It seems likely that a similar translocation of Mn(III) in MnxG would provide ready access to chelating agents, accounting for the facile production of Mn(III)PP in solution.
Fig. 7.

Structure of human ceruloplasmin (pdb #:1KCW) showing mononuclear type 1 copper site, and “substrate” and “holding” cation-binding sites in domain 6. Arrow indicates a movement of sidechain E935 from the substrate site to the holding site upon iron translocation after oxidation. Adapted from ref. [28]
However, MnO2 production from Mn(III) has no precedent in MCO chemistry. Oxidation to mononuclear Mn(IV) is improbable, since Mn(IV) is a powerful oxidant, and cannot be stabilized by the relatively weak donor ligands available from protein sidechains; these are likely to be carboxylates, as seen in ceruloplasmin [28]. In the absence of strong donor ligands (e.g. porphyrin [35,36]), known Mn(IV) complexes are polynuclear, and are stabilized by oxide bridges [37-39]. We speculate that the MnxG holding site for Mn(III) might accommodate multiple Mn(III) ions, which could then form polynuclear Mn(IV) complexes upon electron transfer. Binuclear binding sites are common in metalloproteins. A pertinent example is Mn catalase [27,38,40], in which a pair of Mn ions cycle between Mn(II) and Mn(III) oxidation states during enzymatic turnover. (A ‘superoxidized’ form of the enzyme contains Mn(III) and Mn(IV), but is not in the catalytic cycle [40,41]).
However, if the trivalent ion holding site is displaced from the type 1 Cu, as in ceruloplasmin, then direct electron transfer from Mn(III) to Cu is unlikely. A possible pathway that circumvents this difficulty is sketched in Fig. 8. In this model, a binuclear site is envisioned adjacent to the holding site. If two Mn(III) ions fill the binuclear site, then a third Mn(III), in the holding site could serve as an electron shuttle. It could abstract an electron from the binuclear site, then migrate back to the Mn(II) site and deliver its electron to the type 1 Cu, permitting successive oxidations by O2. The driving force for electron abstraction from the binuclear Mn(III) complex would be the formation of oxide bridges from coordinated water or hydroxide ions, concerted with Mn(IV) formation. Two electron shuttle rounds would produce a [Mn(IV)2O2]4+ unit, likely bound to carboxylate sidechains, and primed to nucleate MnO2 formation. (A binuclear complex is the minimum structure for oxide bridge formation. Higher nuclearity complexes are possible. There are many synthetic examples of tri- and tetra-nuclear Mn complexes [37,42-44], and the water oxidation complex of photosystem II contains four Mn ions [37,43,45,46]).
Fig. 8. Proposed mechanism of Mn(II) oxidation and MnO2 formation catalyzed by manganese oxidase, MnxG (see text for details).

A precedent for how oxide mineral nucleation may proceed is offered by ferritin, the iron storage protein [47,48]. In ferritin, a hollow protein shell is filled with Fe2O3, after oxidation of Fe(II) at a binuclear ferroxidase site. Recent NMR experiments reveal details of how the binuclear Fe(III) intermediates that are formed at the ferroxidase site migrate along protein channels to nucleation sites within the core [49]. While bacterially produced MnO2 is not confined to a protein core, it is likely that manganese oxidases likewise offer pathways for polynuclear Mn(IV) complexes to migrate toward nucleation sites.
In this proposed mechanism, Mn can be delivered to the enzymes as either Mn(II) or as Mn(III). A conceptual issue is that the Mn(III) oxidation step is actually a disproportionation, conversion to Mn(IV) being accompanied by Mn(II) production. Why then is MnO2 not produced from exogenous Mn(III) in the absence of oxygen? The simplest explanation would be that access to the substrate site is restricted to the channel connecting it to the holding site. Mn(II) could enter through this channel, but would be unable to migrate back to solution once the holding site contains Mn(III). Thus, the disproportionation reaction would be blocked unless the resulting Mn(II) is reoxidized by O2.
An alternative possibility is that Mn(III) oxidation involves direct electron transfer to O2. If O2 could react directly with the putative binuclear Mn(III) intermediate, the products would be [Mn(IV)2O2]4+ and peroxide; the peroxide would additionally oxidize Mn(III). This scenario seems unlikely, since Mn(III) is normally unreactive with O2. Literature reports of Mn(III) oxidation by O2 have involved complexes with electron-rich organic ligands [50-53], unlike the potential ligands available in the protein. On the other hand, a mass-spectrometric oxygen isotope study [54] has indicated that about half the O atoms in bacterially produced MnO2 are derived from molecular O2. This observation is inconsistent with a purely MCO mechanism, since MCOs reduce O2 to water without releasing intermediates. The present results do not rule out a direct reaction of Mn(III) with O2, since, as noted above, azide inhibition may result from complexing at the Mn(III) site, as well as at the MCO trinuclear Cu center. Resolution of this important issue will require further experiments to distinguish reactivity at the Cu and putative Mn(III) sites.
Efforts are underway to produce purified MnxG, in order to clarify the features of this intriguing enzyme.
Conclusions
Bacterial oxidation of Mn(II), a process central to the global manganese cycle, appears to be controlled by manganese oxidases that are MCOs. Since MCOs are one-electron oxidants, this finding raises the issue of how they can catalyze the two-electron transformation of Mn(II) to MnO2. Trapping experiments with PP and exosporium from the Mn-oxidizing Bacillus sp. SG-1 have shown that Mn(III) is the initial oxidation product, consistent with its oxidase being an MCO. However, it has not been clear how the subsequent conversion of Mn(III) to MnO2 is catalyzed.
The present experiments show that this conversion requires O2, and is inhibited by azide. This finding is consistent with MCO catalysis of Mn(III) as well as Mn(II) oxidation. We present a model of how the MnxG active site might be organized to couple multiple electron transfers to the formation of MnO2-precursor polynuclear Mn(IV) complexes via a Mn(II)/Mn(III) shuttle.
However, our results do not rule out an alternative possibility, that O2 might react directly with a putative polynuclear Mn(III) complex, a process that might also be inhibited by azide. While such a reaction seems chemically unlikely, it would explain the otherwise puzzling observation that O2 is incorporated in bacterially produced MnO2. Further experiments, hopefully with purified protein, will be needed to distinguish these possibilities.
Acknowledgments
We thank Radhika Rajendran for developing the linear least squares fitting algorithm in VBA Excel and Satya Chinni for producing some of the exosporium preparations. This work was partially funded by NSF grants OCE-1031200 and OCE-1129553 to BMT.
Abbreviations
- TEM
transmission electron microscopy
- MCO
multicopper oxidase
- PP
pyrophosphate
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
References
- 1.Nelson YM, Lion LW. In: Geochemical and Hydrological Reactivity of Heavy Metals in Soils. Selim HM, Kingerly WL, editors. CRC Press; Boca Raton: 2003. [Google Scholar]
- 2.Tebo BM, Bargar JR, Clement BG, Dick GJ, Murray KJ, Parker D, Verity R, Webb SM. Annu Rev Earth Planet Sci. 2004;32:287–328. [Google Scholar]
- 3.Morgan JJ. Metal Ions in Biological Systems. Marcel Dekker; New York: 2000. [Google Scholar]
- 4.Morgan JJ. Geochim Cosmochim Acta. 2005;69:35–48. [Google Scholar]
- 5.Spiro TG, Bargar JR, Sposito G, Tebo BM. Acc Chem Res. 2010;43:2–9. doi: 10.1021/ar800232a. [DOI] [PubMed] [Google Scholar]
- 6.Brouwers GJ, Vijgenboom E, Corstjens PLAM, de Vrind JPM, de Vrind-de Jong EW. Geomicrobiol J. 2000;17:1–24. [Google Scholar]
- 7.Sunda WG, Kieber DJ. Nature. 1994;367:62–64. [Google Scholar]
- 8.Brouwers GJ, de Vrind JPM, Corstjens PLAM, Cornelis P, Baysse C, de Vrind-de Jong EW. Appl Environ Microbiol. 1999;65:1762–1768. doi: 10.1128/aem.65.4.1762-1768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Waasbergen LG, Hildebrand M, Tebo BM. J Bacteriology. 1996;178:3517–3530. doi: 10.1128/jb.178.12.3517-3530.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridge JP, Lin M, Larsen EI, Fegan M, McEwan AG, Sly LI. Environ Microbiol. 2007;9:944–953. doi: 10.1111/j.1462-2920.2006.01216.x. [DOI] [PubMed] [Google Scholar]
- 11.Larsen EI, Sly LI, McEwan AG. Arch Microbiol. 1999;171:257–264. [Google Scholar]
- 12.Francis CA, Tebo BM. Appl Environ Microbiol. 2002;68:874–880. doi: 10.1128/AEM.68.2.874-880.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francis CA, Casciotti KL, Tebo BM. Arch Microbiol. 2002;178:450–456. doi: 10.1007/s00203-002-0472-9. [DOI] [PubMed] [Google Scholar]
- 14.Okazaki M, Sugita T, Shimizu M, Ohode Y, Iwamoto K, de Vrind-de Jong EW, de Vrind JPM, Corstjens PLAM. Appl Environ Microbiol. 1997;63:4793–4799. doi: 10.1128/aem.63.12.4793-4799.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dick GJ, Torpey JW, Beveridge TJ, Tebo BM. Appl Environ Microbiol. 2008;74:1527–1534. doi: 10.1128/AEM.01240-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corstjens PLAM, de Vrind JPM, Goosen T, de Vrind-de Jong EW. Geomicrobiol J. 1997;14:91–108. [Google Scholar]
- 17.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2605. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 18.Schlosser D, Hofer C. Appl Environ Microbiol. 2002;68:3514–3521. doi: 10.1128/AEM.68.7.3514-3521.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hofer C, Schlosser D. FEBS Lett. 1999;451:186–190. doi: 10.1016/s0014-5793(99)00566-9. [DOI] [PubMed] [Google Scholar]
- 20.Webb SM, Dick GJ, Bargar JR, Tebo BM. Proc Natl Acad Sci (USA) 2005;102:5558–5563. doi: 10.1073/pnas.0409119102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Silva D, Davis-Kaplan S, Fergestad J, Kaplan J. J Biol Chem. 1997;272:14208–14213. doi: 10.1074/jbc.272.22.14208. [DOI] [PubMed] [Google Scholar]
- 22.Allendorf MD, Spira DJ, Solomon EI. Proc Natl Acad Sci (USA) 1985;82:3063–3067. doi: 10.1073/pnas.82.10.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dick GJ, Lee YE, Tebo BM. Appl Environ Microbiol. 2006;72:3184–3190. doi: 10.1128/AEM.72.5.3184-3190.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon J, Liboiron BD, Sarangi R, Hodgson KO, Hedman B, Solomon EI. Proc Natl Acad Sci (USA) 2007;104:13609–13614. doi: 10.1073/pnas.0705137104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole JL, Avigliano L, Morpurgo L, Solomon EI. J Am Chem Soc. 1991;113:9080–9089. [Google Scholar]
- 26.Hirota S, Matsumoto H, Huang HW, Sakurai T, Kitagawa T, Yamauchi O. Biochem Biophys Res Commun. 1998;243:435–437. doi: 10.1006/bbrc.1998.8108. [DOI] [PubMed] [Google Scholar]
- 27.Whittaker JW. Arch Biochem Biophys. 2012 doi: 10.1016/j.abb.2011.1012.1008. [DOI] [Google Scholar]
- 28.Lindley PF, Card G, Zaitseva I, Zaitsev V, Reinhammar B, Selin-Lindgren E, Yoshida K. J Biol Inorg Chem. 1997;2:454–463. [Google Scholar]
- 29.Machonkin TE, Solomon EI. J Am Chem Soc. 2000;122:12547–12560. [Google Scholar]
- 30.Quintanar L, Gebhard M, Wang TP, Kosman DJ, Solomon EI. J Am Chem Soc. 2004;126:6579–6589. doi: 10.1021/ja049220t. [DOI] [PubMed] [Google Scholar]
- 31.Taylor AB, Stoj CS, Ziegler L, Kosman DJ, Hart PJ. Proc Natl Acad Sci (USA) 2005;102:15459–15464. doi: 10.1073/pnas.0506227102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoj CS, Augustine AJ, Zeigler L, Solomon EI, Kosman DJ. Biochemistry. 2006;45:12741–12749. doi: 10.1021/bi061543+. [DOI] [PubMed] [Google Scholar]
- 33.Singh SK, Grass G, Rensing C, Montfort WR. J Bacteriol. 2004;186:7815–7817. doi: 10.1128/JB.186.22.7815-7817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Djoko KY, Chong LX, Wedd AG, Xiao Z. J Am Chem Soc. 2010;132:2005–2015. doi: 10.1021/ja9091903. [DOI] [PubMed] [Google Scholar]
- 35.Weiss R, Gold A, Trautwein AX, Terner J. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Academic Press; Boston: 2000. [Google Scholar]
- 36.Groves JT, Stern MK. J Am Chem Soc. 1988;110:8628–8638. [Google Scholar]
- 37.Manchanda R, Brudvig GW, Crabtree RH. Coord Chem Rev. 1995;144:1–38. [Google Scholar]
- 38.Wu AJ, Penner-Hahn JE, Pecoraro VL. Chem Rev. 2004;104:903–938. doi: 10.1021/cr020627v. [DOI] [PubMed] [Google Scholar]
- 39.Dave BC, Czernuszewicz RS. Inorg Chim Acta. 1994;227:33–41. [Google Scholar]
- 40.Waldo GS, Penner-Hahn JE. Biochemistry. 1995;34:1507–1512. doi: 10.1021/bi00005a006. [DOI] [PubMed] [Google Scholar]
- 41.Khangulov SV, Barynin VV, Voevodskaya NV, Grebenko AI. Biochim Biophys Acta. 1990;1020:305–310. [Google Scholar]
- 42.Mullins CS, Pecoraro VL. Coord Chem Rev. 2008;252:416–443. doi: 10.1016/j.ccr.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukhopadhyay S, Mandal SK, Bhaduri S, Armstrong WH. Chem Rev. 2004;104:3981–4026. doi: 10.1021/cr0206014. [DOI] [PubMed] [Google Scholar]
- 44.Bhula R, Gainsford GJ, Weatherburn DC. J Am Chem Soc. 1988;110:7550–7552. [Google Scholar]
- 45.McEnvoy JP, Brudvig GW. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 46.Cady CW, Crabtree RH, Brudvig GW. Coord Chem Rev. 2008;252:444–455. doi: 10.1016/j.ccr.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Theil EC. Acc Chem Res. 2005;38:167–175. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- 48.Ha Y, Shi D, Small GW, Theil EC, Allewell NM. J Biol Inorg Chem. 1999;4:243–256. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- 49.Turano P, Lalli D, Felli IC, Theil EC, Bertini I. Proc Natl Acad Sci (USA) 2010;107:545–550. doi: 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacDonnell FM, Fackler NLP, Stern C, O'Halloran TV. J Am Chem Soc. 1994;116:7431–7432. [Google Scholar]
- 51.Bossek U, Weyhermuller T, Wieghardt K, Nuber B, Weiss J. J Am Chem Soc. 1990;112:6387–6388. [Google Scholar]
- 52.Larson E, Soo Lah M, Li X, Bonadies JA, Pecoraro VL. Inorg Chem. 1992;31:373–378. [Google Scholar]
- 53.Chandra SK, Chakravorty A. Inorg Chem. 1992;31:760–765. [Google Scholar]
- 54.Mandernack KW, Fogel ML, Tebo BM, Usui A. Geochim Cosmochim Acta. 1995;59:4409–4425. [Google Scholar]