

Abstract
Background and Objectives
Several studies have suggested a difference in clinical features of intellectual ability and psychiatric illness in the Prader–Willi syndrome (PWS) with the 15q11-q13 paternal deletion and maternal uniparental disomy (mUPD). Our objective was to appraise evidence on this association through a meta-analysis.
Methods
The electronic records PubMed and EMBASE from 1956 to 2012 were extracted for meta-analysis. Meta-analyses were performed by using fixed effect model. Mean difference, odds ratio, and 95% confidence interval were calculated.
Results
We retrieved a total of 744 PWS cases from 13 studies. These include 423 cases with paternal 15q11-q13 deletions and 318 cases of mUPD. Compare to the PWS cases with mUPD, PWS patients with the paternal 15q11-q13 deletion associated with significantly lower full scale IQ (FSIQ) [mean difference (MD), -2.69; 95%CI, -4.86 to -0.52; p=0.02] and verbal IQ (VIQ) (MD, -7.5; 95%CI, -9.75 to -5.26; p<0.00001) but higher performance IQ (PIQ) (MD, 4.02; 95%CI, 1.13 to 6.91; p=0.006). In contrast, PWS patients with mUPD are associated with significantly higher risk of psychiatric illness [odds rate (OR), 0.14; 95%CI, 0.08 to 0.23; p<0.00001] and higher risk of bipolar disorder (OR, 0.04; 95%CI, 0.01 to 0.23; p=0.0002).
Conclusions
Significant different clinical features of cognitive development and psychiatric illness are associated with PWS with different molecular defects. These findings provide support for evidence based practice to evaluate and manage the PWS syndrome with different molecular defects.
Introduction
Prader–Willi syndrome (PWS) is a genomic imprinting disorder caused by deficiency of paternally expressed gene or genes in the chromosome 15q11-13 region [1–4]. Most patients are caused by a paternal deletion on the chromosome 15q11–q13 (70%-75%) and a maternal uniparental disomy (mUPD) of chromosome 15 (25%-29%). The imprinting center defect is found in less than 1% of PWS cases. The estimated prevalence of PWS is 1:10,000 to 1:30,000 [5–7]. The clinical features of PWS include neonatal hypotonia, feeding difficulty, respiratory problem at birth, hypogonadism, cognitive disabilities, hyperphagia, childhood onset obesity, obsessive and compulsive behaviors, and psychiatric illness in adult [8,9]. The diagnostic criteria for the Prader–Willi syndrome (PWS) developed in 1993 have proven to be a useful tool in clinic setting [10]. However, the confirmation of clinical diagnosis usually relies on the molecular genetic testing.
Although the majority of clinical features of PWS are well recognized and characterized, the behavioral features associated with PWS has not been fully investigated. The number of new PWS cases, caused by either paternal deletion of 15q11-q13 or mUPD, has been increased significantly in recent years due to available array CGH (comparative genomic hybridization) technique in clinical diagnostic laboratory. The different clinical presentations related to behaviors and mental health have been suggested between individuals with paternal deletion of 15q11-q13 and mUPD [11–13]. These differences appear more prominent in the domains of the neurodevelopment and psychiatric presentations. Individuals with mUPD are more likely to have psychosis [14,15], autism spectrum disorders [16–19], and milder intellectual disability than other causes [20–22]. Individuals with paternal deletion of 15q11-q13 showed a higher frequency of impaired speech articulation and language development [23].
There is, however, no consensus as to whether the difference in the neurodevelopment outcomes between 15q11-q13 paternal deletion and mUPD are evident and clinically significantly. This information, however, is important to counsel family and develop anticipatory care. Therefore, we reviewed published literature on this topic systematically and conducted a meta-analysis to evaluate the level of intellectual disability and frequency of psychiatric illness and their correlations between PWS cases caused by the 15q11-q13 paternal deletion and mUPD.
Methods
Literature Search
A comprehensive literature search was performed to identify the relevant studies in PubMed, the Cochrane Central Register of Controlled Trials and EMBASE from 1956 to May 31, 2012. Using Medical Subject Headings terms, searches were limited to original studies in human. The keyword of Prader–Willi syndrome was used in combination with deletion or UPD.
Study Selection and Data Extraction
The Endnote X4 and NoteExpress2 software were used to compile the data for meta-analysis. The PWS cases confirmed molecularly with either paternal deletion or mUPD were included for meta-analysis. We contacted corresponding authors of the studies for additional information as needed. The studies reported more than one of the outcomes of neurodevelopment were included in the metaanalysis: 1) IQ: full scale IQ (FSIQ), verbal IQ (VIQ) and performance IQ (PIQ); 2) psychiatric illness including depressive psychosis, bipolar disorder or other psychotic illness. The studies were excluded for meta-analysis if required data were incomplete. Two reviewers independently screened all studies for those requiring further retrieval (full text or abstract), and then independently reviewed these studies to identify whether they met the inclusion criteria. Disagreements regarding trial eligibility were discussed and resolved by two reviewers.
Statistical Analysis
We report outcomes using odds rate (OR) with 95% confidence intervals (CIs) for dichotomous data and weighted mean difference (MD) with 95% CI for continuous data. I 2 statistics were used to measure heterogeneity of the studies. If the I 2 value was less than 50%, a fixed effects meta-analysis was applied. If the I 2 value was more than 50%, the random-effects meta-analysis was performed. Potential publication bias was investigated by visual assessment of the funnel plot (plots of effect estimates against sample size). Metaanalyses and funnel plot calculation were performed using Review Manager software version 5.1 (Nordic Cochrane Centre, Copenhagen, Denmark). Statistical significance was determined if the 2-sided P value was less than 0.05.
Results
Search Results and Study Characteristics
A total of 581 records were identified from the electronic database, 331 records from PubMed, 250 records from EMBASE. Seventy-seven reviews, 24 animal studies, and 71 single case reports were immediately removed. A total of 22 articles were removed due to language barrier and 131 articles without Prader-willi in their title were removed. Ninety three studies were duplicated in two databases. One hundred fifty studies did not meet the inclusion criteria or because of incomplete data in the papers. After these careful filtering, a total of 13 studies [11,19,21,24–33] met the inclusion criteria of the meta-analyses. Flow chart of articles screening is presented in Figure 1. There are 744 PWS cases in 13 studies. Among them, 423 PWS patients with 15q11-q13 paternal deletion (56.9%), 318 with mUPD (42.7%), 3 with imprinting center defect. The distribution of three molecular defects is consistent with other studies in general but with a slightly higher percentage of mUPD [34]. The characteristics of included studies are available in Table 1. Six studies [11,21,27,30,32,33] were carried out in the USA, four [26,28,29,31] in the UK, two [24,25] in Netherlands, one [19] in Belgium. Of the studies included in the systematic review, 11 studies [11,19,21,26–33] reported IQ score (Table 2), 5 of these studies [21,26,27,29,30] were using the Wechsler Intelligence Scale, 1 study [11] was using the K-BIT scale, 1 study [28] was using Wechsler and Raren’s, 1 study [33] was using Kaufman or Wechsler, and 3 studies [19,31,32] did not provide detail description for tools used. Five studies [11,24–26,31] reported psychiatric illness: 3 studies [24,26,31] used the ICD-10 criteria for the diagnosis and 2 studies [11,25] used psychiatric diagnosis in medical records. The clinical diagnosis in all cases were confirmed by molecular genetic testing using DNA methylation analysis, fluorescence in situ hybridization (FISH), DNA microsatellite analysis or multiplex ligation-dependent probe amplification (MLPA). In 3 studies [11,27,30], the paternal deletions of 15q11-q13 are further divided into class I and class II deletion II based on the genomic locations of breakpoints as previously described [30,35]. Weighted average method was used to combine the IQ scores of the two deletion subgroups.
Figure 1. Flow chart of articles screening and selection process.

Table 1. Descriptive characteristics of the included studies.
| Author | Year | Country of study | Case Number | Male | Deletion | mUPD | Age (y) M+SD (Range) | PWS diagnosis | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Sinnema et al. [24] | 2011 | Netherlands | 97 | NA | 53 | 44 | 36.2+12.4 (18-66) | MLPA | Psychiatry |
| Mass et al. [25] | 2010 | Netherlands | 79 | 34 | 45 | 33 | 34.4+11.8 (18-65) | DNA methylation studies on the SNURF/SNRPN locus | Psychiatry |
| Dykens et al. [11] | 2008 | USA | 88 | 43 | 55 | 33 | 22.41+11.74 (5-51) | FISH, methylation studies, or MLPA/MS-MLPA | Psychiatry, IQ |
| Soni et al. [26] | 2008 | UK | 46 | 21 | 24 | 22 | 31.2+9.6 (12-51) | microsatellite analysis | Psychiatry, IQ |
| Zarcone et al. [27] | 2007 | USA | 73 | 20 | 42 | 31 | 22.7+9.4 (10-40) | FISH and DNA microsatellite analysis | IQ |
| Descheemaeker et al. [19] | 2006 | Belgium | 59 | 31 | 40 | 18 | 21.2 (2-51) | chromosome examination and DNA methylation | IQ |
| Milner et al. [28] | 2005 | UK | 96 | 51 | 47 | 49 | 16.3+12.4 (3.3-50) | quantitative fluorescent PCR assay, microsatellite analysis | IQ |
| Walley et al. [29] | 2005 | UK | 18 | 11 | 12 | 6 | 23.5+7.8 (16-49) | molecular genetic testing, no detail | IQ |
| Bulter et al. [30] | 2004 | USA | 46 | 21 | 25 | 21 | 23+8.6 | FISH, methylation and microsatellite analysis | IQ |
| Boer et al. [31] | 2002 | UK | 15 | 8 | 9 | 5 | 38.5+5.7 (29-47) | methylation pattern | Psychiatry, IQ |
| FOX et al. [32] | 2001 | USA | 43 | NA | 24 | 19 | 23.3+8.7 | FISH, methylation and microsatellite analysis | IQ |
| Roof et al. [21] | 2000 | USA | 38 | 16 | 24 | 14 | 22.2+9.1 (10-44) | high-resolution chromosome analysis, DNA microsatellite analysis | IQ |
| Dykens et al. [33] | 1999 | USA | 46 | 18 | 23 | 23 | 17+9.42 (6-42) | molecular genetic testing, no detail | IQ |
Note: MLPA, multiplex ligation-dependent probe amplification; FISH, fluorescence in situ hybridization; NA: not available.
Table 2. Characteristics of the IQ reported studies.
| Author | Year | Diagnostic criteria | Perform | FSIQ |
VIQ |
PIQ |
|||
|---|---|---|---|---|---|---|---|---|---|
| DEL (M, SD) | UPD (M, SD) | DEL (M, SD) | UPD (M, SD) | DEL (M, SD) | UPD (M, SD) | ||||
| Dykens et al. | 2008 | K-BIT2 | NA | 62.07, 12.3 (n=55) | 64.96, 13.07 (n=33) | - | - | - | - |
| Soni et al. | 2008 | Wechsler | carry out by the first author | 64.5,8.5 (n=24) | 68.7,11.2 (n=22) | - | - | - | - |
| Zarcone et al. | 2007 | Wechsler | Trained research staff conducted IQ test | 62.3, 55.1 (n=41) | 65.2, 12.1 (n=30) | 63.3, 9.3 (n=41) | 71.3, 11.9 (n=30) | 65.7, 9.3 (n=41) | 63.1, 11.3 (n=30) |
| Descheemaeker et al. | 2006 | NA | NA | 57.2, 16.21 (n=40) | 58.4, 15.56 (n=18) | - | - | - | - |
| Milner et al. | 2005 | Wechsler and Raven’s | the researchers involved were blind to the genetic status of participants. | 70.78, 16.21 (n=45) | 69.49, 16.89 (n=47) | 81.16, 17.4 (n=19) | 79.62, 21.08 (n=21) | 76.53, 14.43 (n=19) | 68.57, 10.44 (n=21) |
| Walley et al. | 2005 | Wechsler | NA | - | - | 71.64, 12.08 (n=12) | 76.50, 8.64 (n=6) | - | - |
| Bulter et al. | 2004 | Wechsler | NA | - | - | 62.52, 9.18 (n=25) | 70, 6.20 (n=21) | - | - |
| Boer et al. | 2002 | NA | NA | 67.0, 12.39 (n=9) | 62.4, 9.86 (n=5) | - | - | - | - |
| Fox et al. | 2001 | NA | NA | 62.7, 9.8 (n=24) | 63.9, 6.4 (n=19) | 62.1, 9.5 (n=24) | 69.7, 5.5 (n=19) | 66.5, 9.6 (n=24) | 61.6, 7.7 (n=19) |
| Roof et al. | 2000 | Wechsler | carry on by a licensed psychological examiner | 61, 9.2 (n=24) | 64.1, 7.9 (n=14) | 60.8, 8.6 (n=24) | 69.9, 6.4 (n=14) | 64.7, 9.3 (n=24) | 62.2, 9.7 (n=14) |
| Dykens et al. | 1999 | Kaufman or Wechsler | two of the university clinics | 62.97, 8.83 (n=23) | 70.93, 11.15 (n=23) | - | - | - | - |
NA: not available
Results of Meta-analysis
FSIQ in the paternal deletion and mUPD subgroup
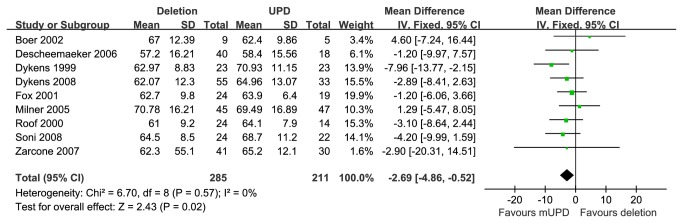
Funnel plot for publication bias about FSIQ is presented in Figure 2. Nine studies [11,19,21,26–28,31–33] provided FSIQ scores of PWS patients with 15q11-q13 paternal deletion or mUPD. These studies encompassed 285 cases with the paternal deletion and 211 cases with mUPD. Compared with two molecular groups, the PWS cases with deletion have statistically significant lower FSIQ scores than that of mUPD [MD =-2.69 (95% confidence interval (CI), -4.86 to -0.52; p=0.02)] and results were homogeneously distributed (p=0.57) (Figure 3).
Figure 2. Funnel plot for publication bias about FSIQ.

Figure 3. Meta analysis of the FSIQ in DEL and UPD groups.
VIQ in paternal deletion and mUPD subgroups
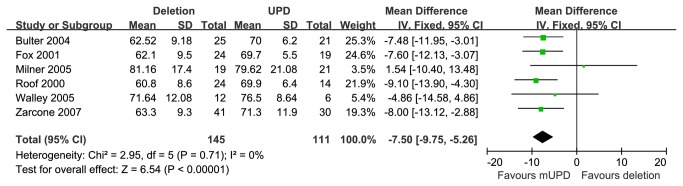
Six studies [21,27–30,32] provided VIQ scores of PWS patients with 15q11-q13 paternal deletion or mUPD. These studies included 145 cases with paternal deletion and 111 cases with mUPD. Compared these subgroups, the PWS cases with deletion showed statistically significant lower VIQ scores than that of mUPD [MD=-7.50 (95% CI, -9.75 to -5.26; p<0.00001)] and results were homogeneously distributed (p=0.71) (Figure 4).
Figure 4. Meta analysis of the VIQ in DEL and UPD groups.
PIQ in paternal deletion and mUPD subgroups
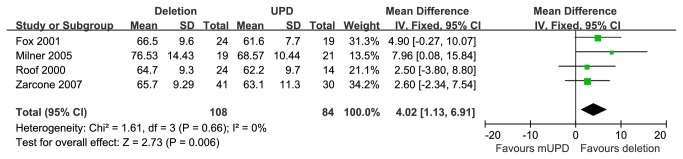
Four studies [21,27,28,32] provided PIQ scores for PWS patients with 15q11-q13 paternal deletion or mUPD. These studies encompassed 108 cases with the paternal deletion and 84 cases with mUPD. Compared with these two subgroups, those with paternal deletion showed statistically significant higher PIQ scores than that of mUPD, [MD=4.02 (95% CI, 1.13 to 6.91; p=0.006)] and results were homogeneously distributed (p=0.66) (Figure 5).
Figure 5. Meta analysis of the PIQ in DEL and UPD groups.
Psychosis in paternal deletion and mUPD subgroups
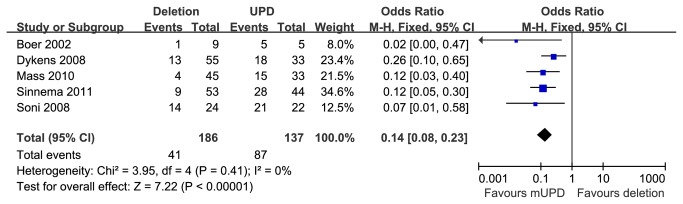
Funnel plot for publication bias about psychiatric illness is presented in Figure 6. Five studies [11,24–26,31] reported prevalence of psychosis in PWS patients with 15q11-q13 paternal deletion or mUPD. These studies encompassed 186 cases with the paternal deletion and 137 cases with mUPD. Compared with the two subtypes, individuals with mUPD were statistically significant more susceptible to psychotic disorder than those with deletion, [OR=0.14 (95% CI, 0.08 to 0.23; p<0.00001)] and results were homogeneously distributed (p=0.41) (Figure 7).
Figure 6. Funnel plot for publication bias about psychosis.

Figure 7. Metaanalysis of the prevalence of psychosis in DEL and UPD groups.
Depressive psychosis in paternal deletion and mUPD subgroups
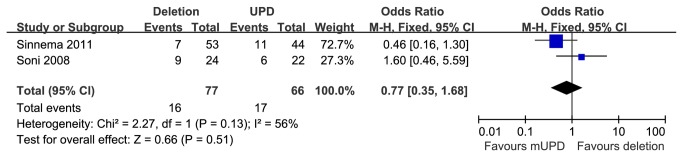
Two studies [24,26] reported prevalence of depressive psychosis in PWS patients with 15q11-q13 paternal deletion or mUPD. These studies encompassed 77 cases with the paternal deletion and 66 cases with mUPD. The frequency of depressive psychosis between the genetic subtypes were not statistically significant [OR=0.77 (95% CI, 0.35 to 1.68; p=0.51)] and results were homogeneously distributed (p=0.13) (Figure 8).
Figure 8. Metaanalysis of the prevalence of depression in DEL and UPD groups.
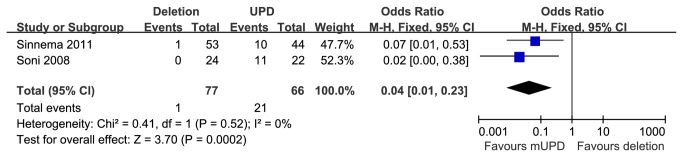
Bipolar disorder in paternal deletion and mUPD subgroups
Two studies [24,26] reported prevalence of bipolar illness in PWS patients with 15q11-q13 paternal deletion or mUPD. These studies encompassed 77 cases with the paternal deletion and 66 cases with mUPD. Compared with the two genetic subsets, individuals with mUPD were statistically significant more susceptible to bipolar illness than those with deletion, [OR=0.04 (95% CI, 0.01 to 0.23; p=0.0002)] and results were homogeneously distributed (p=0.52) (Figure 9).
Figure 9. Metaanalysis of the prevalence of bipolar in DEL and UPD groups.
Discussion
To our knowledge, this is the first meta-analysis to review the psychiatric illness and intellectual abilities in PWS with different types of molecular defects. Our analysis indicates significant differences of clinical presentation related to the neurodevelopment and psychiatric illness between PWS cases with 15q11-q13 paternal deletion and mUPD. Overall, the PWS with paternal deletion have lower FSIQ score than that of mUPD. More specifically, the verbal IQ but not performance IQ is more affected in the cases with 15q11-q13 deletion. Conversely, the psychosis and bipolar disorders are significantly more prevalent in PWS cases with mUPD than that of 15q11-q13 deletion. These findings not only strengthen the previous impression from small scale studies but also provide new insight that has not reported before.
The immediate question is what contributes to these differences between cases with paternal deletion and mUPD. Human chromosome 15q11–q13 is a domain subject to regulation of genomic imprinting, an epigenetic process in which the expression of genes is depending upon the parent-of-origin [36]. It is well known that PWS primarily arises from the deficiency of paternally expressed gene or genes in the 15q11-q13 region. However, it remains a question of debate which genes or genes are responsible for the full spectrum of PWS phenotypes. Three recent reports from analysis of 3 PWS cases have revealed small but overlapping microdeletions including the SNORD116 snoRNA cluster [37–39]. These cases have typical clinical features of PWS. However, the neurobehavioral studies have not been evaluated or described in detail. These data support that the paternally expressed SNORD116 cluster plays a major role in the core features of PWS. However, how the loss of expression non-coding snoRNAs ultimately leads to the clinical presentations of PWS remains unclear. The molecular difference between paternal deletion and mUPD is clear. Summary of the genetic and expression of chromosomal region 15q11, 2-13 are presented in Table 3. In the cases of paternal deletion, the paternally expressed genes in the 15q11-q13 region are predicted to be completely deficient. The maternally expressed genes such as UBE3A and ATP10A are not affected. The non-imprinted genes in the regions are haploinsufficient in paternal deletion cases. In the mUPD cases, the maternally expressed genes are predicted to be two folds of that in paternal deletion cases and one fold higher than that in normal. The non-imprinted genes are not affected. The expression of imprinted gene is expected to be same as paternal deletion cases if the imprinting is complete or 100%. It is reasonable to hypothesize that the differential expression of genes between paternal deletion of 15q11-q13 and mUPD may contribute to the difference in neurocognitive function and prevalence of psychiatric illness. For instance, the higher expression of UBE3A in mUPD cases may play a significant role in psychiatric illness. UBE3A is a maternal and brain-specific imprinting gene in the region [40,41]. UBE3A encodes ubiquitin protein ligase E3A, functions as an E3 ligase in the ubiquitin proteasome pathway and as a transcriptional coactivator [42]. Loss of function of maternal UBE3A causes Angelman syndrome, a neurodevelopmental disorder with intellectual disability, hypotonia and seizures [43,44]. The maternal duplication of 15q11-q13 is one of the most common copy number variants found in autism spectrum disorder [45]. It has been suggested that the UBE3A may be the major player to contribute the ASD in these cases [46,47]. Other gene such as CYFIP1 in the 15q11.2 region may also play a role in contributing to neurodevelopment and psychiatric illness because of copy number variant of 15q11.2 including CYFIP1 has been associated with schizophrenia [48]. CYFIP1 encodes cytoplasmic FMR1 interacting protein 1, and component of the CYFIP1-EIF4E-FMR1 complex which binds to the mRNA cap and mediates translational repression [49].
Table 3. Summary of the genetic and expression of chromosomal region 15q11, 2-13.
| Gene name | Description | Imprinting status | Expression level (Deletion) | Expression level (mUPD) | Comparison in two subtypes |
|---|---|---|---|---|---|
| TUBGCP5 | tubulin, gamma complex associated protein 5 | non-imprinted | haploinsufficiency | normal | different |
| CYFIP1 | cytoplasmic FMR1 interacting protein 1 | non-imprinted | haploinsufficiency | normal | different |
| NIPA2 | non imprinted in Prader-Willi/Angelman syndrome 2 | non-imprinted | haploinsufficiency | normal | different |
| NIPA1 | non imprinted in Prader-Willi/Angelman syndrome 1 | non-imprinted | haploinsufficiency | normal | different |
| GOLGA8E | golgin A8 family, member E, pseudogene | non-imprinted | haploinsufficiency | normal | different |
| MKRN3 | makorin ring finger protein 3 | paternally expressed | no expression | no expression | same |
| MAGEL2 | MAGE-like 2 | paternally expressed | no expression | no expression | same |
| NDN | necdin, melanoma antigen (MAGE) family member | paternally expressed | no expression | no expression | same |
| NPAP1 | nuclear pore associated protein 1 | non-imprinted | haploinsufficiency | normal | different |
| SNRPN | small nuclear ribonucleoprotein polypeptide N | paternally expressed | no expression | no expression | same |
| SNURF | SNRPN upstream reading frame | paternally expressed | no expression | no expression | same |
| snoRNAs | small nucleolar RNA | paternally expressed | no expression | no expression | same |
| UBE3A | ubiquitin protein ligase E3A | maternally expressed | normal | over expression | different |
| ATP10A | ATPase, class V, type 10A | maternally expressed | normal | over expression | different |
| GABRB3 | gamma-aminobutyric acid (GABA) A receptor, beta 3 | paternal biased expression | haploinsufficiency | haploinsufficiency | same |
| GABRA5 | gamma-aminobutyric acid (GABA) A receptor, alpha 5 | paternal biased expression | haploinsufficiency | haploinsufficiency | same |
| GABRG3 | gamma-aminobutyric acid (GABA) A receptor, gamma 3 | non-imprinted | haploinsufficiency | normal | different |
| OCA2 | oculocutaneous albinism II | non-imprinted | haploinsufficiency | normal | different |
| GOLGA8G | golgin A8 family, member G | non-imprinted | haploinsufficiency | normal | different |
The clinical implication of our finding is significant because it provides a basis to develop evidence based practice guideline for evaluating and managing individuals with PWS. In particular, our findings provide evidence to support more diligent care of PWS with psychiatric illness.
Our conclusion from the meta-analysis is convincing and significant. However, several imitations in our study may still be worth for discussion and for improvement in future. First, like all other meta-analysis, because clinic data were collected from different investigators, at different sites, and at different ages, the comparison may be confounded by the variable quality of clinical data. Second, it should be noted that the diagnosis of psychiatric diseases in many reports included in meta-analysis might be made based on self-report but not through a specialized clinical evaluation using DSM IV criteria. These limitations may influence the data quality thereby affect the conclusion. Therefore, more accurate and detailed description about psychiatric symptoms is recommended for further study of PWS to figure out underlying molecular mechanism. Third, although the total number of patients is reasonable, the increase of the sample size may still be desirable in future study. Fourth, the breakpoints for the deletion case are not defined in detail.
Conclusion
Our meta-analysis has revealed significant differences in the severity of neurocognitive impairment and the prevalence of psychiatric illness between PWS cases with paternal deletion of 15q11-q13 and mUPD. The PWS with paternal deletion have lower FSIQ and VIQ score than that of mUPD, but PWS with mUPD are more susceptible to psychosis, bipolar disorders than that of paternal deletion. These findings may provide a framework for future study to elucidate the genetic basis of psychiatric illness. Our finding will also provide rationale to develop evidence based practice for evaluating and caring individuals with PWS.
Supporting Information
PRISMA 2009 Checklist.
(DOC)
Funding Statement
Grant sponsor: The National Basic Research Program (The National 973 Project) (No. 2009CB941704). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS et al. (1981) Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med 304: 325-329. doi:10.1056/NEJM198102053040604. PubMed: 7442771. [DOI] [PubMed] [Google Scholar]
- 2. Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M (1989) Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 342: 281-285. doi:10.1038/342281a0. PubMed: 2812027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S et al. (1995) Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet 9: 395-400. doi:10.1038/ng0495-395. PubMed: 7795645. [DOI] [PubMed] [Google Scholar]
- 4. Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M (2008) Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab 93: 4183-4197. doi:10.1210/jc.2008-0649. PubMed: 18697869. [DOI] [PubMed] [Google Scholar]
- 5. Butler MG (1990) Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet 35: 319-332. doi:10.1002/ajmg.1320350306. PubMed: 2309779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burd L, Vesely B, Martsolf J, Kerbeshian J (1990) Prevalence study of Prader-Willi syndrome in North Dakota. Am J Med Genet 37: 97-99. doi:10.1002/ajmg.1320370122. PubMed: 2240051. [DOI] [PubMed] [Google Scholar]
- 7. Whittington JE, Holland AJ, Webb T, Butler J, Clarke D et al. (2001) Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet 38: 792-798. doi:10.1136/jmg.38.11.792. PubMed: 11732491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB (2001) The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 108: E92. doi:10.1542/peds.108.5.e92. PubMed: 11694676. [DOI] [PubMed] [Google Scholar]
- 9. Horsthemke B, Wagstaff J (2008) Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A 146A: 2041-2052. doi:10.1002/ajmg.a.32364. PubMed: 18627066. [DOI] [PubMed] [Google Scholar]
- 10. Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR et al. (1993) Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 91: 398-402. PubMed: 8424017. [PMC free article] [PubMed] [Google Scholar]
- 11. Dykens EM, Roof E (2008) Behavior in Prader-Willi syndrome: relationship to genetic subtypes and age. J Child Psychol Psychiatry 49: 1001-1008. doi:10.1111/j.1469-7610.2008.01913.x. PubMed: 18665884. [DOI] [PubMed] [Google Scholar]
- 12. Grugni G, Giardino D, Crinò A, Malvestiti F, Ballarati L et al. (2011) Growth hormone secretion among adult patients with Prader-Willi syndrome due to different genetic subtypes. J Endocrinol Invest 34: 493-497. PubMed: 20651469. [DOI] [PubMed] [Google Scholar]
- 13. Bonati MT, Russo S, Finelli P, Valsecchi MR, Cogliati F et al. (2007) Evaluation of autism traits in Angelman syndrome: a resource to unfold autism genes. Neurogenetics 8: 169-178. doi:10.1007/s10048-007-0086-0. PubMed: 17415598. [DOI] [PubMed] [Google Scholar]
- 14. Holland AJ, Whittington JE, Butler J, Webb T, Boer H et al. (2003) Behavioural phenotypes associated with specific genetic disorders: evidence from a population-based study of people with Prader–Willi syndrome. Psychol Med 33: 141-153. PubMed: 12537045. [DOI] [PubMed] [Google Scholar]
- 15. Soni S, Whittington J, Holland AJ, Webb T, Maina E et al. (2007) The course and outcome of psychiatric illness in people with Prader-Willi syndrome: implications for management and treatment. J Intellect Disabil Res 51: 32-42. doi:10.1111/j.1365-2788.2006.00895.x. PubMed: 17181601. [DOI] [PubMed] [Google Scholar]
- 16. Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J et al. (2004) Prader–Willi syndrome - A study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry 13: 42-50. doi:10.1007/s00787-004-0354-6. PubMed: 14991431. [DOI] [PubMed] [Google Scholar]
- 17. Veltman MW, Craig EE, Bolton PF (2005) Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet 15: 243-254. doi:10.1097/00041444-200512000-00006. PubMed: 16314754. [DOI] [PubMed] [Google Scholar]
- 18. Whittington J, Holland A, Webb T, Butler J, Clarke D et al. (2004) Cognitive abilities and genotype in a population-based sample of people with Prader–Willi syndrome. J Intell Disabil Res 48: 172-187. doi:10.1111/j.1365-2788.2004.00556.x. [DOI] [PubMed] [Google Scholar]
- 19. Descheemaeker MJ, Govers V, Vermeulen P, Fryns JP (2006) Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A 140: 1136-1142. PubMed: 16646032. [DOI] [PubMed] [Google Scholar]
- 20. Dykens EM, Cassidy SB, King BH (1999) Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am J Ment Retard 104: 67-77. doi:10.1352/0895-8017(1999)104. PubMed: 9972835. [DOI] [PubMed] [Google Scholar]
- 21. Roof E, Stone W, MacLean W, Feurer ID, Thompson T et al. (2000) Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res 44(1): 25-30. doi:10.1046/j.1365-2788.2000.00250.x. PubMed: 10711647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hartley SL, MacLean WE, Butler MG, Zarcone J, Thompson T (2005) Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am J Med Genet A 136A: 140-145. doi:10.1002/ajmg.a.30771. PubMed: 15940679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Torrado M, Araoz V, Baialardo E, Abraldes K, Mazza C et al. (2007) Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A 143: 460-468. PubMed: 17163531. [DOI] [PubMed] [Google Scholar]
- 24. Sinnema M, Boer H, Collin P, Maaskant MA, van Roozendaal KEP et al. (2011) Psychiatric illness in a cohort of adults with Prader-Willi syndrome. Res Dev Disabil 32: 1729-1735. doi:10.1016/j.ridd.2011.02.027. PubMed: 21454045. [DOI] [PubMed] [Google Scholar]
- 25. Maas APHM, Sinnema M, Didden R, Maaskant MA, Smits MG et al. (2010) Sleep disturbances and behavioural problems in adults with Prader-Willi syndrome. J Intell Disabil Res 54: 906-917. doi:10.1111/j.1365-2788.2010.01306.x. PubMed: 20636465. [DOI] [PubMed] [Google Scholar]
- 26. Soni S, Whittington J, Holland AJ, Webb T, Maina EN et al. (2008) The phenomenology and diagnosis of psychiatric illness in people with Prader-Willi syndrome. Psychol Med 38: 1505-1514. PubMed: 18177526. [DOI] [PubMed] [Google Scholar]
- 27. Zarcone J, Napolitano D, Peterson C, Breidbord J, Ferraioli S et al. (2007) The relationship between compulsive behaviour and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J Intell Disabil Res 51: 478-487. doi:10.1111/j.1365-2788.2006.00916.x. PubMed: 17493030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Milner KM, Craig EE, Thompson RJ, Veltman MW, Thomas NS et al. (2005) Prader-Willi syndrome: intellectual abilities and behavioural features by genetic subtype. J Child Psychol Psychiatry 46: 1089-1096. doi:10.1111/j.1469-7610.2005.01520.x. PubMed: 16178933. [DOI] [PubMed] [Google Scholar]
- 29. Walley RM, Donaldson MD (2005) An investigation of executive function abilities in adults with Prader-Willi syndrome. J Intell Disabil Res 49: 613-625. doi:10.1111/j.1365-2788.2005.00717.x. PubMed: 16011554. [DOI] [PubMed] [Google Scholar]
- 30. Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T (2004) Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 113: 565-573. doi:10.1542/peds.113.3.565. PubMed: 14993551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boer H, Holland A, Whittington J, Butler J, Webb T et al. (2002) Psychotic illness in people with Prader Willi syndrome due to chromosome 15 maternal uniparental disomy. Lancet 359: 135-136. doi:10.1016/S0140-6736(02)07340-3. PubMed: 11809260. [DOI] [PubMed] [Google Scholar]
- 32. Fox R, Yang GS, Feurer ID, Butler MG, Thompson T (2001) Kinetic form discrimination in Prader-Willi syndrome. J Intell Disabil Res 45: 317-325. doi:10.1046/j.1365-2788.2001.00326.x. PubMed: 11489053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dykens EM, Cassidy SB, King BH (1999) Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am J Ment Retard 104: 67-77. doi:10.1352/0895-8017(1999)104. PubMed: 9972835. [DOI] [PubMed] [Google Scholar]
- 34. Mitchell J, Schinzel A, Langlois S, Gillessen-Kaesbach G, Schuffenhauer S et al. (1996) Comparison of phenotype in uniparental disomy and deletion Prader-Willi syndrome: sex specific differences. Am J Med Genet 65: 133-136. doi:10.1002/(SICI)1096-8628(19961016)65:2. PubMed: 8911605. [DOI] [PubMed] [Google Scholar]
- 35. Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR et al. (1995) Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet 57: 40-48. PubMed: 7611294. [PMC free article] [PubMed] [Google Scholar]
- 36. Ferguson-Smith AC (2011) Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet 12: 565-575. doi:10.1038/nrm3175. PubMed: 21765458. [DOI] [PubMed] [Google Scholar]
- 37. Sahoo T, Del GD, German JR, Shinawi M, Peters SU et al. (2008) Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 40: 719-721. doi:10.1038/ng.158. PubMed: 18500341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE et al. (2009) A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet 18: 3257-3265. doi:10.1093/hmg/ddp263. PubMed: 19498035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S et al. (2010) Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 18: 1196-1201. doi:10.1038/ejhg.2010.102. PubMed: 20588305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hogart A, Leung KN, Wang NJ, Wu DJ, Driscoll J et al. (2009) Chromosome 15q11-13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet 46: 86-93. PubMed: 18835857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet 17: 111-118. PubMed: 17940072. [DOI] [PubMed] [Google Scholar]
- 42. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75: 495-505. doi:10.1016/0092-8674(93)90384-3. PubMed: 8221889. [DOI] [PubMed] [Google Scholar]
- 43. Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH et al. (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15: 74-77. doi:10.1038/ng0197-74. PubMed: 8988172. [DOI] [PubMed] [Google Scholar]
- 44. Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15: 70-73. doi:10.1038/ng0197-70. PubMed: 8988171. [DOI] [PubMed] [Google Scholar]
- 45. Cook EJ, Lindgren V, Leventhal BL, Courchesne R, Lincoln A et al. (1997) Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet 60: 928-934. PubMed: 9106540. [PMC free article] [PubMed] [Google Scholar]
- 46. Smith SE, Zhou YD, Zhang GP, Jin Z, Stoppel DC et al. (2011) Increased Gene Dosage of Ube3a Results in Autism Traits and Decreased Glutamate Synaptic Transmission in Mice. Sci Transl Med 3:103ra97 PubMed: 21974935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hogart A, Leung KN, Wang NJ, Wu DJ, Driscoll J et al. (2009) Chromosome 15q11-13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J Med Genet 46: 86-93. PubMed: 18835857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. St CD (2009). Copy number variation and schizophrenia. Schizophr Bull 35: 9-12 [Google Scholar]
- 49. Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL (2001) A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci U S A 98: 8844-8849. doi:10.1073/pnas.151231598. PubMed: 11438699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PRISMA 2009 Checklist.
(DOC)